
Abstract
Familial pancreatic cancer (FPC) is a rare hereditary tumor entity with broad phenotypic heterogeneity, including colorectal carcinoma (CRC) in some families. The underlying factors for this co-occurrence are still not well evaluated. FPC families in the National Case Collection of Familial Pancreatic Cancer with an additional occurrence of CRC were analyzed regarding the phenotype, genotype and recommendation for a clinical screening program. The total cohort of 272 FPC families included 30 (11%) families with at least one CRC case. The proportion of affected family members with PDAC was 16.1% (73/451) compared to 9.3% of family members with CRC (42/451, p < 0.01). Females were affected with PDAC in 49% (36/73) and CRC in 38% (16/42). The median age of PDAC was 63 compared to 66 years in CRC, whereas 8 (26.6%) of families had an early onset of PDAC and 2 (6.7%) of CRC. Seventeen families had 2 or more affected generations with PDAC and 6 families with CRC. Eleven (9.6%) of affected patients had both PDAC and CRC. Potentially causative germline mutations (2 ATM, 1 CDKN2a, 1 MLH1, 1 PALB2) were detected in 5 of 18 (27.7%) analyzed cases. These findings provide a step forward to include the phenotypic and genotypic characteristics of FPC-CRC families for the genetic counseling and management of these families. Nevertheless, results need to be verified in a larger patient cohort beforehand.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of the most aggressive and lethal tumor entities, with an average five-year survival rate of less than 9% [1]. This poor prognosis is mainly attributed to the late onset of subtle or nonspecific symptoms, in combination with limited treatment options at the time of diagnosis [1]. Despite recent breakthroughs for various types of cancer, early diagnosis and screening of PDAC remains challenging, due to the extensive heterogeneity of low-frequency genetic mutations [2]. Nevertheless, having two or more first-degree relatives with PDAC defined as so-called familial pancreatic cancer (FPC) syndrome [3, 4] is still one of the major risk factors (10% of incidences) for developing PDAC [3]. Therefore, several international tumor registries and collections were founded, including the North American National Familial Pancreatic Tumor Registry (NFPTR), the German National Case Collection of Familial Pancreatic Cancer and the European Registry of Hereditary Pancreatitis and Familial Pancreatic Cancer (EUROPAC) [5,6,7]. The common goal is to identify signature gene clusters and related phenotypes in affected families for an improvement in genetic counseling and earlier detection in a prospective PDAC screening program [8]. Recent studies failed to identify general driver mutations for FPC susceptibility, while even less is known about the phenotypic variants in FPC families [2, 9]. Nevertheless, registered families can be roughly divided into groups of “pure” FPC and those with additional co-occurrence of other tumours, such as breast, colon, lung, or prostate cancer [5, 10]. Within this area of study, we recently detected some predisposing low penetrance genes that may relate to an additional susceptibility to breast cancer in some cases of the FPC families [5]. The current study aimed to further fill the knowledge gaps of undefined phenotypic and genotypic factors as well as support future studies for prospective PDAC screening in FPC associated with colorectal cancer (FPC-CRC).
Patients and methods
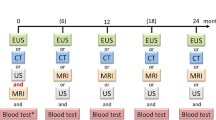
The German National Case Collection of Familial Pancreatic Cancer (FaPaCa) is a tumor collection funded by the Deutsche Krebshilfe in 1999 [5]. It was established to investigate the phenotype and genotype of families with two or more first-degree relatives with PDAC, referred to as FPC families. Moreover, the FaPaCa collection offers a screening program for these FPC family relatives. Initially, the screening age started at 40, until it was set back to 50 years in 2016. Alternatively, the screening of family members can begin at the age corresponding to 10 years before the earliest recorded onset of PDAC within the family history. An exclusion criteria for the screening program is the documented evidence of another inherited tumor syndrome. An FPC family member carrying a predisposing mutation, e.g. BRCA2, will be classified as an individual at risk (IAR), if at least one relative was affected by a PDAC. This screening program entails an annual physical examination, collection of blood samples, determination of serum HbA1c, amylase, GOT, GPT, bilirubin, and CA19-9, and imaging with MRI plus magnetic resonance cholangiopancreatography (MRCP) and endosonography [11,8]. The FPC family members were referred by their physicians, or by contacting the FaPaCa study office based on contact information on the FaPaCa website starting July 1999 (http://www.fapaca.de). As part of the registration process, all eligible families were genetically counseled, and a three-generation family pedigree was constructed.
