
Abstract
Production and accumulation of the major greenhouse gas nitrous oxide (N2O) in surface groundwater might contribute to N2O emissions to the atmosphere. We report on a 15N tracer study conducted in the Fuhrberger Feld aquifer in northern Germany. A K15NO3 tracer solution (60 atom%) was applied to the surface groundwater on an 8 m2 measuring plot using 45 injection points in order to stimulate production of 15N2O by denitrification and to detect its contribution to emissions at the soil surface. Samples from the surface groundwater, from the unsaturated zone and at the soil surface were collected in regular intervals over a 72-days period. Total N2O fluxes at the soil surface were low and in a range between −7.6 and 29.1 μg N2O-N m−2 h−1. 15N enrichment of N2O decreased considerably upwards in the profile. In the surface groundwater, we found a 15N enrichment of N2O between 13 and 42 atom%. In contrast, 15N enrichment of N2O in flux chambers at the soil surface was very low, but a detectable 15N enrichment was found at all sampling events. Fluxes of groundwater-derived 15N-N2O were very low and ranged between 0.0002 and 0.0018 kg N2O-N ha−1 year−1, indicating that indirect N2O emissions from the surface groundwater of the Fuhrberger Feld aquifer occurring via upward diffusion are hardly significant. Due to these observations we concluded that N2O dynamics at the soil–atmosphere interface is predominantly governed by topsoil parameters. However, highest 15N enrichments of N2O throughout the profile were obtained in the course of a rapid drawdown of the groundwater table. We assume that such fluctuations may enhance diffusive N2O fluxes from the surface groundwater to the atmosphere for a short time.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The trace gas nitrous oxide (N2O) is known to contribute to global warming (Mosier et al. 1998) and catalyses the destruction of stratospheric ozone (Crutzen 1981). Its global atmospheric concentration has increased since pre-industrial times by about 18% (Intergovernmental Panel on Climate Change 2007) and continues to do so. A significant amount of N2O emissions originates from agricultural ecosystems (Mosier et al. 1998). In aquifers of agricultural catchments, high concentrations of N2O were found at the groundwater surface (Spalding and Parrott 1994; Well et al. 2005a; Deurer et al. 2008). Thus groundwater N2O was assumed to be a potential significant source of N2O emissions to the atmosphere (Mosier et al. 1998; Rice and Rogers 1993; Ronen et al. 1988). These indirect emissions from groundwater are associated with nitrogen that leaves agricultural fields via leaching and runoff to adjacent systems (Nevison 2000; Groffman et al. 2002; Well et al. 2005b). The Intergovernmental Panel on Climate Change (IPCC) methodology provides a concept of N2O emission factors, containing an emission factor EF5-g for indirect N2O emissions from groundwater and drainage ditches in order to construct national inventories for these emissions (Mosier et al. 1998). This concept is introduced and discussed in detail by Well et al. (2005b) and Clough et al. (2007). Firstly, the EF5-g default factor was defined as 0.015 kg N2O-N kg−1 of N leached (Mosier et al. 1998) and based on very few data which were available for its validation (Groffman et al. 2002). Recent studies have emphasized uncertainties with the magnitude of the EF5-g and suggested a substantial downward revision (Hiscock et al. 2002, 2003; Reay et al. 2005; Sawamoto et al. 2005). Taking the results of these studies into account, the EF5-g default value was corrected to 0.0025 kg N2O-N kg−1 of N leached (IPCC 2006). However, the knowledge of indirect N2O emissions from groundwater is still limited because few studies have tried to relate subsurface N2O concentrations to N leaching from soils (Clough et al. 2005). Furthermore, the question how much N2O that was produced in the surface groundwater can finally reach the atmosphere is a subject of uncertainty and the controls governing the balance between N2O production and consumption are not well understood (Clough et al. 2007). Upward diffusive N2O fluxes from the aquifer surface have been estimated from concentration gradients (Ronen et al. 1988; Hiscock et al. 2003; Deurer et al. 2008). However, until now these estimations have not been evaluated by direct measurements. So far, emission factors for groundwater-derived N2O like the IPCC EF5-g are based on N2O concentrations assuming that degassing of all dissolved N2O occurs after convective groundwater flow to wells, springs or streams (Mosier et al. 1998). Principally, the vertical diffusive fluxes from the aquifer surface should be added to the potential total groundwater-derived emission (Deurer et al. 2008; Hiscock et al. 2003). Solid estimates of diffusive fluxes are thus needed in order to check if the inclusion of this path leads to higher emission factors.
Production and consumption of N2O are simultaneously running reactions during denitrification. This well documented microbiological process occurs in O2-depleted layers of aquifers with available electron donors (Korom 1992; Böttcher et al. 1990; Ross 1995). Unfortunately, a number of difficulties in measuring denitrification exist. Diverse approaches to the problem were reviewed by Groffman et al. (2006). In situ tracer tests were conducted in order to study the fate of nitrate (NO3 −) and potential denitrification rates in subsoils and groundwater using 15N labeled NO3 − (Tobias et al. 2001; Addy et al. 2002; Well et al. 2003; Kim et al. 2005). For these “push–pull methods”, a test solution containing 15N-labeled NO3 − is injected (“pushed”) into the soil matrix or groundwater, respectively. After an incubation period, the mixture of test solution and groundwater is extracted (“pulled”) and products of denitrification are determined to quantify the process. However, tracer studies focussing on the analysis of the occurrence of the intermediate denitrification product N2O are rare. Van Groenigen et al. (2005) applied 15N-labeled fertilizer to a sandy soil and traced soil N2O concentrations and fluxes over a 1 year period. They concluded that most of the N2O was formed in the subsoil during the winter, but this did not result in corresponding increases in N2O fluxes from the topsoil to the atmosphere. Overall, total topsoil N2O fluxes were very low, and amounted to 0.06% of the applied N fertilizer, suggesting that emissions of N2O via diffusion upwards through the profile were negligible. However, other authors mentioned the possibility of considerable indirect emissions through supersaturated drainage water (Heincke and Kaupenjohann 1999; Reay et al. 2003).
