
Abstract
Genetic diversity of Cambodian melons was evaluated by the analysis of 12 random amplified polymorphic DNA (RAPD) and 7 simple sequence repeat (SSR) markers using 62 accessions of melon landraces and compared with 231 accessions from other areas for genetic characterization of Cambodian melons. Among 62 accessions, 56 accessions were morphologically classified as small-seed type with seed lengths shorter than 9 mm, as in the horticultural groups Conomon and Makuwa. Gene diversity of Cambodian melons was 0.228, which was equivalent to those of the groups Conomon and Makuwa and smaller than those of Vietnamese and Central Asian landraces. A phylogenetic tree constructed from a genetic distance matrix classified 293 accessions into three major clusters. Small-seed type accessions from East and Southeast Asia formed clusters I and II, which were distantly related with cluster III consisting of large-seed type melon from other areas. All Cambodian melons belonged to cluster I (except three accessions) along with those from Thailand, Myanmar, Yunnan (China), and Vietnam (“Dua thom” in the northwest), thus indicating genetic similarity in these areas. In addition, the Cambodian melons were not differentiated among geographical populations. Conomon and Makuwa were classified into cluster II, together with melon groups from the plains of Vietnam. The presence of two groups of melons in Southeast Asia was also indicated by population structure and principal coordinate analysis. These results indicated a close genetic relationship between Cambodia and the neighboring countries, thus suggesting that Cambodian melons are not directly related to the establishment of Conomon and Makuwa.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Reflecting its long history of cultivation, various usage, and adaptation in various parts of the world, melon (Cucumis melo) is known as the most diversified among Cucurbitaceae crops. Indeed, great diversity is reported in its morphological traits, such as fruit weight and seed length ranging from 50 to 15 kg and 4.5 to 15.0 mm, respectively (Nuñez-Palenius et al. 2008; Akashi et al. 2002). C. melo is divided into 19 horticultural groups based primarily on flower and fruit traits: Agrestis, Kachri, Chito, Tibish, Acidulus, Momordica, Conomon, Makuwa, Chinensis, Flexuosus, Chate, Dudaim, Chandalak, Indicus, Ameri, Cassaba, Ibericus, Inodorus, and Cantalupensis (Pitrat 2016).
Fujishita and Nakagawa (1973), focusing on seed size variation, indicated that melon is classified into large-seed (≥ 9.0 mm) and small-seed (< 9.0 mm) types. Melon accessions of groups Cantalupensis and Inodorous are mostly classified as large-seed type and groups Agrestis, Conomon, and Makuwa as small-seed type according to Fujishita (1983) and Akashi et al. (2002). In Asia, groups Dudaim, Flexuosus, Chandalak, Ameri and Inodorus of large-seed type are mainly distributed in West Asia, Central Asia and India, whereas groups Chinensis, Conomon, Makuwa and Acidulus of small-seed type are distributed in Southeast and Far-East Asia and India (Stepansky et al. 1999; Akashi et al. 2002; Yashiro et al. 2005; Tanaka et al. 2007; Pitrat 2008; Tanaka et al. 2013).
Among Asian melons, small- and large-seed types have proven to be diversified in independent maternal lineages, Ia and Ib, respectively, according to the analysis of chloroplast genome (Tanaka et al. 2013). India and Africa are now considered as domestication centers of melon (John et al. 2013; Endl et al. 2018; Gonzalo et al. 2019), where small- and large-seed types and Ia and Ib types are frequently distributed. However, the landraces of Conomon and Makuwa are not reported in India. Conomon and Makuwa are mostly distributed from China to Japan and are considered to be diversified in small-seed melon with Ia type cytoplasm in areas somewhere lying between India and China (Akashi et al. 2002; Tanaka et al. 2007; Serres-Giardi and Dogimont 2012). In contrast to India and Far-East Asia, little is known about melon landraces in Southeast Asia.
It was proven that Myanmar melon landraces first investigated by Yi et al. (2009) possess large genetic variation as in Indian melon. Myanmar melons consist of both small- and large-seed types, and Yi et al. (2009) classified small-seed type accessions as Conomon, Momordica, or Agrestis. However, most Myanmar Conomon accessions were clustered separately from the Conomon and Makuwa of Far-East Asia. Although one accession was regarded as Makuwa, they could not rule out the possibility of its recent introduction from other countries such as China and Japan.