The current report analyzed the genotype and phenotype of FPC families with an additional occurrence of CRC. The age of diagnosis of PDAC and CRC was retrieved from the three-generation pedigrees and early age of onset was defined as the occurrence of PDAC or CRC prior to the age of 50 years in a family [8, 12,13,14]. All PDAC and CRC diagnoses were confirmed by review of medical records and death certificates. Furthermore, the pathology report defined the pancreatic lesions of operated patients PDAC, pancreatic intraepithelial neoplasia type (PanIN) 1–3 or intraductal papillary mucinous neoplasia (IPMN) with low or high-grade dysplasia.
All patients with PDAC and/or FPC-CRC families with available blood samples, given their informed consent was collected, were subjected to mutation analysis by multi-cancer gene panel, including cancer predisposition genes [9, 15,16,17,18,19,20]. In addition, resected PDACs of affected patients from FPC-CRC families also underwent immunohistochemical analysis of mismatch repair genes MLH1, MSH2, PMS2 and PMS6. If loss of expression was detected in one of these genes, Sanger sequencing of the respective gene was performed in the corresponding germline. If a deleterious germline mutation was identified in the tested patient, genetic counseling and a predictive genetic test of this mutation were offered to all family members. The results were explained to the family members in a follow-up genetic counseling. The FaPaCa collection and screening program, as described here, was approved by the Ethics Committee of the Philipps-University of Marburg (36/1997, last amendment 9/2010), while all participating family members provided their written informed consent.
Statistics
All descriptive information from the FaPaCa family members was compiled. These included age, gender, number of family members with PDAC, earliest age of onset in the family and known germline mutations. The early-onset was defined as the diagnosis of PDAC as well as CRC prior to the age of 50 years [8, 12,13,14].
The chi-square, Fisher’s exact test, t-test and Wilcoxon rank sum test were completed for categorical and numerical variables, to compare the characteristics of patients. Two-tailed p values < 0.05 were considered to be statistically significant. Analyses were performed with Prism GraphPad Software, Inc.
Results
Based on the FaPaCa collection of 272 FPC families, we identified 30 (11%) families with at least one CRC case in their family history. During follow-up screens, only one of these 30 families emerged to fulfill the criteria of Hereditary nonpolyposis colorectal cancer (HNPCC) [21]. Of the 451 registered family members of these FPC-CRC 30 families, 73 (16.1%) had PDAC and 42 (9.3%) had developed CRC.
We further investigated the distinct phenotypes of the two groups. The proportion of affected female patients with 49% (36/73) with PDAC in FPC families was similar to the general population (48% females) [22]. Furthermore, CRC occurrence in females of FPC families was 38% (16/42, p < 0.01), compared to CRC occurrence in the general population with 41% females [23, 24]. However, the median age at the time of diagnosis was lower in PDAC at 63 years (37–83), while CRC demonstrated a median age of 66 (44–90) years (p < 0.01). Furthermore, 8 (26.6%) families had an early-onset of PDAC < 50 years compared to only 2 families (6.7%) with an early onset of CRC < 50 years. Seventeen (57%) families had two or more affected generations with PDAC and 6 (20%) families with CRC (p > 0.05). Eleven of 115 (9.6%) affected patients had both PDAC and CRC. The characteristics of FPC-CRC families are summarized in Table 1. A representative FPC-CRC family is shown in Fig. 1.

Pedigree of a representative FPC-CRC
In the next step, we performed mutation analysis of 18 FPC-CRC family members, where patient consent and tissues were readily available, including 4 individuals with co-occurrence of FPC and CRC. Here, we identified potentially causative germline mutations (2 ATM, 1 CDKN2a, 1 MLH1, 1 PALB2) in 5 (27%) individuals (Table 2). This mutation spectrum is distinct from other previously described gene mutations in FPC (BRCA1, BRCA2, CHEK2, MSH2, MSH6), which remained undetected in the analyzed 18 FPC-CRC family members.
