
Abstract
Polycyclic aromatic hydrocarbons (PAHs) are a class of compounds containing multiple aromatic rings formed during incomplete combustion. Since many of them are known mutagens and carcinogens, PAHs found in the particulate matter (PM) from the wildfire smoke may pose significant health risks to the wildland firefighters. It is pivotal to determine the levels of PAHs in the PM to evaluate the health effects of their inhalation exposure. However, the determination of PAHs using the conventional chromatographic approaches is often time-consuming and laborious. Herein, we describe a novel method for screening nonpolar and polar PAHs in the PM of smoke by direct analysis in real-time mass spectrometry (DART-MS). PM2.5 and PM10 samples were collected on the quartz filters with a sampling system consisting of a cascade impactor with a portable sampling pump. Various indoor and outdoor experiments from biomass burns were conducted to evaluate the PM sampling systems. PAHs were analyzed by DART-MS and gas chromatography-mass spectrometry (GC–MS) methods. The PM samples were collected in California during the wildfire season of fall 2020, and significant levels of multiple nonpolar PAHs and polar PAHs were detected. Overall, the DART-MS method has shown promising ability for high-throughput screening of PAHs in the PM of smoke. Further studies are currently under way to apply this method to study the particulate phase PAH exposures of wildland firefighters during their firefighting activities.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
In 2020 alone, over 10 million acres of land in the United States were burned in wildfire events [1]. These wildfire events pose serious health risks to firefighters and the public not only because of the inherent danger of the fire itself but also because of the toxicants from the smoke. Particulate matter (PM) is considered the most prominent health hazard from wildfire smoke consisting of organic matter, metals, and dust or soil particles [2]. Larger particles can irritate the nose, throat, and eyes; however, the most dangerous of these toxicants are particles smaller than 10 µm, especially particles 2.5 µm or smaller, referred to as PM2.5, pose the greatest health threat because they can enter and irritate the lungs, and are difficult to be removed from lungs and respiratory tracks [3, 4]. The US Environmental Protection Agency (EPA) has estimated that between 1500 and 2500 deaths annually can be attributed to short-term exposure to PM2.5 from wildfire smoke. Long-term exposure to these toxicants year after year can result in many adverse effects, including neurological effects such as memory loss, depression, and learning disorders, and it can also cause pulmonary diseases such as asthma and chronic obstructive pulmonary disease (COPD) and cardiac disease [2].
One particularly hazardous type of toxicants in PM is polycyclic aromatic hydrocarbons (PAHs), a class of compounds formed by two or more fused aromatic rings containing only carbon and hydrogen [5]. PAHs are formed by the incomplete combustion of organic material and were one of the first pollutants found in the atmosphere to be carcinogenic [5]. High molecular weight PAHs are highly toxic, while low molecular weight PAHs are less hazardous but can react with nitrogen oxides and ozone to form highly toxic polar PAHs [6]. Typically, PAHs are lipophilic, so they can easily bind to airborne carbonaceous particulates, enabling them to persist in the environment and enter the body through breathing. Firefighters generally have a higher exposure to PAHs than workers in most other occupations via inhalation and the transdermal absorption from the contamination of their working environment [7]. The damage imposed on wildland firefighters by PM is increased during the fire suppression time due to the strenuous nature of direct suppression because nasal breathers will switch to oral-nasal breathing during hard labor. This causes less effective filtering of air and a higher deposit of toxicants in the distal airway [8]. Even those fighting structural fires (wearing respiratory personal protective equipment, PPE) have a high risk of exposure to PAHs via dermal and inhalation routes [9]. These dangers are exacerbated by the complete lack of, in most cases, breathing protection for wildland firefighters. Therefore it is critical to analyze the PAH levels from PM to evaluate their effect of exposure to firefighters.
Gas chromatography has traditionally been the most commonly used analytical technique to analyze PAHs. Liquid chromatography (LC) has also been utilized and is especially preferred for the analysis of PAHs in aqueous samples [10]. For ambient air samples, the gas chromatography-mass spectrometry (GC–MS) based method, EPA TO-13A, is commonly used because of its selectivity, sensitivity, and ability to separate PAHs in complex samples [11]. EPA TO-13A method also provides air sampling and sample extraction recommendations for PAH analysis. Briefly, the ambient air is drawn through the filter using an air sampler. The PAHs are extracted from the filter using Soxhlet extraction, which includes the use of methylene chloride as the extraction solvent, and the sample is concentrated using an evaporator before its injection for GC–MS analysis. Other methods have also required laborious sample preparation procedures. For example, solid-phase microextraction was coupled with GC–MS to extract and determine PAH concentrations in air samples [12]. Nevertheless, these methods are labor-intensive and require hours to complete the extraction of PAHs on the filters and the analysis by the analytical instruments (i.e., GC–MS). A high throughput method for fast quantitative analysis of fire-derived PAHs in PM samples will be desirable.
Direct analysis in real-time mass spectrometry (DART-MS) has become an important analytical technique for trace analysis since it came into use in the early 2000s [13]. DART-MS has recently been seen as a screening tool for ignitable liquid residues [14, 15], automotive paints [16], and plant materials [17,18,19]. The main advantages to the DART-MS over more traditional methods such as GC–MS and LC–MS include the versatility of analyzing a variety of sample types, the high throughput of methods, sensitive detection of both polar and nonpolar compounds, and the simple sample preparation [20].
