
Abstract
A radish panel of 152 accessions with diverse root shapes was assembled from the National Plant Germplasm System (NPGS) collection based on weighted geographical sampling. Accessions represented 35 countries and were categorized into eight geographical regions—North America, Europe, Africa, West Asia, Central Asia, South Asia, East Asia, and Southeast Asia. Multiple plants per accession were assayed using genotyping by sequencing (GBS), and 8539 high-quality polymorphisms were discovered. Average observed heterozygosity (Ho) of the markers consistently ranged from 5 to 7% in all geographical regions. Central Asia was most diverse in terms of average expected heterozygosity (He = 0.107), while Southeast Asia was the least diverse (He = 0.050). Europe and Southeast Asia were the most divergent from each other (pairwise FST = 0.199), while Europe and North America were the least divergent (pairwise FST = 0.022). The clustering of genotypes based on principal components analysis (PCA) illustrated an east/west geographical pattern. Large numbers of private alleles and a substantial proportion (3%) of markers showing signals of selection based on allele frequency differences indicate that allelic variation is available for cultivar development. European and North American gene pools could be expanded by crossing with East Asian and Southeast Asian germplasm, and vice-versa. Both could potentially benefit from South Asian sources, while the addition of West Asian germplasm could increase East Asian and Southeast Asian diversity. This resource for molecular marker development combined with an understanding of global genetic relationships will inform the continued germplasm conservation and genetic improvement of radish.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Radish (Raphanus sativus L.) is an ancient, globally distributed crop with multiple diverse uses. Radish is consumed as a root vegetable (hypocotyl plus true root), a leafy vegetable, sprouts, seeds, or as seed pods and is also used as a cover crop, forage crop, or for extraction of oil from its seed. Consumer preferences are regionally based, resulting in morphological diversity combined with adaptation to local conditions. In human diets radish provides a rich source of vitamin A, vitamin C, minerals, and carbohydrates. Breeding improvement for many cultivars and F1 hybrids has focused on size, color, and shape of the root as well as resistance and horticultural traits.
The crop has traditionally been classified into five morphotypes (Yamagishi 2017), namely, European small (Raphanus sativus L. var. sativus), East Asian long (Raphanus sativus var. sativus), black (R. sativus var. sativus), oil (R. sativus var. oleiformis Pers.) and rat-tail (Raphanus sativus var. mougri H.W.J. Helm). Wild relatives of the crop are native to the Mediterranean coast as well as. Coastal areas of China, Korea, and Japan (Lee et al. 2018 and references therein). Four wild taxa are currently recognized, namely, Raphanus raphanistrum L., Raphanus raphanistrum subsp. landra (Moretti ex DC.) Bonnier & Layens, Raphanus raphanistrum L. subsp. raphanistrum, and Raphanus raphanistrum subsp. rostratus (DC.) Thell. In addition to morphotypes, classification of cropping types such as autumn, spring, or summer is used in some production regions. These types are adapted in terms of temperature stress resistance and bolting tendency (Kaneko and Matsuzawa 1993). Various studies of mtDNA and cpDNA have shown that cultivated radish may have originated from multiple, independent domestication events (Yamane et al. 2009; Yamagishi 2017; Kobayashi et al. 2020).
Approximately 5200 active accessions of Raphanus are conserved in germplasm repositories in Asia, Europe, and the USA (https://www.genesys-pgr.org). The largest collections are held in the UK and USA with 1393 and 1164 accessions, respectively. For the current study, a Plant Genetic Resources Unit (PGRU), National Plant Germplasm System (NPGS) radish diversity set was assembled based on a weighted geographical sampling of provenance from the 685 active R. sativus accessions held in Geneva, NY. DNA from multiple seedlings per accession was isolated and genotyped using genotyping by sequencing (GBS). The resultant single nucleotide polymorphism (SNP) markers represent an unbiased, broad, and random sampling of the radish genome. The major objectives of this study were to: (i) assemble and genotype a cultivated radish (R. sativus L.) geographical diversity set, (ii) map the high-quality SNPs to the R. sativus L. draft genome (Zhang et al. 2015), (iii) analyze radish genetic diversity and partitioning of variation among defined hierarchical levels of geographical region, accession, and individual, (iv) describe global patterns of genetic relatedness, (v) screen the SNPs for signals of potential balancing or directional selection, and (vi) deposit DNA sequence data into a public database as a resource for developing SNP markers for applications in research, germplasm conservation, and crop improvement.