A study with soil columns and 15N labeled NO3 − as a tracer was conducted by Clough et al. (1999). After labeling the bottom of the columns, they observed a decrease in 15N-N2O/15N-N2 ratios during upward diffusion of the labeled gases. Furthermore, they stated that this effect may be caused by dilution of the 15N-N2O pool with soil derived N2O, or by consumption of 15N-N2O in the topsoil. To detect the fate of N2O after its occurrence in subsurface environments, Clough et al. (2007) introduced 15N2O enriched groundwater into the groundwater–subsoil matrix of a salt marsh and a forested riparian zone. Whereas added 15N2O behaved in a conservative manner at the salt marsh, it was partly and significantly consumed in the saturated zone of the riparian zone. In conclusion, the authors emphasized complexity and variability of the fate of N2O applied or produced in groundwater and that these quantities should be considered in the development of improved IPCC inventory calculations.
In this study, we examine the occurrence of 15N-N2O following the application of a tracer solution containing 15N-labeled NO3 − directly to the groundwater surface and its emission at the soil surface. To achieve this, we analysed 15N-N2O and 15N-NO3 − in the surface groundwater and 15N-N2O in different depths of the unsaturated zone and at the soil surface over a 72 days time period. The objectives in detail are:
-
1.
to initiate 15N-N2O production at the groundwater surface stimulated by weekly application of a 15N-NO3 tracer solution
-
2.
to evaluate the 15N enrichment of N2O and to detect groundwater-derived N2O in the system groundwater/unsaturated zone/atmosphere
-
3.
to measure diffusive fluxes of groundwater-derived N2O to assess its significance as a component of agricultural N2O emissions from groundwater.
Materials and methods
Research site
The experimental field plot was located within the catchment of the Fuhrberger Feld aquifer (FFA) in northern Germany, which is situated about 30 km northeast of the city of Hannover. The aquifer is unconfined and consists of 20–40 m of pleistocene, highly permeable carbonate—free sands and gravels underlain by impermeable cretaceous clays. More information about the research site is given by Frind et al. (1990) and Deurer et al. (2008). In the FFA, substantial microbially mediated processes and reactions like denitrification and desulfurication occur, strongly influencing groundwater chemistry. Autotrophic denitrification with reduced sulphur as an electron donor is the dominant process in the deeper aquifer (Böttcher et al. 1992). In the surface groundwater, heterotrophic denitrification with organic carbon as an electron donor has replaced the autotrophic process due to exhaustion of the reduced sulphur compounds (Kölle et al. 1983; Deurer et al. 2008). The heterotrophic process close to the groundwater table is characterised by low nitrate removal efficiency (Weymann et al. 2008). The organic carbon content of the aquifer sands is typically in a range between 0.5 and 0.8 g kg−1. Dissolved organic carbon (DOC) was found to be between 20 and 70 mg l−1 in groundwater samples. Our study was conducted from July–September 2007 on an 8 m2 measuring field plot situated at the south of the Fuhrberger Feld aquifer (52°32′N, 9°51′E). Until July 2005, tillage has been the dominant land use for years. The last cultivated species was Festuca rubra. Afterwards, the field had been kept under fallow for experimental purposes. Despite a stronger influence of denitrification and groundwater-derived N2O is likely during the winter months, we decided to conduct the experiment in summer in order to study the effects of a groundwater drawdown which could be expected during that time.
Experimental setup
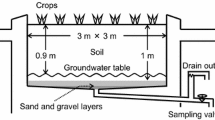
To apply the tracer solution, the plot was divided into a raster of 0.5 by 0.5 m. This yielded 45 raster points, where we installed polyvinyl chloride (PVC) pipes (30 mm inner diameter) with their endings direct above the groundwater table. To assess 15N enrichment gradients of N2O, we conducted measurements in groundwater, in the unsaturated zone and at the soil surface. Four multilevel sampling wells (Böttcher et al. 1985) were installed at the plot in order to collect samples from near the groundwater surface from defined depths (1.5, 1.6 and 1.7 m below soil surface). The soil atmosphere was sampled using gas probes installed in four replications in the unsaturated zone at depths of 0.3, 0.6 and 0.9 m, respectively. Surface emissions were monitored using eight static flux chambers. The arrangement of the PVC pipes, the wells, the gas probes and the static flux chambers at the measuring field are represented in Fig. 1.

Measuring field with its elements and dimensions
Tracer application and sampling schedule
We prepared a tracer solution on site using raw water from a drinking water well of the waterwork of the Fuhrberger Feld aquifer. Although parts of the aquifer receive a strong influx of NO3 − from agricultural activities, the raw water contains only traces of NO3 − due to intensive denitrification (Duijnisveld et al. 1989). The raw water was filled into two canisters and K15NO3 (60 atom%) was added. The NO3 − concentration of the tracer solution was 12.5 mg N l−1, which is close to the mean NO3 − concentration in seepage water of the arable land. The tracer solution was transferred into 45 plastic buckets (10 l). Afterwards, 10 l of the tracer solution were carefully applied to the groundwater surface via each PVC pipe. Pieces of silicone tubing (diameter of 6 mm, length of 1 m) were used as siphons to transfer the tracer solution from the buckets to the PVC pipes. Great care was taken to exclude contamination of the soil surface with tracer solution in order to avoid labeling of the topsoil. In order to assure a permanent labeling of surface groundwater throughout the experiment, the tracer solution was applied weekly. A single injection was typically completed within 15 min. We conducted the first tracer application on July 4, 2007 and the last on August 29, 2007. Altogether, nine tracer applications took place.
Prior to the first tracer application, we measured NO3 −-N and N2O-N concentrations in the surface groundwater. From July 11 to September 14, 2007, the surface groundwater, the soil atmosphere and samples from the flux chambers were collected weekly. Sampling was conducted at the days of labeling while the tracer solution was applied. Total N2O and its 15N enrichment were analysed in the surface groundwater, in the soil atmosphere and in the samples from the flux chambers. NO3-N and 15N enrichment of N2 and NO3 − were analysed in the groundwater samples.