In this study, GC–MS and DART-MS were utilized to analyze PAHs found in the particulate phase of smoke from biomass burns, and their results were compared. This paper demonstrates that the DART-MS allows rapid, simultaneous analysis of multiple PAHs in the PM collected on the quartz filters. The personal PM sampling systems were also tested in different smoke conditions, and the PAHs in PM2.5 and PM10 were analyzed.
2 Experimental
2.1 Materials
A certified reference standard with analyte concentrations of 2.0 mg/mL in dichloromethane was used for quantitative calibration of 18 PAHs from AccuStandard (New Haven, Connecticut, USA), including acenaphthene, acenaphthylene, anthracene, benzo(a)anthracene, benzo(a)pyrene, benzo(b)fluoranthene, benzo(g,h,i)perylene, benzo(k)fluoranthene, chrysene, dibenzo(a,h)anthracene, fluoranthene, fluorene, indeno(1,2,3-cd)pyrene, naphthalene, phenanthrene, pyrene, 1-methylnaphthalene, and 2-methylnaphthalene. For the polar PAHs, 2-naphthol, 1-naphthol, 9-fluorenone were acquired from Sigma-Aldrich (St. Louis, MO, USA), 1-nitropyrene and 2-nitrofluorene were obtained from Santa Cruz Biotechnology (Dallas, TX, USA), and 1-hydroxypyrene, 1,2-benzanthraquinone, and 1-nitronaphthalene were purchased from Tokyo Chemical Industry (TCI, Tokyo, Japan). The solution containing 7 deuterated PAHs at 200 µg/mL each [e.g., acenaphthylene (D8, 98%), benzo(a)pyrene (D12, 98%), benzo(g,h,i)perylene (D12, 98%), fluoranthene (D10, 98%), naphthalene (D8, 98%), phenanthrene (D10, 98%), and pyrene (D10, 98%)] was purchased from Cambridge Isotope Laboratoruies, Inc. (Tewksbury, MA, USA). 9-Bromophenanthrene (Alfa Aesar, Haverhill, MA, USA) was used as an internal standard for the DART-MS experiment. The blank ultra-fine dust (RM8632) was purchased from the National Institute of Standards and Technology, USA.
2.2 Instruments
The GC–MS system consisted of a Shimadzu QP2010S mass spectrometer with an autosampler (Shimadzu, Kyoto, Japan). The analytes were separated on an SHRCI-5MS, 30 m × 0.25 mm I.D. capillary column chemically bonded with 5% diphenyl/95% dimethyl polysiloxane at 0.1 µm film thickness (Shimadzu Scientific Instruments Inc. Columbia, MD, USA). A constant helium flow of 1 mL/min was used, of which a fixed split of 1:5 entered the column. The injection temperature was 270°C. The temperature program started at 100°C and was held for 2 min. The temperature was then raised by 8°C/min to 210°C and then 2°C/min to 280°C. Finally, the temperature was increased by 17°C/min to 340°C and held at 340°C for 6 min. The total run time for this method was 60.28 min. The transfer line and ion source temperatures were maintained at 300°C and 280°C, respectively. Selected ion monitoring (SIM) mode was used to monitor 25 PAHs and 7 isotopic internal standards.
For DART-MS experiments in this study, a Thermo LTQ XL MS (Thermo Scientific, Darmstadt, Germany) was coupled with a DART ion source (IonSense inc., Saugus, MA, USA). The mass range was set in an m/z range of 50–1000 and operated in positive-ion mode. The DART heater was set at 500°C. The IonRocket temperature gradient system (Biochromato, San Diego, CA, USA) was used for the thermal desorption DART-MS analysis. The following temperature gradient was used: the starting temperature was 30°C, which was held for 12 s, and then the temperature was raised at a heating rate of 100°C/min until 600°C with the final temperature hold time of 30 s.
2.3 Sample Collection
The SKC AirCheck TOUCH sampling pumps with personal modular impactors (SKC PM coarse, SKC, Inc., Eighty Four, PA, USA) were used in all PM collections. The Omega PTFE filter and PALLFLEX quartz filter from SKC, Inc. were tested, and the quartz filter was selected for better recovery of PAHs on the filter (average recoveries for 14 tested PAHs: 77 ± 11% for PTFE filter and 86 ± 15% for quartz filter). Two sizes of quartz filters (i.e., 25-mm and 37-mm diameters, SKC, Inc., Eighty Four, PA, USA) were used for analysis: a 37-mm filter for the collection of PM2.5 and a 25-mm filter for PM10. All filters were weighed on a microbalance with a precision of 0.5 µg (Mettler Toledo XPR6UD5, Columbus, OH, USA). The airflow for the sampling pump was set at 3 L/min with ± 5% flow rate accuracy, calibrated individually before deployment using the SKC chek-mate Calibrator with a calibration adapter (SKC, Inc., Eighty Four, PA, USA). The collection time varied among different experiments and was recorded in the sampling pumps and the laboratory notebooks. The pumps were either set up on tripods or put into firefighters' pockets, and the impactors were attached to firefighters’ jackets in the breathing zone region.