More recently, Nhi et al. (2010) and Duong et al. (2021) analyzing Vietnamese melon landraces showed that all except one accession were small-seed type. Vietnamese melon consists of seven cultivar groups, including “Dua le” and “Dua vang” regarded as Makuwa and “Dua bo” and “Dua gang-andromonoecious” as Conomon. In contrast, “Dua thom” and “Montok” showed genetic similarities with Indian and Myanmar landraces. Based on these results, the presence of groups Conomon and Makuwa was first confirmed in Vietnam. Therefore, to identify the origin of groups Conomon and Makuwa, melon landraces of Cambodia, Laos, and Thailand should be investigated.
We have conducted germplasm collection expeditions in Cambodia since 2014 and successfully introduced germplasms of melon landraces (Matsunaga et al. 2015; Tanaka et al. 2016, 2017, 2019). Genetic resources of Cambodian melon are not available in major Genebanks other than NARO Genebank, National Agriculture and Food Research Organization, Japan, so they are expected to contribute to breeding for disease resistance and to the analysis of diversification of melon in Asia. More specifically, genetic analysis of Cambodian landraces is essential for understanding the distribution of groups Conomon and Makuwa and their genetic relationships with Vietnamese landraces. Therefore, in this study, we aimed to identify the genetic diversity and genetic structure of Cambodian melons using RAPD and SSR markers, and to discuss genetic diversification in Asian melons.
Materials and methods
Plant materials
Among the Cambodian melon landraces collected during 2014–2017, 62 accessions were selected to cover most of Cambodia and divided into five geographical groups: east, center, west, north, and south. Details of the materials used are summarized in Table 1 and Fig. 1. These accessions have been introduced to NARO Genebank, Japan, with SMTA (Standard Material Transfer Agreement). A total of 229 accessions were used as references, including Conomon (9), Makuwa (11), Agrestis (6), Cantalupensis (10), Inodorus (8), and accessions from different areas (Vietnam: 58, Myanmar: 36, Thailand: 5, Yunnan (China): 5, Xinjiang (China): 24, Central Asia: 14, Pakistan: 11, Afghanistan: 9, Iran: 10, Spain: 8, and the USA: 5). Two accessions of wild cucumber C. sativus var. hardwickii were also used as the outgroup. Seed length and width were measured for 10 seeds of each accession, and accessions were classified as large-seed (≥ 9.0 mm in length) or small-seed (< 9.0 mm in length) types, according to Akashi et al. (2002).

Map of Cambodia showing 19 provinces where melon landraces were collected
DNA extraction
Seeds of each accession were germinated on wet filter paper in a Petri dish and later transferred into pots, and grown in an incubator maintained at 30 °C with a 16 h light- 8 h dark cycle at a light intensity of 46.5 µMs−1-m−2. After 2 weeks, cotyledons from one seedling of each accession were ground individually in the liquid nitrogen. Total DNA was extracted using the cetyl-trimethyl-ammonium bromide (CTAB) method (Murray and Thompson 1980) with minor modifications. The quality and quantity of each DNA sample were determined with a spectrophotometer.
RAPD analysis
Ten random primers which were selected for their ability to detect polymorphism and for their stability in PCR amplification were used for RAPD analysis according to Nhi et al. (2010) (Table 2). PCR amplification was carried out in a 10 µl mixture containing 50 ng genomic DNA, 1 μl PCR buffer (Sigma®, St. Louis, MO, USA: 10 mM Tris–HCl; pH 8.3, 50 mM KCl), 2.5 mM MgCl2, 0.1 mM dNTP, 0.5 μM primer, and 0.25 U Taq polymerase (Sigma) by using an iCycler (Bio-Rad Laboratories, Hercules, CA, USA). The PCR cycle was started with an initial denaturing step at 95 °C for 3 min, 40 cycles at 93 °C for 1 min, 40 °C for 2 min, and 72 °C for 2 min. The duration of annealing temperature at 40 °C and the duration of extension at 72 °C were modified to 45 s and 1.5 min, respectively, for three primers A31, A57 and B86. The final extension step was at 72 °C for 5 min for all primers. After amplification, samples were electrophoresed on a 1.5% agarose gel (GenePure LE, BM Bio, Tokyo, Japan) at a constant voltage of 100 V using a horizontal gel electrophoresis system (Mupid-2, Cosmo Bio, Tokyo, Japan). Then, the PCR products were visualized with ethidium bromide staining, and their polymorphisms were evaluated.