From these 30 FPC-CRC families, 25 Individuals at risk (IAR) with a median age of 49 (36–62) years participated in a prospective annual PDAC screening program with a median of 2 (1–18) screening visits. Upon imaging, small (< 10 mm) cystic lesions were detected in 20 (80%) of IAR and 2 (8%) IARs underwent surgery because of suspicious lesions upon imaging in the pancreatic head. A 56 year old male underwent pylorus-preserving pancreaticoduodenectomy (PPPD) and pathology revealed pancreatic intraductal papillary mucinous neoplasms (BD-IPMN) with multifocal PanIN2 lesions. This IAR was not a carrier of the predisposing PDAC-germline mutation. Another 83 year old male carrying a MLH1 mutation had total pancreatectomy for a solid lesion. Pathology reported a PDAC UICC stage 3 (pT3, N1, M0).
Discussion
There is still an overwhelming knowledge gap in the clinical management of FPC families, since only in about 10–15% of FPC cases predisposing germline mutations have been detected so far [5, 25]. FPC can be roughly divided into two groups, namely pure FPC families (~ 40% of families) and those associated with co-occurrences of other tumor types, most frequently breast cancer (31%), CRC (11%), or melanoma (9.7%) [5, 26]. In this study we sought to take advantage of the FaPaCa collection [5] to analyze, for the first time, the phenotype and genotype of FPC families associated with additional CRC.
A total of 9.3% (42/451) FPC family members developed CRC, compared to the lower incidence of CRC in the general German population with 0.08% (84.8/100,000) as reported in 2019 [24, 27].
Our analysis demonstrated that almost half of the patients in the FPC-CRC families with PDAC were female 49% (36/73), whereas only 38% (16/42) females demonstrated CRC (Table 1). CRC, in general, is a disease strongly influenced by gender and biased towards males [28]. Underlying reasons include behavioral CRC risk factors, e.g., increased consumption of red, processed meat, alcohol, and smoking, in addition to a greater likelihood to deposit visceral fat [29,30,31]. These factors can potentially also play a role in the gender bias in FPC-CRC families.
In this study, the median age of diagnosis was relatively low in FPC-CRC families with 63 years for PDAC and 66 years for CRC, respectively (Table 1). Nevertheless, there was no difference in the previously reported median age of PDAC onset in FPC families with 63 (35–91) years [5] compared to the presented FPC-CRC families with 63 (37–83) years (Table 1). Overall, there were 9.3% (42/451) of FPC family members with CRC in this FPC-CRC cohort. Recently, there was more effort put into context-dependent driver gene discovery, leading to either exclusive or co-occurrence of cancer entities in one patient [32]. Therefore, we further investigated the genetic makeup of FPC-CRC families to analyze the relationship of cross-cancer mutation patterns, where a co-occurrence in both FPC and CRC may indicate synergistic impact on tumorigenesis This was also an opportunity to expand the panel of genes under study and discover new candidate germline genes to be further validated prospectively.
Here, we detected four potentially causative germline mutated genes (2 ATM, 1 CDKN2a, 1 MLH1, and 1 PALB2) in 5 of 18 (27.7%) FPC-CRC families (Table 2). This is a relatively high frequency of gene mutations, compared to the rate of detected potentially deleterious germline mutations in reported pure FPC families about 10% [5, 9, 33,34,35,36,37]. The ATM, CDKN2A, MLH1, and PALB2 mutations in FPC-CRC families have separately been associated with a high to moderate risk of CRC [38]. ATM and MLH1 are classified as high penetrance modifiers in CRC [39], while CDKN2A and PALB2 have been reported as moderate-risk CRC susceptibility genes [40]. In brief, ATM is a key player in the maintenance of genomic integrity during DNA repair. Previous studies identified a connection between ATM mutations and an increased predisposition in solid tumors, including pancreas, breast, gastric, lymphoid, CNS, skin, and others [41]. Furthermore, ATM mutations may result in resistance to chemotherapeutic therapies. To date, its potential role as a predictive and prognostic biomarker has not been fully investigated. Further pathogenic gene mutations of MLH1 and CDKN2A have been recently described in pancreatic and upper gastrointestinal tract tumors but have not been evaluated in the setting of familial FPC-CRC predisposition [42]. Whereas BRCA2 mutations, have been reported to play a role in neoplasia in hereditary breast and ovarian cancer (HBOC) and may be involved in up to half of hereditary breast cancer [43]. Instead of BRCA2 itself, this study detected mutations in FPC-CRC kindreds in PALB2, a co-localizer and partner gene to BRCA2, which is also proposed to be involved in FPC [44]. Consequently, ATM, CDKN2A, MLH1, and PALB2 mutations in FPC-CRC families might biologically and clinically relevant for both PDAC and CRC development. This should be further investigated in a larger cohort of familial FPC-CRC patients. Since the general issue in the field is the relatively small number of families and samples, the establishment of multi-center cohorts would be beneficial to increase the cohorts for statistical power and relevance for clinical practice.