The live-fire smoke samples were collected with the help of both the Mount Pleasant Fire Department and the Murfreesboro Fire and Rescue Department in Tennessee. The first set of samples was collected from a prescribed burn using wooden boards placed inside an enclosed fire training house. Two PM sampling pumps were set up on tripods inside the house, while the other two pumps were worn by the firefighters controlling the burn. The sampling lasted about 10 min, including activities of firing, staging, and attacking. The second outdoor prescribed fire took place in Mount Pleasant, TN. The burning material consisted of biomass common to Middle Tennessee, including branches, leaves, and grass. One pump was placed onto a tripod about 10 feet away from the fire, while two pumps were worn by firefighters controlling the fire. The three pumps collected samples for one hour. No accelerant was used to start either fire.
Another set of samples was collected during two California wildfire events, the Silverado fire, which burned from October 26, 2020, till November 7, 2020, and the Bond fire, which burned from December 2, 2020, till December 10, 2020. Three sampling sites were used, all of which had varying distances from the fires (Figure S1). The first site was 4.0 miles from the fires. The second site was 8.0 miles from the fires, and the third site was 26.5 miles from the fires. The sampling devices were placed on tripods for about 16 h. The exact sampling time was recorded on the sampling pump and was used to calculate the toxicant concentrations in the air. The filters were put into filter holders and shipped to the Department of Chemistry, Middle Tennessee State University, for chemical analysis.
Small-scale fires were set for the GC–MS and the DART-MS method development. These fires were set in 1-L aluminum cans, and holes were placed in the bottom of these cans for better airflow. Biomass common to Middle Tennessee, such as pine needles, leaves, and dried grasses, was placed in the can and lit using a lighter with no accelerant. The samples were collected using three pumps placed in a triangle formation on tripods around the can. The impactors were placed about 2 feet above the fire. The samples were collected over one hour.
2.4 Sample Preparation
The filters were weighed and then cut in half for organic chemical analysis and heavy metal analysis (not reported in this study), respectively. For GC–MS analysis, 50 μL of the internal standard (1.00 µg/mL) was added and allowed to dry on the bench for ten minutes. The filter half was placed in 10 mL of an extraction solution (methanol and methylene chloride 1:2, v/v). The solution was then vortexed for 20 s and then sonicated for 20 min. It was then vortexed again, and the filter was removed. The solution was then dried under nitrogen. It was then reconstituted using 2 mL of the extraction solution and vortexed again for 20 s. The solution was filtered using a 0.22 μm nylon syringe filter (13 mm diameter, Celltreat Scientific Products, Pepperell, MA, USA), and 200 μL of the filtered solution was removed for the analysis of both polar and nonpolar PAHs using GC–MS.
An 8-mm filter disc was punched out of the filter for DART-MS analysis. Five μL of an internal standard (9-bromophenanthrene) with a concentration of 500 ng/mL was spiked onto the filter and dried for 5 min. The filter was then placed onto a copper pot, which was heated by a thermal desorption system with the temperature gradient described in Session 2.2. The desorbed chemicals were then analyzed by the DART-MS.
2.5 Method Validation
For the GC–MS, the calibration standards were prepared with the concentrations of 0.02, 0.05, 0.10, 0.20, 0.50, and 1.00 μg/mL in the methanol and methylene chloride mix (1:2, v/v). For the DART-MS, a five-point calibration curve was created using the concentrations of 0.05, 0.10, 0.20, 0.50, and 1.00 μg/mL. Twenty μL of calibration standard solution and 5 μL of internal standard solution were spiked onto the filters, which were dried in the fume hood for 10 min following the sample preparation procedures described in Sect. 2.4. All calibration samples were prepared and analyzed in triplicate. Calibration curves were based on peak area ratios of PAHs to their respective internal standards for the calibration standards. Linearity was assessed by linear least-squares regression.
Twenty µL of solutions with PAH concentrations of 0.05, 0.20, and 0.5 μg/mL were spiked on the filter, extracted, and analyzed in triplicate by both GC–MS and DART-MS. The precision [as relative standard deviation (RSD)] was calculated. The recovery for the GC–MS method was evaluated by comparing the peak area of PAHs with/without the extraction. For the recovery of the DART-MS method, the known concentrations of analytes on the filters were compared against the samples without filters. The analysis was performed in triplicate. The simulated certified dust samples were prepared by spiking 20 µL of the PAH solutions at the concentrations of 0.05, 0.20, and 0.5 μg/mL into about 5 mg blank ultra-fine dust (NIST-RM8632) weighed out on a filter. The filter with the dust was analyzed by the GC–MS and DART-MS methods described above. The results from both analyses were compared to the known concentrations, and then the relative error (RE) was calculated.