Sampling methods and analytical techniques
Surface groundwater samples for measuring NO3 − concentrations, N2O concentrations and the 15N enrichments of NO3 −, N2O and N2 were collected using partially evacuated (−0.53 bar) serum bottles (118 ml) sealed with gas-tight butyl rubber septa and crimp caps. 50-ml samples of groundwater were collected with a plastic syringe from the multilevel sampling wells and were transferred into the partially evacuated serum bottle without any air contact. The first 20 ml of every depth were discarded. The samples were stored upside down in water at 4°C and were measured within 10 days. Prior to the gas measurements, liquid and gas phase were equilibrated at constant temperature (25°C) by agitating on a horizontal shaker for 3 h. Concentrations of N2O in the gas phase of the serum bottles were directly analysed using a gas chromatograph (Fisons GC 8000, Milan, Italy) equipped with a split-injector and an electronic capture detector and a HP-PLOT Q column (30 m length × 0.32 mm ID; Agilent Technologies, Santa Clara, USA) kept at 30°C. The split ratio was 1:8 and Ar–CH4 (95/5) was used as carrier and make-up gas. Samples of 300 μl were injected using an autosampler (model GC-PAL, CTC-Analytics, Zwingen, Switzerland). Precision as given by the SD obtained from four injections of a standard gas was typically 1.5%. Dissolved N2O was calculated from the gas phase concentrations of the water samples using the Bunsen absorption coefficient of N2O (Weiss and Price 1980). 15N-N2 was analysed following the method specified in Well et al. (1998) and (2003). The gas concentrations of the sample solution (dissolved N2) were calculated according to Henry’s law from the headspace concentrations using the Bunsen absorption coefficient of N2 (Weiss 1970).
NO3 − in the groundwater samples was determined photometricly using a continuous flow analyser (Skalar, Erkelenz, Germany). The measurements precision was 5%.
Gas probes for sampling the soil atmosphere in the unsaturated zone were modified from a soil atmosphere sampling device as described by Schack-Kirchner et al. (1993). PVC pipes were permanently installed in defined soil depths (0.3, 0.6 and 0.9 m below the soil surface), containing an aerated macropore, which was connected with the soil surface using an 1 mm-ID stainless steel tubing. We collected the soil atmosphere by using a plastic syringe, which was connected with the tubing via a plastic three-way luer-lock stop cock (Braun, Melsungen, Germany). Prior to the sample collection we discarded the first 5 ml to flush the volume of the tubing with soil air. Afterwards, the soil atmosphere sample was transferred into evacuated serum bottles (118 ml) sealed with gas-tight butyl rubber septa and crimp caps. The samples were analysed within 10 days using the gas chromatograph as described above.
Surface N2O emissions were measured using a static chamber technique. Eight PVC base collars, 24 cm in diameter and covering an area of 0.0434 m2, were cut into the soil to a depth of 6 cm and remained in place until the end of the study. Plexiglass covers were prepared with a vent tube to allow pressure equilibration (Mosier 1989) and were covered with aluminium foil to avoid heating during the measurements. Prior to the collection of gas samples, the covers were attached to the PVC-collars using a rubber seal, resulting in a total volume of the static chambers of 14 l. Four gas samples were taken with a syringe at 20 min intervals over a 1 h period (0, 20, 40, 60 min). At 0 and 60 min, gas samples were transferred into evacuated serum bottles (118 ml) sealed with gas-tight butyl rubber septa and crimp caps, in order to allow GC and IRMS analysis. At 20 and 40 min, gas samples were transferred into evacuated gas-tight 12-ml ExetainersTM (LABCO, High Wycombe) for GC-analysis only. The samples were analysed within 1 week.
15N enrichment of N2O was analysed using an isotope ratio mass spectrometer (Delta XP IRMS, Thermo-Finnigan, Bremen, Germany). The IRMS was connected to a modified Precon (Thermo-Finnigan, Bremen, Germany) equipped with an autosampler (model Combi-PAL CTC-Analytics, Zwingen, Switzerland) as described by Casciotti et al. (2002). 15N enrichments are reported as atom% 15N or δ15N versus air-N2 (in ‰). Typical analytical precision for δ15N was 0.6‰, the detection limit for N2O-N was 0.5 nM.
15N enrichment of NO3− was determined using the method described by McIlvin and Altabet (2005). This method is based on the reduction of NO3− to nitrite (NO2−) with spongy cadmium and a further reduction of NO2− to N2O using sodium azide in an acetic acid buffer. Both reduction steps are assumed to be complete. 15N enrichment of N2O was analysed using the instrumentation as described above.
Groundwater level, precipitation and soil water content
The groundwater level was continuously recorded during the experiment in order to assess the distance between the groundwater surface and the level of the injection pipes. This was done with a water depth gauge (Keller Druckmesstechnik GmbH, Jestetten, Germany). The measurement accuracy was ±0.01 m. Precipitation was measured to estimate the occurrence of potential seepage during the experiment which might cause dilution of the 15N tracer by NO3 − leaching to the groundwater surface. For recording of precipitation, we installed a rain gauge with a tilting balance (Lambrecht, Göttingen, Germany). The rain gauge was connected with a data logger (DL2e, Delta-T Devices Ltd, Cambridge, UK) which recorded the rainfall every hour. The minimal resolution was 0.1 mm m−2. To assess possible effects of soil moisture and soil temperature on N2O fluxes at the soil surface, these soil properties were measured using time domain reflectrometry (TDR) (Easy Test, Poland). We installed two TDR probes in a depth of 0.2 m. The measurement accuracy was 2% for soil moisture and 0.8°C for the temperature. Water filled pore space (WFPS) was subsequently calculated from the bulk density and soil moisture, assuming a particle density of 2.65 g cm−3.
Flux calculations and the mass of groundwater-derived N2O in surface emissions
Fluxes of total N2O-N at the soil surface were calculated by linear regression from four samples taken from the flux chambers (atmospheric air before enclosure; samples 20, 40 and 60 min after enclosure). The slope of the temporal change of N2O concentrations within the closed chamber had to show r 2 > 0.8 derived from linear regression analysis to be accepted as significant. Otherwise, fluxes were considered zero. Negative fluxes were counted in the data set, because those fluxes have been measured at the soil surface under a large range of conditions Chapuis-Lardy et al. (2007).
The mass of groundwater-derived N2O (cN2Ogw) [ppmv] in the chamber atmosphere after 1-h enrichments was calculated using a mixing equation for the three components groundwater, unsaturated zone and surface emissions, respectively:
what leads to
where δ mix, chamber is the 15N enrichment of N2O measured in the flux chambers after 60 min. cN2Osoil, cN2Oatm and cN2Ochamber are concentrations of N2O in the soil atmosphere of the unsaturated zone (means of all investigated sampling depths), in atmospheric air and in the flux chambers after 60 min in ppmv, respectively. δ gw, δ soil and δ atm denote the 15N-enrichment of N2O in the groundwater (means of the investigated sampling depths), in the soil atmosphere of the unsaturated zone and in atmospheric air.
