
Abstract
Super Basmati is an elite variety with extra-long grains and superior quality but its yield is severely affected by water stress. Recombinant inbred lines (RILs) developed from the cross of Super Basmati and IR55419-04 (a coarse grained variety with high yield potential) were used to identify and map yield-related quantitative trait loci (QTLs) under normal field conditions. Genotypes for 244 KASP markers were obtained from 188 F9 RILs and a linkage map constructed of 1369.4 cM, with average marker interval of 10.5 cM. Eleven agronomic traits were phenotyped in RILs and parents. Composite interval mapping and inclusive composite interval mapping model identified 21 common QTLs related to nine agronomic traits; of the QTLs identified, twelve mapped at novel positions. LOD scores ranged from 2.51 for number of filled grains (qFG1.1) to 16.8 for plant height (qPH1.1). Four major effect QTLs could explain 20% of variation related for plant height, hundred-grain weight and grain width. Alleles from IR55419-04 improved grain filling, grain weight and grain width, while alleles from Super Basmati contributed to longer grains, panicles and flag leaves. Putative candidate genes were identified for 18 yield-related QTLs. These results validate the use of KASP genotyping for QTL mapping of yield-related traits in a bi-parental segregating population. SNPs in the QTLs identified in this study can be used in marker assisted selection for yield components to improve Basmati rice.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pakistan is among the top ten rice producing countries. Its cultivated area has increased by 11% since 2019 and production by 20%, with a 5 years average (2018/19–2022/23) of 3.1 million hectares producing 7.5 million metric tons (USDA 2023). Rice is Pakistan’s second major crop in terms of cultivation and export, contributing about 70.2% to the agricultural economy of Pakistan (Abbas and Mayo 2021). Pakistan contributes about 32% of global basmati export (Hussain et al. 2014). Breeders developed elite basmati varieties by crossing between high-yielding, short statured non-basmati varieties with basmati varieties including Super Basmati (SB) (Nagaraju et al. 2002).
Super Basmati has superior market value with extra-long grains, aroma and soft texture with intermediate amylose content (Sabar et al. 2019), however, it suffers approximately 90% paddy yield reduction when exposed to water stress. Rice yield components are highly complex traits that contribute towards overall yield; these include spikelet per panicle, fertility, grain number per panicle, panicle number, grain filling and weight. Grain length and width contribute towards appearance quality of grain (Li et al. 2021).
Quantitative traits having continuous variation are often controlled by multiple genes (Khan 2015) thus it is necessary to locate and map quantitative trait loci (QTLs). Usually parents with contrasting characters are crossed and recombinants are produced which are further inbred to achieve homozygosity hence, providing a reliable source for trait mapping (Pollard 2012). QTLs identified in mapping populations can be used in marker-assisted selection (MAS) breeding programs to develop elite varieties. However, the higher cost of genotyping with DNA markers can limit the use of MAS (Steele et al. 2018). Next generation sequencing (NGS), array-based genotyping and genotype by sequencing (GBS) all provide high resolution for efficient QTL mapping but they lack the flexibility and ease provided by SSRs (simple sequence repeats) which are often the preferred genotyping method of public sector breeders (Yang et al. 2015). An alternative but similarly flexible genotyping marker system is Kompetitive allele-specific PCR (KASP™) technology of LGC Biosearch Technologies, UK (He et al. 2014). KASP can genotype either SNP or InDel loci, although the majority of assays are designed to target SNPs. KASP genotyping costs can be up to 60% lower than SSRs (Steele et al. 2018), with a more efficient workflow. KASP genotyping has been used for screening of rice stripe virus (RSV) resistance genes at early growth stage (Kang et al. 2020), high throughput genotyping of varieties tolerant to abiotic germination stress (Lee et al. 2021), QTL mapping of bakanae disease (BD) resistance in rice (Cheon et al. 2019), genotyping and screening of candidate alleles in maize (Mideros et al. 2014) and discrimination between commercial Basmati rice varieties (Steele et al. 2021). Previously, about 2015 KASP markers were developed by International Rice Research Institute (IRRI) covering the rice genome but only a few KASP were reported as functional or linked to traits (Pariasca-Tanaka et al. 2015).