3 Results and Discussion
3.1 Optimization of the GC–MS and DART-MS Methods
This study included all the 16 PAHs on the United States Environmental Protection Agency (US EPA) priority pollutant list with the other 9 PAHs commonly found in airborne PM samples (Figure S2). The GC–MS method was optimized to lower the limit of detection and to enhance separation. The first method used a 20-min temperature gradient with a start temperature of 80°C, held for 2 min, and raised to 280°C at a rate of 17°C/min, which had poor separation of the "heavy" PAHs with five to seven aromatic rings. The second method was modified to analyze a sample within 53 min, which showed improved separation. It had a start temperature of 100°C, which was held for 2 min and then raised to 210°C at a rate of 8°C/min. Finally, the temperature was raised to 280°C at a rate of 2°C/min. While this method had better separation than the first method, both methods only eluted 15 of the 18 nonpolar PAHs. The three PAH that were not eluted were indeno(1,2,3-cd) pyrene, dibenzo(a,h)anthracene, benzo(g,h,i)perylene. These three PAHs have higher molecular weights and are less volatile. A final method was developed based on the 53-min method with an additional temperature increase to 340°C by a rate of 17°C/min and held for 6 min. This method allowed the elution of the last three PAHs. The selective ion monitoring mode (SIM) was used to monitor 25 PAHs and 7 deuterated PAHs as internal standards. Their corresponding ions and retention times are listed in Table S1. Figure 1 shows the representative SIM chromatograms for the 25 PAHs with well-resolved GC peaks due to the unique combination of their characteristic ions and retention times.

Typical SIM chromatograms from GC–MS analysis for the 25 PAHs investigated in this study
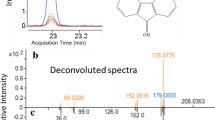
The DART-MS method was also optimized prior to PM sample analysis. Both positive and negative modes were tested, and it was found that the sensitivity for the polar PAHs under the negative mode was very low, so the positive mode DART-MS was used in the present work. Mild separation was achieved for both polar and nonpolar compounds using a temperature gradient, but coelution of the analytes, especially the isomers such as phenanthrene and anthracene, was inevitably observed. Selected ions were used to monitor the PAHs. Table S2 lists the polar and nonpolar PAHs, their desorption times, and their characteristic ions used in this study. Figure 2 shows the retention times and the selected ions for nonpolar and polar PAHs using the positive mode with the thermal desorption DART-MS method. Naphthalene, 1-methylnaphthalene, 2-methylnaphthalene, acenaphthylene, acenaphthene, fluorene, and 1-nitronaphthalene were not included because of the poor reproducibility due to their high volatility.

The extracted ion chromatograms (EIC) for the nonpolar PAHs (left) and polar PAHs (right) from the DART-MS method
3.2 Analytical Method Performance
Both external standard calibration and internal standard calibration methods for GC–MS and DART-MS analysis were tested in the initial study, and it was found that the results calculated using the internal standard calibration method showed improved precision for both GC–MS and DART-MS methods. The sample preparation for GC–MS analysis involved multiple steps, including sonication, drying, reconstitution, and filtration, and the internal standards were effective in correcting for the variability in this multi-step process. Though the sample preparation for DART-MS was minimal, volatilization of PAHs with lower molar masses cannot be avoided due to the thermal desorption and ionization of analytes taking place in the open space under ambient conditions. Therefore, an internal standard was used to compensate for the possible loss of analytes and to reduce the impacts of the analytical instrument variations. In terms of the internal standard, the stable isotope analogs based on deuterated PAHs were used for the GC–MS method. A structural analog, 9-bromophenanthrene, was chosen as the internal standard for the DART-MS method on the basis of its similarity to the PAH analytes in the characteristics of desorption, recovery, and ionization.
Calibration curves were linear over the concentration ranges 0.02–1.00 µg/mL and 0.05–1.00 µg/mL for the respective GC–MS and DART-MS methods with correlation coefficients (r2) > 0.98 (mostly r2 > 0.995, Table S3 and S4).
The accuracy of GC–MS and DART-MS methods was evaluated by analyzing the simulated certified dust with three concentrations in triplicate. For the GC–MS method, the relative errors of all the measurements are in the range of − 16.9 ~ 18.6%, as shown in Table 1. Similar results were achieved by using the DART-MS method, with relative errors between − 15% and 15% (Table 2). Overall, the results indicate that all PAHs compounds investigated did not significantly influence the analysis of the PAHs on the filters and in the dust samples.