Materials and methods
Germplasm and DNA sequencing
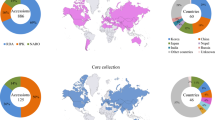
Radish germplasm accessions originated from 35 countries and were received by NPGS between 1934 and 2013 (Online Resource Table S1). Root shapes were categorized into one of ten different phenotypes (IBPGRI 1990) based on digital images of whole roots and longitudinal sections (https://npgsweb.ars-grin.gov/gringlobal). DNA samples from each of five seedlings per 152 accessions (Online Resource Table S2) were isolated from 50 mg frozen leaf sample using Qiagen DNeasy (96) Plant Kit (Qiagen, Valencia, CA). GBS libraries were prepared at Cornell University’s Genomic Diversity Facility using the restriction enzyme PstI and genotyped on an Illumina HiSeq 2000 in 96-plex format using standard protocols (Elshire et al. 2011). One blank well was included as a negative control on each of the eight sequenced plates. All raw GBS data are available through the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) (BioProject PRJNA691871 and BioSample numbers SAMN17305744–SAMN17306511)
Bioinformatics and statistics
TASSEL5 pipeline (Glaubitz et al. 2014) was used to call SNPs using Raphanus sativus L. cultivar XYB36-2 (GenBank assembly accession GCA_002197605.1) as the reference genome (Zhang et al. 2015). VCF software (Danecek et al. 2011) was used to filter the sites using the following criteria: minGQ = 98, max missing = 0.10, bi-allelic only and minimum allele frequency (MAF) = 0.06. TASSEL 5 GUI (Bradbury et al. 2007) was used to estimate summary statistics of sites and samples, and to perform principal components analysis (PCA). Arlequin ver. 3.5.2.2 (Excoffier and Lischer 2010) was used for an Analysis of Molecular Variance (AMOVA) and to estimate pairwise genetic distances, FST, between populations; 1023 random permutations of the data were used to test statistical significance of results. Diversity statistics: observed heterozygosity (Ho), unbiased expected heterozygosity (He), number of private alleles, i.e., unique alleles, (np) and inbreeding coefficient (F) were estimated using GENALEX ver. 6.502 (Peakall and Smouse 2006). BayeScan ver. 2.1 (Foll and Gaggiotti 2008) was used to identify SNP loci that rejected the null hypothesis of selective neutrality based on differences in allele frequencies among eight populations defined by geographical origins North America (NA), Europe (EU), Africa (AF), West Asia (WA), Central Asia (CA), South Asia (SA), East Asia (EA) and Southeast Asia (SE). For BayeScan results indicating log10(Posterior Odds) greater than 0.5, which indicated substantial to decisive evidence of selection (Foll 2012), NCBI standard nucleotide BLAST searches against the nucleotide collection nr/nt database (Stover and Cavalcanti 2017) were performed on 100 nt genomic sequence stretches with the SNP site at position 50.
Results
Quality control of GBS sequencing runs showed a pass_rate of 0.999 for all eight plates, where pass_rate = pass_count/read_count, pass_count = number of reads starting with (barcode + cut site remnant) sequence and not containing any N bases and read_count = number of reads starting with (barcode + cut site remnant) sequence (may contain N bases). Pass_count ranged from 2.37 × 108 to 2.59 × 108 across the eight plates. SNP calling and quality filtering yielded 8539 sites × 760 plant samples with overall means of 0.02 missing data, 0.06 heterozygosity and 0.06 minor allele frequency (Online Resource Table S3).