Prior to the calculation of the mass of groundwater-derived N2O, we compared δ mix, chamber with background δ 15N of N2O in atmospheric air and in the soil atmosphere of a control plot. The background of δ 15N of N2O was between 2 and 8‰, the calculated mean value was 4.4‰ and the SD was 1.2‰, respectively, (data not shown). A confidence interval analysis with 94 samples using a confidence coefficient (α) of 0.001 resulted in an upper interval limit of 4.78‰. We only used samples with δ 15N of N2O higher than 4.78‰ for the calculations of the mass of groundwater-derived N2O and groundwater-derived N2O fluxes. Otherwise, the mass of groundwater-derived N2O and groundwater-derived N2O fluxes were considered zero.
To calculate the flux of groundwater-derived N2O to the flux chambers, we converted cN2Ogw [ppmv] into the mass of groundwater-derived N2O in the chamber, N2Ogw [μg N2O-N] by using molar weight and molar volume of N2O-N and the volume of the flux chambers. Finally, we used the following equation for the calculations of groundwater-derived fluxes:
where N2Ogw is the mass of groundwater derived N2O in the surface emission during a 1-h enrichment interval under the flux chamber in μg N h−1. A chamber is the area that is covered by the collars in m2, T denotes the collection time in h and F is the flux of groundwater derived N2O in μg N2O-N h−1 m−2.
Results
Groundwater level, soil moisture and meteorological conditions
During July and August 2007, the precipitation was 104.3 and 102.9 mm, respectively. The groundwater level varied between 1.09 and 1.42 m below the soil surface (Fig. 2a). If the groundwater levels at the start and at the end of the measuring period in Fig. 2a are compared, it can be seen that no effective decrease of the groundwater level occurred. This was contrary to our expectations, because a decrease of the groundwater level during the summer is representative and has been observed earlier (Deurer et al. 2008).

Groundwater level below the soil surface, precipitation (a) and water-filled pore space as well as soil temperature at 0.2 m below the soil surface (b) during the measurement period
However, there were two stages with stronger groundwater fluctuation: a drawdown of 0.26 m from day 12 to day 20 after the first application of the tracer solution and a phreatic rise of 0.16 m within 2 days from day 48 to day 50. The drawdown occurred during a dry period between day 9 and day 17. This period was also characterized by lowest WFPS and highest soil temperatures in the topsoil (0.2 m below the soil surface, Fig. 2b). The abrupt phreatic rise from day 48 to day 50 followed a major precipitation event at day 48 with a daily rainfall of 34 mm (Fig. 2a). Correspondingly, we observed a WFPS of 49% at day 49, which was the highest during the measurement period (Fig. 2b).
NO3 − concentrations and 15N enrichment of NO3 − in the surface groundwater
The time course of NO3 − concentrations in the surface groundwater (1.5–1.7 m below soil surface) is represented in Fig. 3a. Prior to the tracer application, NO3 − concentrations were low, ranging from 0.38 to 0.82 mg N l−1. During the labeling phase with K15NO3 tracer solution (12.5 mg NO3-N l−1), mean NO3 − concentrations in each depth increased until day 21 to a level of approximately 6–10 mg N l−1. After this NO3 − concentrations in 1.5 and 1.6 m remained nearly constant until the last sampling event, whereas NO3 − concentrations in 1.7 m decreased temporarily to 3.5 mg N l−1 and reincreased from day 42 to the last sampling event to the concentration level in 1.5 and 1.6 m depth.

NO3 −-N concentrations (a) and 15N enrichment of NO3 − (b) in the surface groundwater. Error bars denote the SD of four samples collected from multilevel wells. The dates of the tracer applications are indicated by arrows
The time course of the 15N enrichment of NO3 − in the surface groundwater is shown in Fig. 3b. At day 7, the labeling of the surface groundwater decreased rapidly with depth. The 15N enrichment of NO3 − was 21.1, 14.6 and 4.2 atom% in 1.5, 1.6 and 1.7 m, respectively. In contrast, we observed a substantial and relatively homogenous labeling of the surface groundwater from day 15 until the end of the measuring period. Both, the increased NO3 − concentrations as well as the stable labeling of the surface groundwater since day 15 show that the label had been successfully distributed at the groundwater surface.
N2O and 15N-N2 concentrations in the surface groundwater, N2O concentrations in the unsaturated zone and total N2O fluxes at the soil surface
Time courses of the mean N2O concentrations and of the N2O fluxes at the soil surface are represented in Fig. 4. In the surface groundwater (Fig. 4c), highest N2O concentrations between 14.0 and 15.8 μg N2O-N l−1 occurred at day 21. At all the other sampling events, N2O concentrations were comparatively low, ranging between 0.9 and 4.9 μg N2O-N l−1 with a tendency to lowest concentrations at the beginning and at the end of the labeling time. The lowest value is very close to a concentration of 0.88 μg N2O-N l−1, which reflects the N2O concentration in water equilibrated with atmospheric air at groundwater temperature. Generally, we found no significant differences in N2O concentrations between the sampling depths at the same sampling day. Mean 15N-N2 concentrations in the groundwater were 56.14 (±50.42) μg l−1 and within the range of the concentrations that were previously measured in the saturated zone of hydromorphic soils (Well et al. 2003). The mean N2O-to-N2 ratio was found to be 0.10 (±0.11).

N2O fluxes at the soil surface measured in static flux chambers after an enrichment time of 60 min (a), N2O concentration in the soil atmosphere in 0.3, 0.6 and 0.9 m depth below the soil surface (b) and dissolved N2O concentration in the surface groundwater in 1.5, 1.6 and 1.7 m depth below soil surface (c). Error bars denote the SD from eight static flux chamber measurements (a) and four samples collected from defined depths in the unsaturated zone (soil atmosphere, b) and from surface groundwater (c), respectively. The arrows indicate the date of the tracer applications
In the soil atmosphere of the unsaturated zone (Fig. 4b), N2O concentration ranged between 297 and 427 ppbv and was thus close to ambient concentration for the entire measurement period. However, highest N2O concentration levels occurred at day 7 and day 49 after the first tracer application and were connected with significant precipitation rates several days running or a major precipitation event at day 48, respectively. In contrast, we found lowest N2O concentrations in the soil atmosphere at day 15 during a period of several days without rainfall (Figs. 2, 4b). Significant differences in N2O concentrations between the three sampling depths were not detected, with the exception of the sampling event at day 35, where we measured a N2O concentration in 0.9 m depth that was up to 56 ppbv higher than those in 0.3 and 0.6 m depth.