The precision and the recovery results of the PAHs on the filters by GC–MS and DART-MS methods were summarized in Table S5–S6. The mean spiked recovery for the PAH measured on quartz filter by GC–MS method was 96%, with individual PAH recoveries ranging from 86% to 125%, whereas the mean spiked recovery for the PAH measured on quartz filter by DART-MS method was 105%, with individual PAH recoveries ranging from 84% to 131%. In general, the GC–MS method has shown a better reproducibility than the DART-MS method with all RSD values ≤ 20.3%, especially for low concentration PAH samples (i.e., 0.10 µg/mL). It is worth noting that the DART-MS analysis did not involve chromatographic separation; therefore, the PAHs peaks in the EIC plots of DART-MS were apparently broader since the thermal profiles of the desorption/ionization process are not as well defined as the chromatographic partition of analytes between mobile and stationary phases. The peak areas for lower concentration samples were significantly affected by the baseline fluctuations, which might contribute notable variations to the analysis from run to run. GC–MS has been used as a standard technique for PAH analysis over the years, and it has been well recognized for its performance in the separation and detection of environmental pollutants such as PAHs [11]. However, the DART-MS method provided a unique opportunity to estimate the quantity and identity of PAHs in a high throughput fashion because it does not require labor-intensive solvent extraction and lengthy chromatographic separation like the GC–MS method. In addition, the minimal consumption of reagents, solvents, and energy, as well as less production of waste by DART-MS, would cause little concern regarding the toxicity and possible pollution to the environment. The DART-MS method only consumes a small portion of the filter samples, which allows the remaining portion of the filters to be used for a more comprehensive screening along with various other methods, such as scanning electron microscopy and energy-dispersive X-ray analysis (SEM/EDX), infrared microanalysis, and ICP-MS analysis of heavy metals. Overall, the DART-MS method can be used as an effective tool for the initial assessment of the PAHs types and levels on the PM filter samples, and the results will assist in the decision about whether a detailed examination of the sample is needed, and this will reduce the turnaround time necessary for the toxicant analysis of batch samples. Given the potentially high demands of GC–MS or LC–MS analysis times and constrained resources in most laboratories, a fast and economically and environmentally sustainable method such as DART-MS will benefit the fire safety research.
3.3 Study of PAH Distribution on Filter
When removing the filters from the filter holders, it was noticed that the particles were not evenly distributed throughout the 25-mm filter for PM10. There were four darker circular spots with apparently more PM concentrated. When analyzing the sample by the GC–MS, this uneven concentration was not an issue as the entire half filter was used for the extraction; however, when analyzing the sample by the DART-MS, where only an 8-mm hole was punched out, it was important to determine the percent distribution of PAHs on the filter in various places. Four samples for the 25-mm PM10 filters and four samples for the 37-mm PM2.5 filters were collected from small-scale fire experiments. Four 8-mm filter discs were punched out of the 25-mm sample filters, and three discs were punched out of the 37-mm sample filters. The PAHs on the discs were extracted and analyzed by GC–MS. Figure 3 shows the placement of discs for both types of filters and their represented total PAH percentages of the entire filter. As shown in Figures. 3a and c, spot D, which was the darkest portion of the PM10 filter, had 15% of the total PAH on the filter, while spot B represented 12% of total PAHs. The darkest spot had the most PAH, as expected, but spot B also had a high percentage due to its placement close to two of the dark spots. Spots O and M both had less than 8% because they were further away from the dark spots. Figures 3b and d show that the 37 mm PM2.5 filter had the highest concentration at the center of the filter, and as the edges approached, the concentration decreased slightly. Overall, we recommend punching out the darkest spot of the 25-mm PM10 filter for DART-MS analysis, representing approximately 14% of total PAHs in the PM10, and sample the center area of the 37-mm PM2.5 filter for DART-MS analysis which represents about 5.4% of the total PAHs in the PM2.5.

The placement of the discs punched out from the 25-mm filter (a) and the 37-mm filter (b) and their respective total PAH concentrations on the discs (c, d)
3.4 Sampling PM by Firefighters Under Light and Severe Biomass Smoke Conditions
The personal modular impactor (PMI) sampling device was tested for PM collection from biomass smoke in our study before deploying the sampling systems to the firefighters. Since the toxic effects of PM are size-dependent [21], the cascade impactor (i.e., PMI coarse) was used in our study, allowing us to investigate the PAHs exposure from both PM2.5 and PM10 in the biomass fire smoke. The PMI coarse is a multi-stage impactor with two filters for PM2.5 and PM10, respectively, and a 25-mm pre-oiled impaction disc to remove particles larger than 10 µm and reduce particle bounce [22]. The AirChek TOUCH pump can compensate for the increased pressure drop due to the particle load increases on the filter during sampling, ensuring constant airflow throughout the sampling event. The dimensions for the pump and impactor are 4.1 × 3.7 × 2.8 inches (10.4 × 9.4 × 7.1 cm) and 2.1 × 1 inches (5.1 × 2.6 cm), respectively, with the approximate total weight of 22 oz (624 g) for the sampling system. The small size and lightweight of the sampling system should cause minimal interruption to the regular firefighting operation.
The outdoor biomass fire was conducted on an open field with two sampling systems equipped by firefighters who controlled the fire and one sampling system placed on a tripod. The white and thin smoke was observed during the 60-min sampling period. The indoor fire study was conducted by burning wooden boards in an enclosed space of the fire training facility. Much thicker smoke was observed in this experiment. Two PM sampling systems were set up on tripods inside the house about 10 ft away from the fire with a 5 ft height during the entire sampling period. After 3 min of burning, the temperatures of the ceiling and floor of the facility and PMI head were measured to be 600 °C, 175 °C, and 260 °C, respectively. After 6 min, the PMI base started to melt, and the sampling systems were taken out of the house. The other two sampling systems were worn by the firefighters controlling the burn. The firefighters stayed in the house for 7 min in total only during the firing and extinguishing fire with a fire hose, and they waited outside of the house for 5 min while the fire was burning when they were exposed to minimal smoke. The two sampling systems on firefighters remained working properly throughout the process.