SSR analysis
Seven SSR markers, developed by Ritschel et al. (2004) and Fukino et al. (2007) and showing high polymorphism in Vietnamese landraces as selected by Nhi et al. (2010), were used for the analysis of Cambodian melon landraces (Table 2). The PCR mixture was the same as used in the RAPD analysis, but the primer concentration was changed to 0.25 μM each. The PCR cycle was started with an initial denaturing step at 95 °C for 3 min, 35 cycles at 95 °C for 1 min, 60 °C for 1 min, and 72 °C for 2 min. The final extension step was at 72 °C for 5 min. PCR products were electrophoresed on 10% nondenatured polyacrylamide gel at a constant voltage of 260 V. Gels were stained in the same way as for RAPD analysis, and the size of the smallest band of each landrace was compared among the landraces.
Genotyping of CmACS7
The CmACS7 genotype was determined for sex expression analysis using the Cleaved Amplified Polymorphic Sequences (CAPS) marker developed by Boualem et al. (2008). The details of the PCR condition were given in Duong et al. (2021).
Data analysis
DNA fragments were scored as present (1) or absent (0) for RAPD markers. For SSR, DNA fragments were scored based on their size from the smallest (1) to the largest. From these data, the polymorphic information content (PIC) was calculated following Botstein et al. (1980). Genetic similarity (GS) was calculated after Apostol et al. (1993), using the formula GS = (N11 + N00)/T, where N11 and N00 were the number of positive and null bands, respectively, shared between two accessions and T was the total number of bands scored. The genetic distance (GD) between them was calculated using the formula GD = 1 − GS. Gene diversity (D) within each group and genetic distance (GD) between each group were calculated according to Weir (1996) and Nei (1972), respectively. A dendrogram was constructed by PHYLIP version 3.5.c (Felsenstein 1993), based on the GD matrix, with the unweighted pair group with arithmetic mean (UPGMA) method. Principal coordinate analysis (PCO; Gower 1966) based on the GS matrix was performed to show multiple dimensions of each population in a scatter-plot.
The population structure and the degree of admixture were estimated using the Bayesian clustering procedure of the STRUCTURE 2.3.4 (Pritchard et al. 2000). First the program was run to assume the number of distinct populations defined as K using the admixture model with correlated allele frequencies. The parameter sets were configured as follows: the length of the burn-in period and the number of Markov chain Monte Carlo (MCMC) repetitions after burn-in were 10,000, and 50,000, respectively, for each K ranging from 1 to 10 and running 20 iterations. The most plausible number of clusters was determined in the Structure Harvester program (Earl and von Holdt 2012) by calculating the distribution of the ∆K statistics as described by Evanno et al. (2005) and the performance of cluster matching across different K values was implemented through Cluster Markov Packager Across K (CLUMPAK) (Kopelman et al. 2015).
Results
Characterization of Cambodian melon landraces
Most Cambodian melons (56/62) were small-seed type as in those from Yunnan (China), Myanmar, Vietnam, Conomon, Makuwa and Agrestis (Table 3). In contrast, large-seed type was common in areas from Xinjiang (China) to the USA and also in Cantalupensis and Inodorus, while large- and small-seed types were common in areas from Central Asia to Iran.
Sex expression type, determined by CAPS analysis of CmACS7, also differed among melon populations (Table 3). All of the Cambodian melon accessions (62/62) proved to be monoecious. The monoecious type is also common in neighboring countries, such as Yunnan (China), Myanmar, and Vietnam (Dua thom). In contrast, andromonoecious type was common in Conomon, Makuwa, Vietnam (except Dua thom), in areas from Xinjiang (China) to the USA, and in Cantalupensis and Inodorus. Both monoecious and andromonoecious types were frequent in areas from Central Asia to Afghanistan.
RAPD and SSR analysis
Ten RAPD markers produced 12 polymorphic bands whose size ranged from 700 to 1520 bp, and the average number of marker bands per primer was 1.2 (Table 2). The average of PIC index was 0.297 and ranged from 0.047 (B32_700) to 0.452 (B99_1400). SSR analysis with 7 primer pairs generated 70 bands polymorphic in 291 accessions of melon and the average number of marker bands per primer was 10.0. The PIC index ranged from 0.664 (CMN04-07) to 0.891 (CMBR2) and was almost doubled that of the RAPD markers (Table 2).