Despite the small patient cohort, the assumption that the co-occurrence between FPC and CRC is merely a coincidence is relatively unlikely, since 11 (15%) of 73 patients with PDAC also had CRC, but only 1 of these patients belonged to a hereditary non-polyposis colorectal cancer (HNPCC) family. In comparison, HNPCC is one of the most common familial aggregations of hereditary cancer in the gastrointestinal tract [45], but its association with PDAC was reported between 1.3 and 6%, and is not as striking [46,47,48]. Similar low co-occurrences are seen in Familial Adenomatous Polyposis [21], which is related to the germline mutations of the APC gene [49]. Initial studies from 1993 suggested that FAP patients have a 4.5 times increased risk of developing PDAC compared to the general population [50]. However, a more recent study from the Johns Hopkins Polyposis registry found only 4 (0.3%) patients with PDAC in their cohort of 1391 patients with FAP [50, 51], while Driffa et al. did not report any cases of PDAC in their cohort of 127 FAP patients between 1990 and 2009 [52]. Therefore, the FPC-CRC cases are relatively frequent in comparison to FPC-HNPCC or FPC-FAP cases, and potentially more than just an arbitrary coincidence.
Recent expert guidelines recommend a PDAC screening in IAR of FPC families [33], especially in mutation carriers of predisposing genes. During screening, cystic lesions are frequently detected in 40 to 60% of IAR [5, 21, 33, 53, 54]. Twenty-five IAR from our 30 FPC-CRC families participated in a board approved controlled screening program and 20 (80%) showed cystic lesions (Table 3), which are somehow pathognomonic for FPC [55]. The diagnostic yield of screening, defined as the detection of high grade pancreatic intraepithelial neoplasia (PanIN) or PDAC lesions, is reported overall between 1.5 and 8% [5, 33, 53, 56] and up to 9.3% in carriers of BRCA1/2, CDKN2a and ATM mutation carries [53]. In the present study, 2 of 25 (8%) IAR under screening had histopathological proven relevant pancreatic lesions (Table 3), which is a comparably high rate compared to mutation carriers of predisposing genes [10, 26]. This could be a characteristic of FPC-CRC families, which might be considered during counselling and for inclusion of these IAR into PDAC screening programs.
We observed interesting phenotypic and genotypic findings for FPC-CRC families, but a major limitation of our study was the relatively small number of families, as well as only partial availability of all patient material for mutational sequencing analysis. The same issue holds true for the limited number of IARs enrolled in the screening program. Therefore, our observations should be interpreted with caution and the results need to be verified by a larger number of FPC-CRC families to determine, whether these reported characteristics can be confirmed and can be translated into the management of these families.