The comparison of polar and nonpolar PAH levels in PM2.5 and PM10 from the indoor and outdoor fire experiments is shown in Figure 4. In general, both polar and nonpolar PAH concentrations from the indoor experiment were much higher than those from the outdoor experiment. The PAHs in the PM2.5 were present at comparable levels relative to those in the PM10 with no significant difference (based on the unpaired two-tailed Student's t-tests at 95% confidence level). One of the sampling systems on the tripod inside the fire training house stopped collecting samples after 4 min because the solid particles overloaded the filter substrate. The airflow was choked due to the blockage of the particles, and the sampling pump stopped as the pressure compensation system was not able to provide sufficient pressure for the desired airflow rate. The indoor and outdoor experiments suggested that the sampling systems worked well for the light and moderate smoke conditions, and the system did not cause significant interruptions to the firefighting operations. Additional cautions are necessary during the sampling under severe smoke conditions, and the sampling times are limited by the buildup of particulate deposits on the filter substrates. The long exposure to extreme heat may also cause damage to the PMI. Data logging capability for the sampling pump is also highly recommended so that the flow rate and sampling time information can be recovered from the pump's internal data system when accidental failures happen.

Polar and nonpolar PAH levels on PM2.5 and PM10 from indoor (severe smoke condition) and outdoor (light smoke condition) experiments
3.5 PAHs in PM from Air Sampling During Wildfire Periods
PM samples from the ambient air were collected during the wildfire events in the region of Orange County, California: Silverado Fire started on 10/26/2020 and was extinguished on 11/07/2020, and Bond Fire started on 12/02/2020 and was extinguished on 12/10/2020 [23]. Three sampling sites in the residential area were selected to collect PM samples and air samples during October 26, 2020, and January 25, 2021 (Figure S1a). Five sampling experiments (Exp. 1–5) were carried out, among which four sampling experiments (Exp. 1–4) were conducted during the wildfire events, and one (Exp. 5) was performed approximately 45 days after the Bond Fire to estimate the atmospheric background levels of PAHs originating from sources such as residential, industrial, and traffic emissions. An exemplary sampling setup is shown in Figure S1b. All the PM sampling systems worked properly in these activities.
Total nonpolar and polar PAHs in PM2.5 and PM10 samples are shown in Figure 5. The average nonpolar PAHs in PM2.5 and PM10 samples were 71 ± 38 and 80 ± 35 ng/m3, respectively, during the wildfire incidents, which were significantly higher than the regular PAHs’ background levels (27 ± 2 and 31 ± 5 ng/m3 for PM2.5 and PM10). Polar PAHs, including nitrated and oxygenated PAHs, were reported with even higher toxicity than their related parents, regular PAHs, and are also found in airborne PM [24]. Eight polar PAHs were studied in our study. The average polar PAHs in PM2.5 and PM10 samples were 63 ± 47 and 63 ± 39 ng/m3, respectively, during the wildfire incidents, which were also significantly higher than the polar PAHs background levels (14 ± 1 and 16 ± 4 ng/m3 for PM2.5 and PM10).

Total nonpolar and polar PAHs in PM2.5 (a) and PM10 (b) samples during the California wildfire incidents (Exp. 1–4) and one and a half months after the wildfire incidents (Exp. 5). Note: S1-S3 refers to the sampling sites 1–3 in Figure S1, and Exp. 1–5 refers to the sampling experiments on dates of October 26, 2020, October 28, 2020, November 06, 2020, December 04, 2020, and January 25, 2021
The PAH levels in PM samples from Exp. 3 (11/06/2020) were lower than the samples from the other two sampling activities (Exp. 1–2, or 10/26/2020 and 10/28/2020) during the Silverado fire incident. The Silverado Fire was extinguished on 11/07/2020, and this sampling activity was close to the end of the fire incident, so the respective toxicant levels are expected to be lower than those when the wildfires were burning more intensively earlier. Since Sampling Site 2 was further away from the fire than Sampling Site 1, which also affected the PM concentrations in the air. During the wildfire incidents, the lowest PAH levels were observed from Exp. 4 at Sampling Site 3, which was over the greatest distance to the fire. Significant PAH levels were detected in Exp. 5 (background levels), which indicates PAHs and PM from sources other than wildfire (e.g., fuel and combustion processes) should not be overlooked [25]. It is worth noting that the atmospheric levels of PAHs could vary significantly on a daily basis. For example, one study reported the total PAH concentrations in the outdoor air surrounding 19 homes in Los Angeles, CA, ranged from 4.2 ng/m3 to 64 ng/m3 from June 1999 to May 2000 [26], which suggested a longer and more frequent ambient air sampling would be needed for a more accurate estimate of background PAH levels in the air. Nevertheless, the total PAH levels detected during the wildfire events in this study were higher than days without wildfire (i.e., Exp. 5) and values reported in the aforementioned literature.