Proportion heterozygosity per site ranged from zero to 0.98 (Online Resource Table S4) and per plant ranged from 0.01 to 0.12 (Online Resource Table S5). The number of plant samples within each geographical region were: n = 45 NA, n = 80 EU, n = 15 AF, n = 299 WA, n = 20 CA, n = 150 SA, n = 131 EA and n = 20 SE (Online Resource Table S2). Sampling captured a broad range of root shape phenotypes with all eight geographic regions showing a diversity of root shapes and only 14 accessions with unknown shape (Online Resource Table S6). The most common root shapes were elliptic (n = 38), triangular (n = 33) and horn (n = 21). Apically bulbous (n = 5), branched (n = 2) and inverse triangle (n = 2) were the least common shapes. The other shapes were moderately represented, ranging from n = 10 to n = 16.
SNP diversity estimates for the eight geographic regions showed CA as most diverse (n = 16.952 ± 0.027, HO = 0.069 ± 0.002, He = 0.107 ± 0.002) and SE as least diverse (n = 19.854 ± 0.006, HO = 0.048 ± 0.001, He = 0.050 ± 0.001) (Table 1). He is a function of allele frequencies rather than sample size, and a good indicator of relative diversity for comparative purposes. HO fell within a consistent narrow range across regions from approximately 5–7%. The coefficient of inbreeding, F, estimates the deviation from random mating (Peakall and Smouse 2006). If HO = He, F = 0; positive values of F indicate inbreeding while negative values indicate excess heterozygosity due to assortative mating or heterosis. Estimates of F ranged from 0.040 ± 0.004 in SE to 0.244 ± 0.005 in CA (Table 1). All eight regions contained private alleles ranging from np = 28 (AF, mean n = 14.813) to np = 1104 (WA, mean n = 291.858). This count will increase with increasing sample size so is not a good measure for direct comparisons between the regions. Noticeably, CA contained a large number of private alleles relative to its small sample size (np = 771, n = 20).
PCA showed EA and SE to be separated from the other regions along PC1, SA to be largely clustered based on PCs 1 and 2, and WA, CA, NA, EU and AF to be dispersed along PC1 (Fig. 1). EA and SE showed little overlap with SA and WA. SA and WA showed moderate overlap with each other and were distinct at their extremes along PCs 1 and 2. The overall pattern of clustering corresponded with an east to west differentiation across the globe from NA/EU to EA/SE (Fig. 1). One CA accession (PI 502584) appeared as an outlier with values of PC2 > 6.0 for all five plants. The original seed of PI 502584 was described as cultivated, used as a vegetable, and was obtained from a market during a plant exploration expedition in Uzbekistan in 1982. It is classified as Raphanus spp. but there are no images of this accession to check whether it shows wild species traits (https://npgsweb.ars-grin.gov/gringlobal/accessiondetail?id=1397520).

a Countries of origin of 152 radish accessions categorized into eight geographical regions. b Principal Components of five radish plants for each of 152 accessions coded by region of origin. PC1 and PC2 explained 8.7 and 2.9 % of the total variation, respectively
Pairwise genetic distances between populations showed SE and EU to be most distant (FST = 0.199) and EU and NA to be least distant (FST = 0.022) from each other (Table 2). When examining all pairwise comparisons, SE was highly divergent from all others with the exception of EA. This is illustrated as a tight cluster of most of the individuals from the four SE accessions towards extreme negative values of PC1, and nested within EA (Fig. 1). An AMOVA showed that the highest proportion of variation was within individuals (77.49%) and the lowest was among individuals within accessions (1.00%). Only 6.51% of the variation was among geographical regions, and 14.99% was among accessions within geographical regions (Table 3).