Throughout the study period, we carried out a total of 71 measurements of total N2O fluxes at the soil surface using the closed chamber method. In 37 cases, fluxes were not significant (r 2 of the slopes <0.8). Furthermore, we observed significant positive fluxes in 25 cases and significant negative fluxes in 9 cases.
Mean N2O fluxes of the sampling events ranged between −7.6 and 29.1 μg N2O-N m−2 h−1 (Fig. 4a). We measured highest fluxes of 29.1 and 18.2 μg N2O-N m−2 h−1 at day 49 and day 72, respectively. At the other sampling events, fluxes were very low and close to zero. Low negative fluxes occurred at the days 15, 21, 28 and 42 (Fig. 4a).
15N enrichment of N2O in the surface groundwater, in the unsaturated zone and at the soil surface
Figure 5 illustrates the time courses of the mean 15N enrichment of N2O. Generally, 15N enrichment of N2O in the profile rapidly decreased upwards from the surface groundwater via the unsaturated zone to the soil surface.

15N enrichment of N2O: (a) at the soil surface measured in flux chambers after an enrichment time of 60 min, (b) in soil atmosphere in 0.3, 0.6 and 0.9 m depth below soil surface and (c) in surface groundwater in 1.5, 1.6 and 1.7 m depth below soil surface. Error bars denote the SD from eight static flux chamber measurements (a) and four samples of each depth in the unsaturated zone (soil atmosphere, b) and four samples of each depth from the surface groundwater (c), respectively. The arrows indicate the date of the tracer applications
In the surface groundwater (Fig. 5c), we observed average δ 15N between 56,000 and 207,000‰ corresponding to 13 and 42 atom% 15N (mean of 3 sampling depths per sampling event, respectively), i.e. less than the 15N enrichment of the applied tracer solution of 60 atom%. At day 7, no significant 15N enrichment of the groundwater in 1.7 m depth occurred suggesting that a single application of the tracer solution caused only substantial labeling of the groundwater in 1.5 and 1.6 m depth. We found similar results for the pattern of the 15N enrichment of NO3 − in the surface groundwater (Fig. 3b).
In the soil atmosphere of the unsaturated zone (Fig. 5b), N2O was already significantly enriched with 15N at the first sampling date after the first tracer application. Mean δ 15N of N2O was 35, 63 and 92‰ in 0.3, 0.6 and 0.9 m depth, respectively. δ 15N of each sample was significantly higher than the background δ 15N of N2O in the soil atmosphere and atmospheric air, which we determined at a control plot giving a range from 2 to 8‰ and a mean value of 4.4‰ (data not shown). At all sampling days, δ 15N of N2O in the soil atmosphere depended on sampling depth and followed the order 0.3 m < 0.6 m < 0.9 m. At day 21, a clear peak of the mean δ 15N of N2O occurred at each depth (Fig. 5b).
At the soil surface, mean δ 15N of N2O was substantially lower than in the soil atmosphere. Mean δ 15N of N2O was within the range of background δ 15N of N2O at four out of nine sampling events (days 15, 35, 42 and 49). In contrast, mean δ 15N of N2O was higher and out of the background range at the other five sampling events. The highest mean δ 15N of N2O was 38‰ at day 21 (Fig. 5a).
Groundwater derived N2O in surface emissions
Mass of groundwater derived N2O in the chambers per sampling event covered an interval between 0.0002 and 0.0009 μg N2O-N (Table 1) after an enrichment time of 60 min in the flux chambers. These proportions reflect very low N2O fluxes from the surface groundwater via the unsaturated zone to the soil surface. We calculated mean fluxes from 0.0022 to 0.0207 μg N2O-N m−2 h−1 per sampling event (Eqs. 1, 2, 3; Table 1) which is equivalent to 0.0002–0.0018 kg N2O-N ha−1 year−1. Only 0.04–0.28% (depending on sampling date, averaged 0.13%) of the total positive N2O fluxes at the soil surface originated from groundwater-derived N2O (Table 1; Fig. 4a).
Discussion
Groundwater labeling
Was the extent of groundwater labeling sufficient for detecting fluxes of 15N2O that was produced in the surface groundwater by denitrification? In contrast to push-and-pull tracer studies carried out at single groundwater wells (Addy et al. 2002; Well et al. 2003; Kim et al. 2005), we performed a tracer experiment with labeling of an extended area of the surface groundwater where an even distribution of the label was approximated by using multiple injection points. Because concentrations of NO3 − and N2O in the groundwater were very low before the first tracer application (Figs. 3, 4c) and the NO3 − concentration of the tracer solution and its 15N enrichment were comparatively high, we achieved a high enrichment of NO3 − within the groundwater layer which was sampled by the multilevel wells. This was shown by both, increasing concentrations of NO3 − as well as the significant 15N enrichment of the groundwater at all multilevel wells. Consequently, the prerequisite for using δ 15N of N2O to monitor formation of N2O and its emission at the soil surface was fulfilled. However, temporal and spatial variation of labeled N2O and NO3 − was relatively high at all sampling events, showing that there was variable mixing of the tracer solution with the original groundwater.
Identifying the origin of N2O
To which extent was the variability of N2O concentrations in the soil atmosphere of the unsaturated zone and of N2O fluxes at the soil surface caused by N2O produced in the surface groundwater? With a few exceptions, the variability of N2O concentrations in the soil atmosphere of the unsaturated zone and of fluxes at the soil surface was relatively low. Peak N2O concentrations in the groundwater occurring at day 21 did not enhance N2O concentrations in the soil atmosphere and surface fluxes, respectively. This is in line with observations of Van Groenigen et al. (2005), who did not find increases in N2O fluxes from the topsoil as a result of increased N2O concentrations in the subsoil. Furthermore, highest surface fluxes and concentrations in the soil atmosphere measured at day 49 did not coincide with elevated groundwater N2O concentration but with the major precipitation event at day 48 and with the highest value for WFPS in the topsoil measured during the study. Our observation of lowest N2O concentrations in the soil atmosphere and at the soil surface during dry periods with comparatively low values of WFPS and high soil temperatures is in agreement with the frequently reported moisture effect on N2O emissions, which typically leads to low fluxes under these conditions (Granli and Bøckman 1994; Dobbie et al. 1999). According to these findings and to the results represented in this study, it can be concluded that N2O concentrations in the soil atmosphere and N2O fluxes measured at the soil surface were more affected and controlled by factors like precipitation, soil moisture and temperature of the topsoil but obviously less by groundwater N2O dynamics.