Gene diversity (D) in Cambodian melon ranged from 0.105 (center) to 0.246 (south), and was 0.228 in 62 accessions (Table 3). This value was smaller compared with those in neighboring countries such as Vietnam (0.333 in 58 accessions), Myanmar (0.428), and Thailand (0.328), and nearly equivalent to those of Conomon and Makuwa (0.227 and 0.168).
Genetic relationship between Cambodian melon and reference accessions
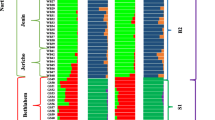
Genetic distance (GD) among 291 accessions ranged from 0.000 (CAn33 vs CA n44 etc.) to 0.947 (P142 vs X8), while the maximum GD was 0.474 (CAs46 vs CAe9 etc.) in Cambodian accessions (data not shown). A dendrogram was constructed based on these GD values, and 291 accessions were grouped into 3 major clusters that were further separated into 24 subclusters (Fig. 2). Cluster I comprised 115 accessions, which included landraces from Cambodia (59/62) and neighboring countries such as Vietnam (Dua thom), Yunnan (China), Thailand, and Myanmar (Table 4). Accessions of Conomon, Makuwa, and Agrestis formed cluster II (68 accessions), together with landraces from Vietnam except Dua thom. Cluster III (108 accessions) consisted of Cantalupensis, Inodorus, and accessions from Xinjiang (China) to the USA. Landraces of Myanmar were classified into three clusters in good accordance with the highest D value (0.428).

Genetic relationship between 293 accessions revealed by unweighted pair group method with arithmetic mean (UPGMA) cluster analysis based on genetic distance, and population structure inferred by STRUCTURE using the admixture model
Structure analysis indicated the presence of 3 main populations in 291 accessions studied (Fig. 2), based on the ∆K value, and 61, 101, and 93 accessions were assigned to populations P1, P2, and P3, respectively, using a Q-value threshold of 70% (Table 4). The remaining 36 accessions were regarded as admixture type. The classifications of 3 clusters and 3 populations were highly associated, as 86.1, 88.2, and 85.2% of accessions in clusters I, II, and III were assigned to P1, P2, and P3, respectively. Accessions of admixture type were mostly found in marginal subclusters such as If, IIg, and IIIa. Accordingly, Cambodian accessions belonged to P2 together with those from Vietnam (Dua thom), Yunnan (China), Thailand, and Myanmar, and the genetic relationship detected by cluster analysis was reproduced by structure analysis. A genetic relationship was also visualized on the scatter plot of PCO1 (21.3%) and PCO2 (16.5%) (Fig. 3). Accessions of three populations were distinctly separated and admixture accessions were located among three groups.

Distribution of 293 accessions on the first two principal coordinates. Accessions are indicated with symbols unique to each population; P1 (◆), P2 (●), P3 (▲), and Admixture (○)
Three clusters were also characterized by seed length and sex expression type (Tables 3 and 4). Most of accessions of clusters I (111/115) and II (65/68) commonly had seeds shorter than 9 mm in length and were classified as small-seed type. In contrast, the sex expression type differed between clusters I and II, and monoecious type and andromonoecious type were common in clusters I (108/115) and II (56/68), respectively. Cambodian melon proved to be small-seed type and monoecious type (56/62), similar to those of Vietnam (Dua thom), Yunnan (China), and Myanmar, thus showing close similarity among these populations. Accessions of cluster III were mostly large-seed type (80/108) and andromonoecious type (87/108).
Genetic relationship between 24 melon populations
The genetic distance (GD) between 24 populations calculated from the RAPD and SSR data ranged from 0.018 (Cambodia-North vs Cambodia-South) to 0.984 (Makuwa vs Xinjiang (China) (Table 5). GD among 5 populations of Cambodian melon ranged from 0.018 to 0.097, and was 0.062 on average. These values were nearly equivalent to those among 3 Vietnamese populations of Dua bo, Dua gang and Dua le (0.035–0.082), among Conomon and Makuwa (0.047) and among 5 populations from Xinjiang (China) to Iran (0.044–0.112), indicating that Cambodian melons were not genetically differentiated among geographical populations.
The genetic relationship between 24 populations was visualized by cluster analysis (Fig. 4) and by PCO analysis (Fig. 5). In accordance with the results of accession-based analysis, 24 populations were separated into 3 groups and Cambodian populations related closely to those in neighboring countries.