The individual nonpolar PAH levels on PM samples are shown in Figure S3. Several high molecular weight PAHs (5–6 fused benzene rings) such as benzo[b]fluoranthene, benzo[k]fluoranthene, and benzo[a]pyrene were present at significantly high levels in PM samples during the first two sampling events when the Silverado Fire actively burned; and they were not detected in the background PM samples. In addition, low molecular weight PAHs such as naphthalene exist at relatively low levels in most PM samples. It is worth noting that the higher molecular weight PAHs are usually of principal concern due to their recalcitrance, persistence, bioaccumulation, carcinogenicity, genotoxicity, and mutagenicity [27, 28]. A recent study on occupational exposure to PAHs of wildland firefighters at wildland fires reported that low levels of high molecular weight PAHs were detected in the gas phase samples. This finding, together with the determination of particulate phase PAHs, is important in evaluating PAHs exposure to firefighters due to the dynamic equilibria of PAHs partitioning between the two phases [29, 30]. The individual polar PAH levels in PM samples are shown in Figure S4. Three oxygenated PAHs, such as 1-naphthol, 2-naphthol, and 9-fluorenone, and two nitrated PAHs including 1-nitronaphthalene and 2-nitrofluorene were present at significantly high levels in PM2.5 and PM10 samples during the active fire periods; and their concentrations were much lower in the background PM samples.
4 Conclusion
In this study, we reported a novel thermal desorption DART-MS method to screen the PAHs in PM samples collected from biomass smoke. The method requires minimal sample preparation and provides qualitative and quantitative information within 10 min. Since only a small portion of the PM filter was used for the screening PAHs by the DART-MS method, the remaining filter could be used for other chemical analyses. The personal PM sampling system was evaluated under various smoke conditions and found that the multistage cascade impactors could be used effectively to collect size-fractionated particles in most of the testing scenarios except for the extreme heat and severe smoke conditions in the indoor biomass burns. Both nonpolar and polar PAHs were detected in the PM from the smoke of different biomass burn experiments and the wildfire events in 2020. The results imply that the study of the PM-derived PAH exposure to wildland firefighters is critical and can be conducted using GC–MS and DART-MS methods, which would help firefighters determine if respiratory protection is needed. The PM sampling systems have been successfully deployed to the wildland firefighters during the Monument fire and Windy fire in 2021 to study the toxicant exposures of their firefighting activities, and the results will be reported in our follow-up study.
Change history
19 June 2023
A Correction to this paper has been published: https://doi.org/10.1007/s10694-023-01444-1
References
National Interagency Coordination Center (2020) National Interagency Coordination Center Wildland Fire Summary and Statistics Annual Report. https://www.predictiveservices.nifc.gov/intelligence/2020_statssumm/2020Stats&Summ.html. Accessed 25 October 2022
Fann N, Alman B, Broome RA, Morgan GG, Johnston FH, Pouliot G, Rappold AG (2018) The health impacts and economic value of wildland fire episodes in the US: 2008–2012. Sci Total Environ 610–611:802–09
California Air Resources Board (2021) Camp fire air quality data analysis. https://ww2.arb.ca.gov/resources/documents/camp-fire-air-quality-data-analysis. Accessed 25 October 2022
Epa US (2003) Particle pollution and your health. Particle pollution and your health. Office of Air and Radiation, Maryland
Kim K-H, Jahan SA, Kabir E, Brown RJC (2013) A review of airborne polycyclic aromatic hydrocarbons (PAHs) and their human health effects. Environ Int 60:71–80
Hassan SK (2018) Particle-bound polycyclic aromatic hydrocarbon in the atmosphere of heavy traffic areas in greater Cairo Egypt: status source and human health risk assessment. Atmosphere 46:693–707
Hwang J, Xu A, Agnew RJ, Clifton S, Malone TR (2021) Health risks of structural firefighters from exposure to polycyclic aromatic hydrocarbons: a systematic review and meta-analysis. Int J Environ Res Public Health 18:4209–4226
Navarro KM, Clark KA, Hardt DJ, Reid CE, Lahm PW, Domitrovich JW, Butler CR, Balmes JR (2021) Wildland firefighter exposure to smoke and COVID-19: a new risk on the fire line. Sci Total Environ 760:144296
Keir JLA, Akhtar US, Matschke DMJ, Kirkham TL, Chan HM, Ayotte P, White PA, Blais JM (2017) Elevated exposures to polycyclic aromatic hydrocarbons and other organic mutagens in Ottawa firefighters participating in emergency. On-Shift Fire Suppr, Environ Sci Technol 51:12745–12755
Famiyeh L, Chen K, Xu J, Sun Y, Guo Q, Wang C, Lv J, Tang Y-T, Yu H, Snape C, He J (2021) A review on analysis methods, source identification, and cancer risk evaluation of atmospheric polycyclic aromatic hydrocarbons. Sci Total Environ 789:147741
US EPA (1999) Determination of polycyclic aromatic hydrocarbons (PAHs) in ambient air using gas chromatography/mass spectrometry (GC/MS). In: Determination of polycyclic aromatic hydrocarbons (PAHs) in ambient air using gas chromatography/mass spectrometry (GC/MS).