We assume that decreasing 15N enrichment of N2O upwards from groundwater via the unsaturated zone to the soil surface is mainly caused by dilution of the 15N-N2O pool with soil derived and atmospheric 14N-N2O. Clough et al. (1999) also observed a decrease in the 15N enrichment of N2O as the gas diffuses upwards through the profile. The authors mentioned dilution as the most likely cause for decreasing 15N enrichment of N2O, but their results are also compatible with consumption of 15N-enriched N2O in the upper part of the soil. In our study, 15N enrichment of N2O in the groundwater was comparatively low at day 49 (Fig. 5). It can be assumed that this phenomenon is also a dilution effect caused by the major precipitation event at day 48.
A clear peak in the time courses of the mean 15N enrichments of N2O through the whole profile occurred at day 21 (Fig. 5) and coincided with a substantial drawdown of the groundwater level immediately before sampling. Apparently, a rapid drawdown can cause a rapid release of dissolved N2O into the unsaturated zone (Well 2002, Grant and Pattey 2003) which might cause also an increased 15N-enrichment of N2O near the soil surface. Otherwise, the increasing groundwater level starting with day 48 did not induce dynamics of the 15N-enrichment in the unsaturated zone and at the soil surface. Aeschbach-Hertig et al. (2002) argued that during a rise of the water table soil air can be trapped in pores and fractionally dissolved under an increasing hydrostatic pressure. Relating this concept to our results, we hypothesize that a release of N2O is unlikely during a phreatic rise, because dissolution of soil air is the dominant process. Hence, we assume that dynamics of the 15N-enrichment within the profile is prevented during a rise of the water table .
Fluxes of total N2O at the soil surface and of groundwater-derived N2O to the atmosphere
Compared to other studies reviewed in Chapuis-Lardy et al. (2007), total fluxes of N2O at the soil surface were low. During the measuring period, a majority of these fluxes were close to zero and not significant (Fig. 4a). Similar low fluxes were found by Butterbach-Bahl et al. (2002) for a sandy cambisol under pine forest in northeastern Germany, ranging between −4.1 and 34.1 μg N2O-N m−2 h−1. This suggests that total fluxes of our study are typical for unfertilized sandy soils.
We observed slightly increased emissions at day 49, following the major precipitation event at day 48. In contrast, highest measured N2O concentrations in groundwater at day 21 did not affect the surface fluxes. These observations confirm the assumption that surface fluxes were hardly influenced by the dynamics of the groundwater level and N2O concentrations in groundwater.
The very low groundwater-derived N2O fluxes to the atmosphere indicate that the importance of groundwater-derived N2O for atmospheric emissions was negligible at our research site during the measuring period. This is illustrated by the fact that only 0.04–0.28% of the positive N2O fluxes at the soil surface originated from 15N2O-N that was produced in groundwater.
In a previous study conducted in the Fuhrberger Feld aquifer, Deurer et al. (2008) estimated upward N2O fluxes from the surface groundwater into the unsaturated zone based on measurements of groundwater N2O concentrations at six multilevel wells. These fluxes ranged from 0.0009 to 0.3 kg N2O ha−1 year−1 which is equivalent to a range from 0.0006 to 0.2 kg N2O-N ha−1 year−1. In our study, fluxes of 15N2O from groundwater to the atmosphere were found to be in a range from 0.0002 to 0.0018 kg N2O-N ha−1 year−1, what is similar to the range of the data of Deurer et al. (2008). However, it remains to be determined to what extent potential N2O consumption in the unsaturated zone and in the topsoil allows a comparison between the different approaches. More research is needed to quantify consumption of N2O during its transport via the unsaturated zone to the atmosphere. In contrast to the results presented in this study and in Deurer et al. (2008), Ronen et al. (1988) estimated for the sandy Coastal Plain aquifer of Israel that the N2O flux from groundwater into the unsaturated zone ranges from 3.4 to 7.8 kg N2O-N ha−1 year−1. This is an extremely high emission rate, although groundwater N2O concentration was not substantially different from the highest values measured by Deurer et al. (2008). This discrepancy can be explained by a calculation error in Ronen et al. (1988) caused by using an incorrect unit (mg l−1 instead of μg l−1) for the concentration gradient of N2O. Thus, the N2O flux from the surface groundwater to the unsaturated zone is substantially overestimated by three orders of magnitude. If the correct concentration gradients would be incorporated in the flux calculation, the data of Ronen et al. (1988) were within the range of the N2O fluxes reported by Deurer et al. (2008), but still higher than the N2O fluxes reported in this study according to the different N2O concentrations in the groundwater. Other previous studies tend to confirm our result of relatively low diffusive N2O fluxes from the groundwater to the atmosphere (McMahon et al. 2000; Hiscock et al. 2003). In the light of these studies and of our results, indirect N2O emissions via the diffusive pathway seem to be hardly significant.
Conclusions
We measured N2O fluxes from the surface layer of a sandy aquifer through the unsaturated zone to the atmosphere. For the first time, we could prove fluxes of labeled N2O to the soil surface that was produced in groundwater by denitrification using a 15N tracer in situ approach.
Stable 15N labeling of the surface groundwater for several weeks showed that it was possible to monitor these fluxes over an extended period and thus yielded robust results.
The contribution of groundwater-derived 15N2O to surface emissions was very low within the measuring period and not detectable with the conventional closed chamber method. Using the 15N tracer technique, we observed highest N2O fluxes from groundwater in temporal connection with a rapid decrease of the groundwater table which suggests that diffusive indirect N2O emissions from groundwater can be favoured by such dynamics. Generally, our data support previous assumptions that indirect N2O emissions from groundwater occurring by upward diffusion to the atmosphere are hardly an important part of total N2O emissions. This shows that the neglect of diffusive emissions in previous estimates of emission factors did not lead to a significant underestimation of total groundwater-derived fluxes.