References
Quante AS et al (2016) Projections of cancer incidence and cancer-related deaths in Germany by 2020 and 2030. Cancer Med 5(9):2649–2656. https://doi.org/10.1002/cam4.767
Slater EP et al (2021) Combinations of Low-Frequency Genetic Variants Might Predispose to Familial Pancreatic Cancer Journal of Personalized Medicine, 11(7) DOI: ARTN 63110.3390/jpm11070631
Bartsch DK, Gress TM, Langer P (2012) Familial pancreatic cancer–current knowledge. Nat Rev Gastroenterol Hepatol 9(8):445–453. https://doi.org/10.1038/nrgastro.2012.111
Canto MI et al (2013) International Cancer of the Pancreas Screening (CAPS) Consortium summit on the management of patients with increased risk for familial pancreatic cancer. Gut 62(3):339–347. https://doi.org/10.1136/gutjnl-2012-303108
Bartsch DK et al (2021) The German National Case Collection for familial pancreatic carcinoma (FaPaCa)-Knowledge gained in 20 years. Dtsch Arztebl Int 118(Forthcoming). https://doi.org/10.3238/arztebl.m2021.0004
Applebaum SE et al (2000) Genetic testing - counseling, laboratory, and regulatory issues and the EUROPAC protocol for ethical research in multicenter studies of inherited pancreatic diseases. Med Clin North Am 84(3):575–. https://doi.org/10.1016/S0025-7125(05)70241-8
Yeo TP et al (2009) Assessment of “gene-environment” interaction in cases of familial and sporadic pancreatic cancer. J Gastrointest Surg 13(8):1487–1494. https://doi.org/10.1007/s11605-009-0923-6
Bartsch DK et al (2016) Refinement of screening for familial pancreatic cancer. Gut 65(8):1314–1321. https://doi.org/10.1136/gutjnl-2015-311098
Roberts NJ et al (2016) Whole genome sequencing defines the genetic heterogeneity of familial pancreatic Cancer. Cancer Discov 6(2):166–175. https://doi.org/10.1158/2159-8290.CD-15-0402
Bartsch DK, Mintziras ME, Böhm I, Gercke LB, Bauer N, Figiel C J. and, Slater EP (2020) Characteristics of pure familial pancreatic Cancer families and those with additional breast Cancer. Open Access Journal of Oncology and Medicine 4(1). https://doi.org/10.32474/OAJOM.2020.04.000178
Ellis LM, Samuel S, Sceusi E (2010) Varying opinions on the authenticity of a human midgut carcinoid cell line–letter. Clin Cancer Res 16(21):5365–5366. https://doi.org/10.1158/1078-0432.CCR-10-2550
Tanaka LF et al (2023) The rising incidence of early-onset Colorectal Cancer in Germany. Dtsch Arztebl Int Forthcoming https://doi.org/10.3238/arztebl.m2022.0368
Sinicrope FA (2022) Increasing incidence of early-onset Colorectal Cancer. N Engl J Med 386(16):1547–1558. https://doi.org/10.1056/NEJMra2200869
Patel SG et al (2022) The rising tide of early-onset colorectal cancer: a comprehensive review of epidemiology, clinical features, biology, risk factors, prevention, and early detection. Lancet Gastroenterol Hepatol 7(3):262–274. https://doi.org/10.1016/S2468-1253(21)00426-X
Hahn SA et al (2003) BRCA2 germline mutations in familial pancreatic carcinoma. Gastroenterology 124(4). https://doi.org/10.1016/S0016-5085(03)82772-5. A548-A548 DOI
Murphy KM et al (2002) Evaluation of candidate genes MAP2K4, MADH4, ACVR1B, and BRCA2 in familial pancreatic cancer: deleterious BRCA2 mutations in 17%. Cancer Res 62(13):3789–3793
Bartsch DK et al (2002) CDKN2A germline mutations in familial pancreatic cancer. Ann Surg 236(6):730–737. https://doi.org/10.1097/00000658-200212000-00005
Slater EP et al (2010) PALB2 mutations in european familial pancreatic cancer families. Clin Genet 78(5):490–494. https://doi.org/10.1111/j.1399-0004.2010.01425.x
Roberts NJ et al (2012) ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov 2(1):41–46. https://doi.org/10.1158/2159-8290.CD-11-0194
Schneider R et al (2011) German national case collection for familial pancreatic cancer (FaPaCa): ten years experience. Fam Cancer 10(2):323–330. https://doi.org/10.1007/s10689-010-9414-x
Potjer TP et al (2013) Variation in precursor lesions of pancreatic cancer among high-risk groups. Clin Cancer Res 19(2):442–449. https://doi.org/10.1158/1078-0432.CCR-12-2730
Pijnappel EN et al (2022) Sex, gender and age differences in treatment allocation and survival of patients with metastatic pancreatic Cancer: a Nationwide Study. Front Oncol 12:839779. https://doi.org/10.3389/fonc.2022.839779