BayeScan results showed log10(PO) greater than 0.5 for 256 SNPs (Online Resource Table S7). Associated q-values for these SNPs were 0.10 or lower, implying that 0.10 of the detected outlier loci are expected to be false positives. Alpha values were positive for 14 SNPs, potentially indicative of directional fixation of alleles within populations. Negative alpha values for the remaining 242 SNPs indicate balancing selection, i.e., the maintenance of multiple alleles due to heterozygous advantage or a response to variable environmental conditions, or alternatively, purifying selection. Three closely linked sites on chromosome 9 (S9_9292666, S9_9292671, S9_9292690) were extreme outliers with log10(PO) greater than 2.0 and FST greater than 0.30 (Fig. 2 and Online Resource Table S7). BLAST results showed these SNP sites to significantly match a 100 nt sequence in exon 11 of AJ550021.2 Raphanus sativus D81Rfo restorer-of-fertility (Desloire et al. 2003) genomic region (Max score = 185, total score = 185, query cover = 100%, percent identity = 100%, E value = 3 × 10− 43) (Online Resource Table S8). Two of these polymorphisms were predicted to be synonymous substitutions while the third predicted a conservative amino acid substitution of valine to isoleucine.

BayeScan results for genome-wide scans to detect outlier loci based on locus-specific FST values. Log10(PO) greater than 0.5 indicates the rejection of the neutral model of no selection, 256 markers met this threshold
Discussion
Radish cultivars are often outcrossing, open-pollinated populations with root shape, color, and market class used to classify them (Bae et al. 2015), as well as seasonal cropping type. The majority of accessions in our study (n = 88) had no name other than a collection code, and only 26 of the 152 accessions were associated with a passport data narrative (https://npgsweb.ars-grin.gov/gringlobal/). Therefore, we were unable to classify the accessions into market classes or usage types. This lack of information can limit their practical utility. Furthermore, speciation or domestication hypotheses were not addressed in the current study because wild radish accessions were not sampled.
This study represents the largest number of cultivated radish accessions with multiple individuals (assayed as un-pooled DNA samples) genotyped with SNP markers to date. This sampling strategy provided high resolution at both the accession and individual levels. Our results supported previous studies that detected clustering of genotypes based on geographical origins (Zhang and Wang 2017 and references therein). Cultivated radish is largely consumed raw as a condiment or salad ingredient in Europe and the USA, while it is a key ingredient in the cuisines of Japan, China, Korea, India, and South Asian and Southeast Asian countries. This different emphasis in usage combined with environmental differences has produced genetically distinct types between the two regions.
Groups corresponding to four geographical areas were observed in a double-digest restriction site-associated DNA sequencing (ddRAD-Seq) study of 500 radish accessions, namely, Japan, East Asia, South and Southeast Asia, and The Occident (Europe and The Americas) and Near East (Kobayashi et al. 2020). More than half of the large number of accessions in this study represented Japanese landraces. Whole-genome resequencing analysis of 17 lines showed that Asian cultivated radish was closely related to wild Asian genotypes but was distinct from European/USA cultivated radish; this was due to independent domestication events from different wild taxa in Asia and Europe (Kim et al. 2016). Genotyping of 65 radish accessions using AFLP markers found four clusters related to origins in Europe, Middle East, South Asia and East Asia (Wang et al. 2008). Sixty-five radish accessions from 31 countries clustered into Asian versus European groups based on 36 insertion/deletion (indel) and simple sequence repeat (SSR) markers (Kang et al. 2016). The European group contained subgroups based on root exterior color. Three groups, namely Asian cultivated, European cultivated, and wild radish were identified in a radish core collection of 125 accessions genotyped with 25 sequence-tagged site (STS) indel markers (Lee et al. 2018). In contrast, GBS of 96 radish entries found the optimal number of K clusters to be eight but the groups were of multiple, mixed origins (Chung et al. 2018). This latter observation and the general geographical trend but overlapping groups in the current study underscore the migration between radish gene pools due to human activities.
Indeed, variation among geographical regions was statistically significant but only represented 6.51% of the total variation (Table 3). Because only 1% of the variation was found among individuals within an accession, it is not necessary for breeders to screen multiple individuals within accessions to look for novel sources of alleles. Rather, accessions within the context of geographical region are the most efficient target of study. European and North American gene pools could be expanded by crossing with South Asian, East Asian and Southeast Asian germplasm, and vice-versa. Both could potentially benefit from South Asian sources, while the addition of West Asian germplasm could increase East Asian and Southeast Asian diversity.