References
Addy K, Kellog DQ, Gold AJ, Groffman PM, Ferendo G, Sawyer C (2002) In situ push—pull method to determine ground water denitrification in riparian zones. J Environ Qual 31:1017–1024
Aeschbach-Hertig W, Beyerle U, Holocher J, Peeters F, Kipfer R (2002) Excess air in groundwater as a potential indicator of past environmental changes. In: Study of environmental change using isotope techniques, Proceedings of the IAEA conference, Vienna, 23–27 April 2001, C&S Papers Series 13/P, pp 174–183
Böttcher J, Strebel O, Duijnisveld WHM (1985) Vertikale Stoffkonzentrationsprofile im Grundwasser eines Lockergesteins—Aquifers und deren Interpretation (Beispiel Fuhrberger Feld). Zeitschr dt Geol Ges 136:543–552
Böttcher J, Strebel O, Vörkelius S, Schmidt H (1990) Using isotope fractionation of nitrate-nitrogen and nitrate-oxygen for evaluation of microbial denitrification in a sandy aquifer. J Hydrol (Amst) 114:413–424. doi:10.1016/0022-1694(90)90068-9
Böttcher J, Strebel O, Kölle W (1992) Redox conditions and microbial sulfur reactions in the Fuhrberger Feld sandy aquifer. In: Matthess G, Frimmel FH, Hirsch P, Schulz HD, Usdowski E (eds) Progress in hydrogeochemistry. Springer, Berlin Heidelberg, pp 219–226
Butterbach-Bahl K, Breuer l, Gasche R (2002) Exchange of trace gases between soils and the atmosphere in Scots pine forest ecosystems of the northeastern German lowlands: 1. Fluxes of N2O, NO/NO2 and CH4 at forest sites with different N-deposition. For Ecol Manage 167:123–134
Casciotti KL, Sigman DM, Hastings MG, Böhlke JK, Hilkert A (2002) Measurement of the oxygen isotopic composition of nitrate in seawater and freshwater using the denitrifier method. Anal Chem 74:4905–4912. doi:10.1021/ac020113w
Chapuis-Lardy l, Wrage N, Metay A, Chotte JL, Bernoux M (2007) Soils, a sink for N2O? A review. Glob Change Biol 13:1–17. doi:10.1111/j.1365-2486.2006.01280.x
Clough TJ, Jarvis SC, Dixon ER, Stevens RJ, Laughlin RJ, Hatch DJ (1999) Carbon induced subsoil denitrification of 15N labelled nitrate in 1 m deep soil columns. Soil Biol Biochem 31:31–41. doi:10.1016/S0038-0717(98)00097-2
Clough TJ, Sherlock RR, Rolston DE (2005) A review of the movement and fate of N2O in the subsoil. Nutr Cycl Agroecosyst 72:3–11. doi:10.1007/s10705-004-7349-z
Clough TJ, Addy K, Kellog DQ, Nowicki BL, Gold AJ, Groffman PM (2007) Dynamics of nitrous oxide in groundwater at the aquatic–terrestrial interface. Glob Change Biol 13:1528–1537. doi:10.1111/j.1365-2486.2007.01361.x
Crutzen PJ (1981) Atmospheric chemical processes of the oxides of nitrogen, including nitrous oxide. In: Delwiche C (ed) Denitrification, nitrification and nitrous oxide. Wiley, New York
Deurer M, von der Heide C, Böttcher J, Duijnisveld WHM, Weymann D, Well R (2008) The dynamics of N2O in the surface groundwater and its transfer into the unsaturated zone: a case study from a sandy aquifer in Germany. Catena 72:362–373. doi:10.1016/j.catena.2007.07.013
Dobbie KE, McTaggert IP, Smith KA (1999) Nitrous oxide emissions from intensive agricultural systems: variations between crops and seasons, key driving variables, and mean emission factors. J Geophys Res 104:26891–26899. doi:10.1029/1999JD900378
Duijnisveld WHM, Strebel O, Böttcher J, Kinzelbach W (1989) Longterm prognosis of the groundwater quality as influenced by landuse and landuse changes in the Fuhrberger Feld using numerical solute transport models. In: Kobus HE, Kinzelbach W (eds) Contaminant transport in groundwater. A.A. Balkema, Rotterdam Brookfield, pp 89–96
Frind E, Duijnisveld WHM, Strebel O, Böttcher J (1990) Modeling of multicomponent transport with microbial transformation in groundwater: the Fuhrberg case. Water Resour Res 26:1707–1719
Granli T, Bøckman OC (1994) Nitrous oxide from agriculture. Nor J Agr Sci 12:1–128
Grant RF, Pattey E (2003) Modelling variability in N2O emissions from fertilized agricultural fields. Soil Biol Biochem 35:225–243. doi:10.1016/S0038-0717(02)00256-0
Groffman PM, Gold AJ, Kellog DQ, Addy K (2002) Mechanisms, rates and assessment of N2O in groundwater, riparian zones and rivers. In: Van Ham J, Baede APM, Guicherit R, Williams-Jacobse JGFM (eds) Non-CO2 greenhouse gases: scientific understanding, control options and policity aspects. Millpress, Rotterdam, pp 159–166
Groffman PM, Altabet MA, Böhlke JK, Butterbach-Bahl K, David MB, Firestone MK, Giblin AE, Kana TM, Nielsen LP, Voytek MA (2006) Methods for measuring denitrification: diverse approaches to a difficult problem. Ecol Appl 16(6):2091–2122. doi:10.1890/1051-0761(2006)016[2091:MFMDDA]2.0.CO;2
Heincke M, Kaupenjohann M (1999) Effects of soil solution on the dynamics of N2O emissions: a review. Nutr Cycl Agroecosyst 55:133–157. doi:10.1023/A:1009842011599
Hiscock KM, Bateman AS, Fukada T, Dennis P (2002) The concentration and distribution of groundwater N2O in the chalk aquifer of eastern England. In: Van Ham J, Baede APM, Guicherit R, Williams-Jacobse JGFM (eds) Non-CO2 greenhouse gases: scientific understanding, control options and policity aspects. Millpress, Rotterdam, pp 185–190
Hiscock KM, Bateman AS, Mühlherr IH, Fukada T, Dennis PF (2003) Indirect emissions of nitrous oxide from regional aquifers in the United Kingdom. Environ Sci Technol 37:3507–3512. doi:10.1021/es020216w
Intergovernmental Panel on Climate Change (IPCC) (2006) 2006 IPCC guidelines for national greenhouse gas inventories. Prepared by the national greenhouse gas inventories programme. Egglestone HS, Buendia l, Miwa K, Ngara T, Tanabe K (eds), IGES, Japan
Intergovernmental Panel on Climate Change (IPCC) (2007) Climate change 2007: AR4 synthesis report. IPCC Plenary XXVII, Valencia, Spain, pp 212–217 November 2007