Abancens M et al (2020) Sexual dimorphism in Colon cancer. Front Oncol 10:607909. https://doi.org/10.3389/fonc.2020.607909
Cardoso R et al (2021) Incidence and mortality of proximal and distal colorectal Cancer in Germany-Trends in the era of Screening Colonoscopy. Dtsch Arztebl Int 118(16):281–287. https://doi.org/10.3238/arztebl.m2021.0111
Astiazaran-Symonds E, Goldstein AM (2021) A systematic review of the prevalence of germline pathogenic variants in patients with pancreatic cancer. J Gastroenterol 56(8):713–721. https://doi.org/10.1007/s00535-021-01806-y
Kartal K et al (2022) Familial pancreatic cancer: who should be considered for genetic testing? Ir J Med Sci 191(2):641–650. https://doi.org/10.1007/s11845-021-02572-9
e.V, R.K.-I.a.G.d.e.K.i.D., Krebs in Deutschland. 2017/2018. Berlin. p. www.krebsdaten.de
White A et al (2018) A review of sex-related differences in colorectal cancer incidence, screening uptake, routes to diagnosis, cancer stage and survival in the UK. BMC Cancer 18(1):906. https://doi.org/10.1186/s12885-018-4786-7
Schutze M et al (2011) Alcohol attributable burden of incidence of cancer in eight european countries based on results from prospective cohort study. BMJ 342:d1584. https://doi.org/10.1136/bmj.d1584
Tchernof A, Despres JP (2013) Pathophysiology of human visceral obesity: an update. Physiol Rev 93(1):359–404. https://doi.org/10.1152/physrev.00033.2011
Bassett JK et al (2010) Body size, weight change, and risk of colon cancer. Cancer Epidemiol Biomarkers Prev 19(11):2978–2986. https://doi.org/10.1158/1055-9965.EPI-10-0543
El Tekle G et al (2021) Co-occurrence and mutual exclusivity: what cross-cancer mutation patterns can tell us. Trends Cancer 7(9):823–836. https://doi.org/10.1016/j.trecan.2021.04.009
Goggins M et al (2020) Management of patients with increased risk for familial pancreatic cancer: updated recommendations from the International Cancer of the Pancreas Screening (CAPS) Consortium. Gut 69(1):7–17. https://doi.org/10.1136/gutjnl-2019-319352
Chaffee KG et al (2018) Prevalence of germ-line mutations in cancer genes among pancreatic cancer patients with a positive family history. Genet Med 20(1):119–127. https://doi.org/10.1038/gim.2017.85
Grant RC et al (2015) Prevalence of germline mutations in cancer predisposition genes in patients with pancreatic cancer. Gastroenterology 148(3):556–564. https://doi.org/10.1053/j.gastro.2014.11.042
Abe T et al (2019) Deleterious germline mutations are a risk factor for neoplastic progression among high-risk individuals undergoing pancreatic surveillance. J Clin Oncol 37(13):1070–. https://doi.org/10.1200/Jco.18.01512
Konings IC et al (2017) Prevalence and progression of pancreatic cystic precursor lesions differ between groups at high risk of developing pancreatic Cancer. Pancreas 46(1):28–34. https://doi.org/10.1097/MPA.0000000000000725
Hu C et al (2018) Multigene Hereditary Cancer Panels Reveal High-Risk Pancreatic Cancer susceptibility genes. JCO Precis Oncol 2. https://doi.org/10.1200/PO.17.00291
Randon G et al (2019) Prognostic impact of ATM mutations in patients with metastatic colorectal cancer. Sci Rep 9(1):2858. https://doi.org/10.1038/s41598-019-39525-3
Valle L et al (2019) Genetic predisposition to colorectal cancer: syndromes, genes, classification of genetic variants and implications for precision medicine. J Pathol 247(5):574–588. https://doi.org/10.1002/path.5229
Choi M, Kipps T, Kurzrock R (2016) ATM mutations in Cancer: therapeutic implications. Mol Cancer Ther 15(8):1781–1791. https://doi.org/10.1158/1535-7163.MCT-15-0945
Earl J et al (2020) A comprehensive analysis of candidate genes in familial pancreatic cancer families reveals a high frequency of potentially pathogenic germline variants. EBioMedicine 53:102675. https://doi.org/10.1016/j.ebiom.2020.102675
Grabenstetter A, Lazaro C, Turashvili G (2020) Editorial: Hereditary breast and ovarian Cancer: current concepts of Prevention and Treatment. Front Oncol 10:618369. https://doi.org/10.3389/fonc.2020.618369
Hofstatter EW et al (2011) PALB2 mutations in familial breast and pancreatic cancer. Fam Cancer 10(2):225–231. https://doi.org/10.1007/s10689-011-9426-1