Genetic relationship between 24 populations of melon, revealed by unweighted pair group method with arithmetic mean (UPGMA) cluster analysis based on genetic distance

Distribution of 24 populations of melon on the first two principal coordinates
Discussion
The distribution of Conomon and Makuwa is limited to the eastern part of Asia, including China, Vietnam, Korea and Japan, and thus Conomon and Makuwa were considered as being introduced from the West. Previous research using isozyme and molecular markers have suggested that Conomon and Makuwa originated from small-seed melons with Ia type cytoplasm in areas lying somewhere between India and China (Akashi et al. 2002; Tanaka et al. 2007; Serres-Giardi and Dogimont 2012), and Tanaka et al. (2007) confirmed the presence of landraces, which were clustered together with Conomon and Makuwa, in Northeast India. These results highlight the importance of genetic research of melon landraces in Southeast Asia. However, few accessions are available in the public genebank worldwide. Therefore, we collected melon germplasm from Southeast Asian countries and analyzed genetic structure in Myanmar (Yi et al. 2009) and Vietnam (Nhi et al. 2010; Duong et al. 2021). This research indicated genetic differentiation in melon landraces in Southeast Asian countries, and highlighted the importance of genetic research using landraces from Cambodia and Laos.
The joint expedition team of NARO (Japan) and CARDI (Cambodian Agricultural Research and Development Institute, Cambodia) conducted melon germplasm collection in the western (2014), eastern (2015), central and northern (2016) and southern (2017) regions (2017) of Cambodia during 2014–2017 (Fig. 1). The geographical distribution, horticultural characteristics, and usage of melon landraces in Cambodia were first fully described by their reports (Matsunaga et al. 2015; Tanaka et al. 2016, 2017, 2019). According to their reports, non-netted melon with low sugar content was commonly grown in every part of Cambodia and utilized mature fruit as a desert, and immature fruit as vegetable or pickles. Immature and mature fruits of Conomon are mainly used for making pickles in Japan, suggesting the similarity between Cambodian melons and Conomon. To discuss the origin of Conomon and Makuwa, using part of this collection, we uncovered the genetic diversity of Cambodian melons in the present study, and compared it with those in neighboring countries.
Genetic diversity of cultivated melons is rich in India (Akashi et al. 2002; McCreight 2004) and this country was once considered the secondary center of diversity. However, based mainly on the discovery of wild perennial melon (C. trigonus), India is now considered as one of the domestication centers of melons along with Africa (John et al. 2013; Endl et al. 2018). Moreover, large genetic variations in Indian melons have been reported in various traits (Dhillon et al. 2007, 2009; Fergany et al. 2011; Roy et al. 2012). Seed size variation was also studied, and both large- and small-seed types are commonly distributed in India, with the latter being rather frequent in Northeastern India (Akashi et al. 2002; Tanaka et al. 2007). In contrast, small-seed type is frequent in Myanmar (36/41, Yi et al. 2009), Cambodia (58/58, Table 3), and Vietnam (61/61, Table 3), indicating an increasing trend of small-seed type from India to Vietnam. Genetic diversity also showed a similar geographical cline. D was larger in India compared to Myanmar (Yi et al. 2009), while in the present study (without Indian accessions), it was highest in Myanmar (0.428, Table 3) followed by Vietnam (0.333) and Cambodia (0.228). Although D was rather high in Thailand (0.328), the proportion of small-seed melons was exceptionally low (40%), indicating a necessity of diversity study using more landraces.
In areas from South to Southeast Asia, Myanmar landraces are closely related to Indian landraces (Tanaka et al. 2007; Yi et al. 2009), and as well as those of Thailand, Cambodia, Yunnan (China), and Vietnam (Dua thom) (Table 4, Fig. 4). These results indicated the introduction of non-netted melons from India to Southeast Asia and the decrease of genetic variation by natural and artificial selection. In this area, various ethnic groups of Tibeto-Burman or Austroasiatic language families live intricately across political borders (Clarke 2001). For example, the “Karen” tribe lives in Northeastern India, Myanmar, and Thailand, and the “Hmong” tribe lives in Laos, Thailand, and Vietnam. Local people definitely played a crucial role in the trade of goods (Misra 2005; Shibayama 2019) and the spread of melon from India to the East.