Kim Y, Kim JH, Son BH, Oa SW (2005) A single well push—pull test method for in situ determination of denitrification rates in a nitrate—contaminated groundwater aquifer. Water Sci Technol 52(8):77–86
Kölle W, Werner P, Strebel O, Böttcher J (1983) Denitrifikation in einem reduzierenden Grundwasserleiter. Vom Wasser 61:125–147
Korom SF (1992) Natural denitrification in the saturated zone: a review. Water Resour Res 28:1657–1668. doi:10.1029/92WR00252
McIlvin MR, Altabet MA (2005) Chemical conversion of nitrate and nitrite to nitrous oxide for nitrogen and oxygen isotopic analysis in freshwater and seawater. Anal Chem 77:5589–5595. doi:10.1021/ac050528s
McMahon PB, Bruce BW, Dennehy KF (2000) Occurrence of nitrous oxide in the central high plains aquifer. Environ Sci Technol 34:4873–4878. doi:10.1021/es001233t
Mosier AR (1989) Chamber and isotope techniques. In: Andreae MO, Schimel DS (eds) Change of trace gases between terrestrial ecosystems and the atmosphere. Wiley, Chichester, pp 175–187
Mosier AR, Kroeze C, Nevison C, Oenema O, Seitzinger SP, van Cleemput O (1998) Closing the N2O budget: nitrous oxide emissions through the agricultural nitrogen cycle. Nutr Cycl Agroecosyst 52:225–248. doi:10.1023/A:1009740530221
Nevison C (2000) Review of the IPCC methodology for estimating nitrous oxide emissions associated with agricultural leaching and runoff. Chemosphere Glob Chang Sci 2:493–500
Reay DS, Smith KA, Edwards AC (2003) Nitrous oxide emission from agricultural drainage waters. Glob Chang Biol 9(2):195–203. doi:10.1046/j.1365-2486.2003.00584.x
Reay DS, Smith KA, Edwards AC, Hiscock KM, Dong LF, Nedwell D (2005) Indirect nitrous oxide emissions: revised emission factors. Environ Sci 2:153–158. doi:10.1080/15693430500415525
Rice CW, Rogers KL (1993) Denitrification in subsurface environments: Potential source for atmospheric nitrous oxide. In: Harper et al (eds) Agricultural ecosystems effects on trace gases and global climate change. ASA special publications. American Society of Agronomy, Crop Science Society of America and Soil Science Society of America, Madison, pp 121–132
Ronen D, Magaritz M, Almon E (1988) Contaminated aquifers are a forgotten component of the global N2O budget. Nature 335:57–59. doi:10.1038/335057a0
Ross SM (1995) Overview of the hydrochemistry and solute processes in British wetlands. In: Hughes JMR, Heathwaite AL (eds) Hydrology and hydrochemistry of British wetlands. Wiley, New York, pp 133–182
Sawamoto T, Nakajima Y, Kasuya M, Tsuruta H, Yagi K (2005) Evaluation of emission factors for indirect N2O emission due to nitrogen leaching in agro-ecosystems. Geophys Res Lett 32(3). doi:10.1029/2004GL021625
Schack-Kirchner H, Hildebrandt EE, von Wilpert K (1993) Ein konvektionsfreies Sam-melsystem für Bodenluft. J Plant Nutr Soil Sci 156:307–310. doi:10.1002/jpln.19931560406
Spalding RF, Parrott JD (1994) Shallow groundwater denitrification. Sci Total Environ 141:17–25. doi:10.1016/0048-9697(94)90014-0
Tobias CR, Macko SA, Anderson IC, Canuel EA, Harvey JW (2001) Tracking the fate of high concentration groundwater nitrate plume through a fringing marsh: a combined groundwater tracer and in situ isotope study. Limnol Oceanogr 46(8):1977–1989
Van Groenigen JW, Georgius PJ, van Kessel C, Hummelink EWJ, Velthof GL, Zwart KB (2005) Subsoil 15N-N2O concentrations in a sandy soil profile after application of 15N-fertilizer. Nutr Cycl Agroecosyst 72:13–25. doi:10.1007/s10705-004-7350-6
Weiss RF (1970) The solubility of nitrogen, oxygen and argon in water and sea water. Deep Sea Res 17:721–735
Weiss RF, Price BA (1980) Nitrous oxide solubility in water and sea water. Mar Chem 8:347–359. doi:10.1016/0304-4203(80)90024-9
Well R (2002) Methodical approaches for investigating the role of subsurface environments in the global N2O budget. In: Van Ham J, Baede APM, Guicherit R, Williams-Jacobse JGFM (eds) Non-CO2 greenhouse gases: scientific understanding, control options and policity aspects. Millpress, Rotterdam, pp 167–178
Well R, Becker KW, Langel R, Meyer B, Reineking A (1998) Continuous flow equilibration for mass spectrometric analysis of dinitrogen emissions. Soil Sci Soc Am J 62:906–910
Well R, Augustin J, Meyer K, Myrold DD (2003) Comparison of field and laboratory measurement of denitrification and N2O production in the saturated zone of hydromorphic soils. Soil Biol Biochem 35:783–799. doi:10.1016/S0038-0717(03)00106-8
Well R, Flessa H, Jaradat F, Toyoda S, Yoshida N (2005a) Measurement of isotopomer signatures of N2O in groundwater. J Geophys Res Biogeosci 110:G02006. doi:10.1029/2005JG000044
Well R, Weymann D, Flessa H (2005b) Recent research progress on the significance of aquatic systems for indirect agricultural N2O emissions. Environ Sci 2:143–151. doi:10.1080/15693430500393334
Weymann D, Well R, Flessa H, von der Heide C, Deurer M, Meyer K, Konrad C, Walther W (2008) Groundwater N2O emission factors of nitrate-contaminated aquifers as derived from denitrification progress and N2O accumulation. Biogeosci 5:1215–1226
Acknowledgments
We thank the German Research Foundation for funding this research project. Modification of the gas probes, the instrumentation of the measuring field and sampling would not have been possible without the help of G. Benseler, G. Klump, and F. Trienen. We thank I. Ostermeyer, K. Schmidt and L. Szwec as well as R. Langel (Centre for Stable Isotope Research and Analysis at the University of Göttingen) for always reliable assistance during the laboratory efforts.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Weymann, D., Well, R., von der Heide, C. et al. Recovery of groundwater N2O at the soil surface and its contribution to total N2O emissions. Nutr Cycl Agroecosyst 85, 299–312 (2009). https://doi.org/10.1007/s10705-009-9269-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10705-009-9269-4