Medina Pabon MA, Babiker HM (2022) A Review Of Hereditary Colorectal Cancers, in StatPearls. : Treasure Island (FL)
Kastrinos F et al (2009) Risk of pancreatic cancer in families with Lynch syndrome. JAMA 302(16):1790–1795. https://doi.org/10.1001/jama.2009.1529
Bujanda L, Herreros-Villanueva M (2017) Pancreatic Cancer in Lynch Syndrome Patients. J Cancer 8(18):3667–3674. https://doi.org/10.7150/jca.20750
Sina M et al (2021) Identification and management of Lynch syndrome in the Middle East and North African countries: outcome of a survey in 12 countries. Fam Cancer 20(3):215–221. https://doi.org/10.1007/s10689-020-00211-3
Stec R et al (2010) Colorectal cancer in the course of familial adenomatous polyposis syndrome (“de novo” pathogenic mutation of APC gene): case report, review of the literature and genetic commentary. Arch Med Sci 6(2):283–287. https://doi.org/10.5114/aoms.2010.13911
Giardiello FM et al (1993) Increased risk of thyroid and pancreatic carcinoma in familial adenomatous polyposis. Gut 34(10):1394–1396. https://doi.org/10.1136/gut.34.10.1394
Groen EJ et al (2008) Extra-intestinal manifestations of familial adenomatous polyposis. Ann Surg Oncol 15(9):2439–2450. https://doi.org/10.1245/s10434-008-9981-3
Moussata D et al (2015) Familial adenomatous polyposis and pancreatic cancer. Pancreas 44(3):512–513. https://doi.org/10.1097/MPA.0000000000000295
Overbeek KA et al (2022) Long-term yield of pancreatic cancer surveillance in high-risk individuals. Gut 71(6):1152–1160. https://doi.org/10.1136/gutjnl-2020-323611
Sheel ARG et al (2019) Identification of cystic lesions by secondary screening of familial pancreatic Cancer (FPC) Kindreds is not Associated with the stratified risk of Cancer. Am J Gastroenterol 114(1):155–164. https://doi.org/10.1038/s41395-018-0395-y
Bartsch DK et al (2013) Multiple small “imaging” branch-duct type intraductal papillary mucinous neoplasms (IPMNs) in familial pancreatic cancer: indicator for concomitant high grade pancreatic intraepithelial neoplasia? Fam Cancer 12(1):89–96. https://doi.org/10.1007/s10689-012-9582-y
Vasen H et al (2016) Benefit of Surveillance for Pancreatic Cancer in High-Risk individuals: outcome of long-term prospective Follow-Up studies from three european Expert Centers. J Clin Oncol 34(17):2010–2019. https://doi.org/10.1200/JCO.2015.64.0730
Acknowledgements
We are grateful to all FPC families for participating in the collection. We thank A. Ramaswamy, Marburg, and Günter Klöppel, Munich, for the pathological examination of the resection specimens.
Funding
This study was supported by grant 111092 of the Deutsche Krebshilfe and a generous donation of the GAUFF-Foundation.
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
DKB: principal investigator, responsible for the planning of the project, acquisition, analysis and interpreting of data, drafting the manuscript, obtained funding. BL: molecular biologist, drafting the manuscript, analysis of data. EPS: molecular biologist and methodologist, analysis of data, performed statistical analysis, assisted in drafting the manuscript. TH: genetic analysis of the FPC-CRC cohort. JF: radiologists who scored the MRIs, assisted in the interpretation of results and critically reviewed the manuscript. UD: endosonographer who performed the EUS, assisted in the interpretation of results and critically reviewed the manuscript. EM, NG: acquisition of data, assistance in the interpretation of results and in the critical review of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The FaPaCa collection, including the genetic analyses and the screening program, was approved by the Ethics Committee of the Philipps-University of Marburg (36/1997, last amendment 9/2010). All participants provided written informed consent.
Competing Interests and Funding
None of the authors has competing financial interests nor other conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lehman, B., Matthäi, E., Gercke, N. et al. Characteristics of familial pancreatic cancer families with additional colorectal carcinoma. Familial Cancer 22, 323–330 (2023). https://doi.org/10.1007/s10689-023-00328-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10689-023-00328-1