Southeast Asian melon landraces comprised two genetic groups, as clearly shown in Table 4 and Figs. 3, 4 and 5. The first group consisted of landraces of cluster I and population P2, which are commonly grown in Myanmar, Thailand, Cambodia, Yunnan (China), and mountain area of Vietnam (Dua thom). The second group consisted of landraces of cluster II and population P1, including Conomon and Makuwa, Agrestis (weedy melon) collected in Japan and Korea, and landraces grown in the plains of Vietnam. Landraces of both groups show similarities in several traits, such as smooth skin without net and small seeds shorter than 9 mm, and so should be classified as ssp. agrestis, according to the former systematics by Pitrat (2013). In addition, both groups belong to the same maternal lineage (Ia), as revealed by the analysis of chloroplast genome markers (Tanaka et al. 2013: Tanaka et al. in preparation). However, they also differed in several traits, reflecting their genetic differentiation. The key trait is sex expression type, and the first and second groups showed monoecy and andromonoecy, respectively (Table 3). Flesh sweetness also differs among groups, but the difference is not simple and only Makuwa of the second group has sweet flesh.
It was therefore concluded that melon landraces predominantly grown in most parts of Southeast Asia, including Cambodia, were of the first group and those of the second group were limited to the plains of Vietnam. In Vietnam, the second group is grown in the plains by “Kinh” tribe originally migrated from southern part of China, whereas the first group is grown in mountain areas by ethnic groups “Hmong” and “Thai” in northern part and by ethnic groups “Jarai” and “Hmong” in southern part (Duong et al. 2021). Therefore, the allopatric distribution of ethnic groups was considered as the primary reason for the presence of two genetic groups in Vietnam, though the possibility of severe selection due to pests, insects and so on could not be ruled out. In case of Cambodia, it shares the border with southern part of Vietnam where “Khmer” tribe lives as ethnic group, and thus the introduction of melon landraces across the border was reasonably hypothesized. However, landraces of different genetic groups were grown on both sides of the border, and Conomon and Makuwa, or their ancestral type were not found in Cambodia. Therefore, Cambodian melon is not directly related with the establishment of Conomon and Makuwa, supporting the introduction of Conomon and Makuwa from southern part of China to Vietnam as indicated by Duong et al. (2021). To further confirm this conclusion, landraces from Thailand, Laos, and Yunnan (China) should be studied in detail.
References
Akashi Y, Fukuda N, Wako T, Masuda M, Kato K (2002) Genetic variation and phylogenetic relationships in East and South Asian melons, Cucumis melo L., based on the analysis of five isozymes. Euphytica 125:358–396
Apostol BL, Black WCIV, Miller BR, Reiter P, Beaty PJ (1993) Estimation of the number of full sibling families at an oviposition site using RAPD-PCR markers: applications to the mosquito Aedes aegypti. Theor Appl Genet 86:991–1000
Botstein D, White RL, Skolnick M, Davis RW (1980) Construction of genetic linkage map in man using restriction fragment length polymorphisms. Am J Hum Genet 32:314–331
Boualem A, Fergany M, Fernandez R, Troadec C, Martin A, Morin H, Sari MA, Collin F, Flowers JM, Pitrat M, Purugganan MD, Dogimont C, Bendahmane A (2008) A conserved mutation in an ethylene biosynthesis enzyme leads to andromonoecy in melons. Science 321:836–838
Clarke G (2001) From ethnocide to ethnodevelopment? Ethnic minorities and indigenous peoples in Southeast Asia. Third World Q 22:413–436
Dhillon NPS, Ranjana R, Singh PP (2007) Diversity among landraces of Indian snap melon (Cucumis melo var. momordica). Genet Resour Crop Evol 54:1267–1283
Dhillon NPS, Singh J, Fergany M, Monforte AJ, Sureja K (2009) Phenotypic and molecular diversity among landraces of snapmelon (Cucumis melo var. momordica) adapted to the hot and humid tropics of eastern India. Plant Genet Resour 7:291–300
Duong TT, Dung TP, Tanaka K, Nhi PTP, Shigita G, Imoh ON, Nishida H, Kato K (2021) Distribution of two groups of melon landraces and inter-group hybridization enhanced genetic diversity in Vietnam. Breed Sci 71:564–574
Earl DA, von Holdt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Endl J, Achigan-Dako EG, Pandey AK, Monforte AJ, Pico B, Schaefer H (2018) Repeated domestication of melon (Cucumis melo) in Africa and Asia and a new close relative from India. Am J Bot 105:1662–1671
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Felsenstein J (1993) PHYLIP: phylogeny inference package (version 3.5c). University of Washington, Seattle.