Wang X, Wang Y, Qin Y, Ding L, Chen Y, Xie F (2015) Sensitive and selective determination of polycyclic aromatic hydrocarbons in mainstream cigarette smoke using a graphene-coated solid-phase microextraction fiber prior to GC/MS. Talanta 140:102–108
Cody RB, Laramee JA, Durst HD (2005) Versatile new ion source for the analysis of materials in open air under ambient conditions. Anal Chem 77:2297–2302
Barnett I, Zhang M (2018) Discrimination of brands of gasoline by using DART-MS and chemometrics. Forensic Chem 10:58–66
Barnett I, Bailey FC, Zhang M (2019) Detection and classification of ignitable liquid residues in the presence of matrix interferences by using direct analysis in real time mass spectrometry. J For Sci 64:1486–1494
Maric M, Marano J, Cody RB, Bridge C (2018) DART-MS: a new analytical technique for forensic paint analysis. Anal Chem 90:6877–6884
Lesiak AD, Musah RA, Domin MA, Shepard JR (2014) DART-MS as a preliminary screening method for “herbal incense”: chemical analysis of synthetic cannabinoids. J Forensic Sci 59:337–343
Liang J, Sun J, Chen P, Frazier J, Benefield V, Zhang M (2021) Chemical analysis and classification of black pepper (Piper nigrum L.) based on their country of origin using mass spectrometric methods and chemometrics. Food Res Int 140:109877
Dong W, Liang J, Barnett I, Kline PC, Altman E, Zhang M (2019) The classification of Cannabis hemp cultivars by thermal desorption direct analysis in real time mass spectrometry (TD-DART-MS) with chemometrics. Anal Bioanal Chem 411:8133–8142
Pavlovich MJ, Musselman B, Hall AB (2016) Direct analysis in real time-mass spectrometry (DART-MS) in forensic and security applications. Mass Spectrom Rev 37:171–187
Kumar P, Kalaiarasan G, Porter AE, Pinna A, Kłosowski MM, Demokritou P, Chung KF, Pain C, Arvind DK, Arcucci R, Adcock IM, Dilliway C (2021) An overview of methods of fine and ultrafine particle collection for physicochemical characterisation and toxicity assessments. Sci Total Environ 756:143553
Wang Z, Calderón L, Patton AP, Sorensen Allacci M, Senick J, Wener R, Andrews CJ, Mainelis G (2016) Comparison of real-time instruments and gravimetric method when measuring particulate matter in a residential building. J Air Waste Manag Assoc 66:1109–1120
Cal Fire (2020) 2020 Incident archive. https://www.fire.ca.gov/incidents/2020. Accessed on 25 October 2022
Mirivel G, Riffault V, Galloo JC (2010) Simultaneous determination by ultra-performance liquid chromatography-atmospheric pressure chemical ionization time-of-flight mass spectrometry of nitrated and oxygenated PAHs found in air and soot particles. Anal Bioanal Chem 397:243–256
Kam W, Delfino RJ, Schauer JJ, Sioutas C (2013) A comparative assessment of PM25 exposures in light-rail, subway, freeway, and surface street environments in Los Angeles and estimated lung cancer ris. Environ Sci: Processes Impacts 15:234–43
Naumova YY, Eisenreich SJ, Turpin BJ, Weisel CP, Morandi MT, Colome SD, Totten LA, Stock TH, Winer AM, Alimokhtari S, Kwon J, Shendell D, Jones J, Maberti S, Wall SJ (2002) Polycyclic aromatic hydrocarbons in the indoor and outdoor air of three cities in the US. EnvironSci Technol 36:2552–2559
Xue W, Warshawsky D (2005) Metabolic activation of polycyclic and heterocyclic aromatic hydrocarbons and DNA damage: a review. Toxicol Appl Pharmacol 206:73–93
Marcoux J, Déziel E, Villemur R, Lépine F, Bisaillon JG, Beaudet R (2000) Optimization of high-molecular-weight polycyclic aromatic hydrocarbons’ degradation in a two-liquid-phase bioreactor. J Appl Microbiol 88:655–662
Navarro KM, Cisneros R, Noth EM, Balmes JR, Hammond SK (2017) Occupational exposure to polycyclic aromatic hydrocarbon of wildland firefighters at prescribed and wildland fires. Environ Sci Technol 51:6461–6469
Naeher LP, Brauer M, Lipsett M, Zelikoff JT, Simpson CD, Koenig JQ, Smith KR (2007) Woodsmoke health effects: a review. Inhal Toxicol 19:67–106
Acknowledgements
This research was supported by the FEMA Fire Prevention and Safety (FP&S) Program from the US. Department of Homeland Security (EMW-2018-FP-00668). We are grateful for the assistance of biomass burns in Tennessee by the Murfreesboro Fire Rescue Department, Murfreesboro, TN, and the Mount Pleasant Fire Department, Mount Pleasant, TN. Special thanks to Assistant Chief/Fire Marshal of Murfreesboro Fire Rescue Department, Mr. Carl Peas, for coordinating the indoor fire experiment in the fire department training facility and providing comments and suggestions for the PM sampling process.
Funding
This study was supported by Federal Emergency Management Agency (EMW-2018-FP-00668).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Benefield, V.L., Perna, S., Pham, S. et al. Evaluation of Mass Spectrometric Methods for Screening Polycyclic Aromatic Hydrocarbons in the Particulate Phase of Wildfire/Biomass Smoke. Fire Technol (2022). https://doi.org/10.1007/s10694-022-01327-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10694-022-01327-x