The forces of natural and artificial selection have been extensive during the domestication, dispersal, and ongoing improvement of cultivated radish. A Bayesian method of detecting selection (Foll and Gaggiotti 2008) has been successfully applied to a variety of plant species, including maize (Gouesnard et al. 2017), live oaks (Meireles et al. 2017) and pear (Kumar et al. 2017). The rejection of neutrality based on observed allele frequency differences reflects recent selection leading to population differentiation. Approximately 3% of the polymorphisms in the current study rejected the model of selective neutrality. The majority supported a model of purifying or balancing selection. Notably, the three most extreme outlier GBS markers located within a 9 Mb region of Chromosome 9 indicated directional fixation of alleles and the predicted locus showed a strong match to a restorer of fertility locus in radish (Desloire et al. 2003). Cytoplasmic male sterility (CMS) is used for hybrid seed production in radish and other vegetable crops such as pepper, carrot, and onion (Mishra and Kumari 2018). The three SNPs are likely in strong linkage disequilibrium with functional polymorphisms rather than direct targets of selection. The identification of such markers and their confirmation as informative polymorphisms within functional candidate loci will be valuable for marker assisted selection and genome wide association studies (GWAS) in radish.
Our results quantified germplasm diversity patterns in a globally representative sample of cultivated radish, including relationships within and among accessions. Deliverables included 128 Gbases of raw sequence data for molecular marker development, a densely-genotyped subset of the NPGS collection, and the identification of candidate markers associated with loci showing signals of selection. These will provide a basis for screening germplasm, mining of novel alleles, association mapping, and evaluation trials to continue to develop cultivars with phenotypic traits of economic importance.
Data availability
All raw data are available at the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA); all germplasm is available upon request at https://npgsweb.ars-grin.gov/gringlobal/search.
Code availability
All software applications used for data analyses are freely available from their respective authors.
References
Bae K-M, Sim S-C, Hong J-H, Choi K-J, Kim D-H, Kwon Y-S (2015) Development of genomic SSR markers and genetic diversity analysis in cultivated radish (Raphanus sativus L.). Hortic Environ Biotechnol 56(2):216–224
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23(19):2633–2635
Chung YS, Lee YG, Silva RR, Park S, Park MY, Lim YP, Choi SC, Kim C (2018) Potential SNPs related to microspore culture in Raphanus sativus based on a single-marker analysis. Can J Plant Sci 98(5):1072–1083
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST (2011) The variant call format and VCFtools. Bioinformatics 27(15):2156–2158
Desloire S, Gherbi H, Laloui W, Marhadour S, Clouet V, Cattolico L, Falentin C, Giancola S, Renard M, Budar F (2003) Identification of the fertility restoration locus, Rfo, in radish, as a member of the pentatricopeptide-repeat protein family. EMBO Rep 4(6):588–594
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6:e19379
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecology Resour 10:564–567
Foll M (2012) BayeScan v2. 1 user manual. Ecology 20:1450–1462
Foll M, Gaggiotti O (2008) A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180(2):977–993
Glaubitz JC, Casstevens TM, Lu F, Harriman J, Elshire RJ, Sun Q, Buckler ES (2014) TASSEL-GBS: A high capacity genotyping by sequencing analysis pipeline. PloS ONE 9:e90346
Gouesnard B, Negro S, Laffray A, Glaubitz J, Melchinger A, Revilla P, Moreno-Gonzalez J, Madur D, Combes V, Tollon-Cordet C (2017) Genotyping-by-sequencing highlights original diversity patterns within a European collection of 1191 maize flint lines, as compared to the maize USDA genebank. Theor Appl Genet 130(10):2165–2189