Fergany M, Kaur B, Monforte AJ, Pitrat M, Rys C, Lecoq H, Dhillon NPS, Dhaliwal SS (2011) Variation in melon (Cucumis melo) landraces adapted to the humid tropics of southern India. Genet Resour Crop Evol 58:225–243
Fujishita N, Nakagawa K (1973) About seed (embryo) and fruit development of Cucumis species (1). Effects of growth regulator affected apomixis and parthenocarpy of C. melo. J Japan Soc Hort Sci 42(Suppl.2):186–187 (in Japanese)
Fujishita N (1983) Genetic diversity and phylogenetic differentiation in melon. Curr Top Plant Breed 24:3–21
Fukino N, Sakata M, Kunihisa M, Matsumoto S (2007) Characterization of novel simple sequence repeat (SSR) markers for melon (Cucumis melo L.) and their use for genotype identification. J Hort Sci Biotech 82:330–334
Gonzalo MJ, Diaz A, Dhillon NPS, Reddy UK, Pico B, Monforte AJ (2019) Re-evaluation of the role of Indian germplasm as center of melon diversification based on genotyping-by-sequencing analysis. BMC Genom 20:448
Gower JC (1966) Some distance properties of latent root and vector methods used in multivariate analysis. Biometrika 53:325–338
John J, Scariah S, Nissar VAM, Latha M, Gopalakrishnan S, Yadav SR, Bhat KV (2013) On the occurrence, distribution, taxonomy and genepool relationship of Cucumis callosus (Rottler) Cogn., the wild progenitor of Cucumis melo L. from India. Genet Resour Crop Evol 60:1037–1046
Kopelman K, Naama M, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) CLUMPAK: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15:1179–1191
Matsunaga H, Matsushima K, Tanaka K, Theavy S, Heng SL, Channa T, Takahashi Y, Tomooka N (2015) Collaborative exploration of the Solanaceae and Cucurbitaceae vegetable genetic resources in Cambodia, 2014. Annu Rep Explor Introd Plant Genet Resour 31:169–187
Misra U (2005) Chapter 3. Assam. In: Murayama M, Inoue K, Hazarika S (eds) Sub-regional relations in the eastern South Asia: With special focus on India's north eastern region. Institute of Developing Economies, Japan External Trade Organization, Chiba, Japan, pp 49–64
McCreight JD (2004) Isozyme variation in Indian and Chinese melon (Cucumis melo L.) germplasm collections. J Am Soc Hort Sci 129:811–818
Murray GC, Thompson WF (1980) Rapid isolation of high molecular weight DNA. Nucleic Acids Res 8:4321–4325
Nei M (1972) Genetic distance between populations. Am Nat 106:283–292
Nhi PTP, Akashi Y, Hang TTM, Tanaka K, Aierken Y, Yamamoto T, Nishida H, Long C, Kato K (2010) Genetic diversity in Vietnamese melon landraces revealed by the analyses of morphological traits and nuclear and cytoplasmic molecular markers. Breed Sci 60:255–266
Nuñez-Palenius HG, Gomez-Lim M, Ochoa-Alejo N, Grumet R, Lester G, Cantliffe DJ (2008) Melon fruits: genetic diversity, physiology, and biotechnology features. Crit Rev Biotechnol 28:13–55
Pitrat M (2008) Melon. In: Prohens J, Nuez F (eds) Handbook of plant breeding—vegetables I: Asteraceae, Brassicaceae, Chenopodiaceae, and Cucurbitaceae, pp 238–317
Pitrat M (2013) Phenotypic diversity in wild and cultivated melons (Cucumis melo). Plant Biotechnol 30:273–278
Pitrat M (2016) Melon genetic resources: Phenotypic diversity and horticultural taxonomy. In: Grumet R, Katzir N, Garcia-Mas J (eds) Genetics and genomics of Cucurbitaceae. Springer, Cham, pp 25–60
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multi-locus genotype data. Genetics 155:945–959
Ritschel PS, Lins TCL, Tristan RL, Buso GSC, Buso JA, Ferreira ME (2004) Development of microsatellite markers from an enriched genomic library for genetic analysis of melon (Cucumis melo L.). BMC Plant Biol 4:9
Roy A, Bal SS, Fergany M, Kaur S, Singh H, Malik AA, Singh J, Monforte AJ, Dhillon NPS (2012) Wild melon diversity in India (Punjab state). Genet Resour Crop Evol 59:755–767
Serres-Giardi L, Dogimont C (2012) How microsatellite diversity helps to understand the domestication history of melon. In: Sari N, Solmaz I, Aras V (eds) Cucurbitaceae 2012. In Proceedings of the Xth EUCARPIA meeting on genetics and breeding of Cucurbitaceae, University of Cukurova, Antalya, Turkey, pp 254–263
Shibayama M (2019) The ancient east-west corridor of mainland Southeast Asia. Geoinfomatics International, Thailand
Stepansky A, Kovalski I, Perl-Treves R (1999) Intraspecific classification of melons (Cucumis melo L.) in view of their phenotypic and molecular variation. Plant Syst Evol 217:313–333
Tanaka K, Nishitani A, Akashi Y, Sakata Y, Nishida H, Yoshino H, Kato K (2007) Molecular characterization of South and East Asian melon, Cucumis melo L., and the origin of Group Conomon var. makuwa and var. conomon revealed by RAPD analysis. Euphytica 153:233–247
Tanaka K, Akashi Y, Fukunaga K, Yamamoto T, Aierken Y, Nishida H, Long C, Yoshino H, Sato Y, Kato K (2013) Diversification and genetic differentiation of cultivated melon inferred from sequence polymorphism in the chloroplast genome. Breed Sci 63:183–196
Tanaka K, Duong TT, Yamashita H, Lay Heng S, Sophany S, Kato K (2016) Collection of Cucurbit crops (Cucurbitaceae) from Eastern Cambodia, 2015. Annu Rep Explor Introd Plant Genet Resour 32:109–137
Tanaka K, Shigita G, Sophea Y, Thun V, Sophany S, Kato K (2017) Collection of melon and other Cucurbitaceous crops in Cambodia in 2016. Annu Rep Explor Introd Plant Genet Resour 33:175–205
Tanaka K, Shigita G, Dung TP, Sophea Y, Thun V, Sophany S, Kato K (2019) Collection of melon and other Cucurbitaceous crops in Cambodia in 2017. Annu Rep Explor Introd Plant Genet Resour 35:121–146
Weir BS (1996) Genetic data analysis II. Sinauer Associate Inc. Publisher, Massachusetts
Yashiro K, Iwata H, Akashi Y, Tomita K, Kuzuya M, Tsumura Y, Kato K (2005) Genetic relationship among East and South Asian melon (Cucumis melo L.) revealed by AFLP analysis. Breed Sci 55:197–206
Yi SS, Akashi Y, Tanaka K, Cho TT, Khaing MT, Yoshino H, Nishida H, Yamamoto T, Win K, Kato K (2009) Molecular analysis of genetic diversity in melon landraces (Cucumis melo L.) from Myanmar and their relationship with melon germplasm from East and South Asia. Genet Resour Crop Evol 56:1149–1161
Funding
Open access funding provided by Okayama University. This study was supported by the PGRAsia project (https://sumire.gene.affrc.go.jp/pgrasia/index_en.php) from the Ministry of Agriculture, Forestry and Fisheries of Japan, and a Grant-in-Aid for International Scientific Research of Ministry of Education, Science, Culture and Sports, Japan (No. 26257409), entitled “Genetic assay and study of crop germplasm introduced/originated in East Asia (2nd)”.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Field research and the collection of landraces were prepared and performed by Katsunori Tanaka, Sakhan Sophany, Ouch Sreynech, Gentaro Shigita, Yon Sophea, Ouk Makara, Norihiko Tomooka, and Kenji Kato. DNA experiments were performed by Pervin Mst Naznin, Odirichi Nnennaya Imoh, and Katsunori Tanaka. The first draft of the manuscript was written by Pervin Mst Naznin and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have not disclosed any competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Naznin, P.M., Imoh, O.N., Tanaka, K. et al. Analysis of genetic diversity and population structure in Cambodian melon landraces using molecular markers. Genet Resour Crop Evol 71, 1067–1083 (2024). https://doi.org/10.1007/s10722-023-01677-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10722-023-01677-7