IBPGRI (1990) Descriptors for Brassica and Raphanus. International Board for Plant Genetic Resources, Rome
Kaneko Y, Matsuzawa Y (1993) Radish: Raphanus sativus L. In: Bergh BO, Kalloo G (eds) Genetic improvement of vegetable crops. Pergamon Press, Oxford, pp 487–510
Kang ES, Ha SM, Ko HC, Yu H-J, Chae WB (2016) Reproductive traits and molecular evidence related to the global distribution of cultivated radish (Raphanus sativus L.). Plant Syst Evol 302(10):1367–1380
Kim N, Jeong Y-M, Jeong S, Kim G-B, Baek S, Kwon Y-E, Cho A, Choi S-B, Kim J, Lim W-J (2016) Identification of candidate domestication regions in the radish genome based on high-depth resequencing analysis of 17 genotypes. Theor Appl Genet 129(9):1797–1814
Kobayashi H, Shirasawa K, Fukino N, Hirakawa H, Akanuma T, Kitashiba H (2020) Identification of genome-wide single-nucleotide polymorphisms among geographically diverse radish accessions. DNA Res 27(1):dsaa001. https://doi.org/10.1093/dnares/dsaa001
Kumar S, Kirk C, Deng C, Wiedow C, Knaebel M, Brewer L (2017) Genotyping-by-sequencing of pear (Pyrus spp.) accessions unravels novel patterns of genetic diversity and selection footprints. Hortic Res 4(1):1–10
Lee Y-J, Mun J-H, Jeong Y-M, Joo S-H, Yu H-J (2018) Assembly of a radish core collection for evaluation and preservation of genetic diversity. Hortic Environ Biotechnol 59(5):711–721
Meireles JE, Beulke A, Borkowski DS, Romero-Severson J, Cavender-Bares J (2017) Balancing selection maintains diversity in a cold tolerance gene in broadly distributed live oaks. Genome 60(9):762–769
Mishra S, Kumari V (2018) A review on male sterility-concepts and utilization in vegetable crops. Int J Curr Microbiol Appl Sci 7(2):3016–3034
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Stover NA, Cavalcanti AR (2017) Using NCBI BLAST. Curr Protoc Essent Lab Tech 14(1):11
Wang N, Kitamoto N, Ohsawa R, Fujimura T (2008) Genetic diversity of radish (Raphanus sativus) germplasms and relationships among worldwide accessions analyzed with AFLP markers. Breed Sci 58(2):107–112
Yamagishi H (2017) Speciation and diversification of radish. In: Nishio T, Kitashiba H (eds) The radish genome. Springer, Dordrecht, pp 11–30
Yamane K, Lü N, Ohnishi O (2009) Multiple origins and high genetic diversity of cultivated radish inferred from polymorphism in chloroplast simple sequence repeats. Breed Sci 59(1):55–65
Zhang X, Yue Z, Mei S, Qiu Y, Yang X, Chen X, Cheng F, Wu Z, Sun Y, Jing Y (2015) A de novo genome of a Chinese radish cultivar. Hortic Plant J 1(3):155–164
Zhang L, Wang Q (2017) DNA polymorphism and genetic diversity in Raphanus accessions. In: The radish genome. Springer, pp 71–91
Acknowledgements
We thank S. Srmack and P. Kisly for their excellent technical support. USDA is an equal opportunity provider and employer. The use of trade, firm, or corporation names in this publication is for the information and convenience of the reader. Such use does not constitute an official endorsement or approval by the United States Department of Agriculture of any product or service to the exclusion of others that may be suitable.
Funding
Jie Arro was a participant of the Oak Ridge Institute for Science and Education-Oak Ridge Associated Universities (ORISE-ORAU) Education and Training Program. This research was funded by USDA-ARS CRIS Project Number: 8060-21000-027-00D.
Author information
Authors and Affiliations
Contributions
J. Arro and J.A. Labate contributed to experimental design, data analyses, and writing of this article.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest or competing interests.
Consent for publication
All authors consent to publish.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Arro, J., Labate, J.A. Genetic variation in a radish (Raphanus sativus L.) geodiversity collection. Genet Resour Crop Evol 69, 163–171 (2022). https://doi.org/10.1007/s10722-021-01212-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10722-021-01212-6