In order to select drought tolerance and yield traits, Super Basmati was crossed with a coarse grained indica variety IR55419-04 that has better yield potential and shows only 51% reduction in yield under two weeks water stress which is better than Super Basmati (Sabar and Arif 2014). The F2 introgression lines developed from the cross of Super Basmati and IR55419-04 exhibited drought tolerance due to deeper roots and enhanced root diameter (Sabar et al. 2019). A similar study using an F7 recombinant inbred lines (RIL) population developed from the cross of Super Basmati and Azucena (a japonica cultivar) was used to map traits related to drought response (Waheed et al. 2021). The present study focused on mapping the genomic regions involved in enhancing or controlling yield-related traits in F9 RILs from the contrasting parents Super Basmati and IR55419-04.
The objective of this study was to use KASP markers, designed to target either functional SNPs or SNPs located close to previously published markers associated with agronomic traits, for genetic linkage mapping of traits related to yield in RILs derived from the cross between Super Basmati and IR55419-04. The goal was to validate KASP as an efficient and accurate genotyping system for QTL mapping by comparing the identified QTL locations with already reported QTLs and previously clone genes.
Methods
Plant materials
An F9 generation of 188 RILs developed from the cross of Super Basmati (female parent) and IR55419-04 (male parent) (Sabar and Arif 2014) using single seed descent were used in this study. Both parents, Super Basmati and IR55419-04 were also included with studied population.
Field experiments and traits evaluation
One hundred and eighty-eight RILs along with both parents were planted in an alpha lattice design in two replicates in the research area of the National Institute for Biotechnology and Genetic Engineering (NIBGE), Faisalabad, Punjab, Pakistan (31° ′42′ N 73° ′02′ E), in the 2020 rice growing season. Fifteen plants from each RIL family and parents were planted in each row and data of agronomic traits was recorded from central three plants (usually 7th, 8th and 9th plant) at vegetative and reproductive stage. The soil conditions, fertilizers applied, and irrigation regimes are described in Supplementary File 1, Table S1.
Data of agronomic traits including plant height (PH), number of tillers per plant (PTN), flag leaf length (FLL) was recorded several days before harvest. PH was calculated at maturity stage and height of single plant from the soil surface to the tip of the panicle of the main tiller. PTN were counted for each plant at maturity and FLL was measured at the beginning of the anthesis of main tiller. For panicle length (PL), three panicles per plant were taken and length was calculated from the tip to the neck of panicle after harvest. Number of filled grains per panicle (FG) and number of unfilled grains (UFG) were calculated manually after threshing, as was the ratio of unfilled to total number of spikes, giving spikelet sterility percentage. Spikelet fertility percentage (SF%) was calculated by dividing FG by total number of spikelet. An electronic balance was used for the measurement of hundred grains weight per plant (HGW) and grain yield per plant (GYP) of sun-dried samples having up to 12% moisture content. For the measurement of grain length (GL) and grain width (GW), ten grains from each selected plant were taken and analyzed with SMARTGRAIN software (Tanabata et al. 2012).
Statistical data analysis
Mean values were calculated for each trait from the data of three plants per line. Frequency distribution for each trait was calculated using GraphPad Prism v7.00 (GraphPad Software, Boston, USA). Support for possible shared or pleiotropic effects of co-located QTLs was assessed by testing the correlation between traits using Pearson’s correlation coefficient at significance level p < 0.05. Pearsons’s correlations, Skewness, kurtosis and phenotypic variance were calculated using SPSS 23.0 (SPSS, Chicago, IL, United States). Non-normal data was transformed using box-cox functionality in PAST software (Hammer et al. 2001) for QTL mapping.
KASP marker development and genotyping
A schematic flowchart of the main steps in KASP-marker development, molecular analysis and linkage analysis can be seen in Fig. 1. Prior to this study, novel KASP markers were designed from 50 bp flanking sequences flanking each target SNP using the bioinformatics pipeline described by Steele et al. (2021) by alignment of whole genome sequences against the Shuhui498 (R498) indica reference genome (Du et al. 2017). SNPs flanking SSRs associated with agronomic traits or predicted functional SNPs, were selected on all chromosomes for genotyping with allele specific KASP markers. Each KASP was assigned a working KASP ID relating these selection criteria. The 50 bp flanking sequences for 250 selected SNP sites were sent to LGC in the SNP submission template that they provided.

Steps used for KASP marker development, genotyping and SNP filtering for linkage analysis
The all-inclusive services for plant samples of LGC Biosearch Technologies, UK (https://www.biosearchtech.com/services/genotyping-services/all-inclusive-services) was used for KASP genotyping. The leaf samples from 188 RILs and parents were sent to LGC Biosearch Technologies, UK, following the protocol specified by them.
The genotyping data file received from LGC Genomics, UK was analyzed using SNPViewer™ software (https://www.biosearchtech.com/support/tools/genotyping-software/snpviewer) and a dataset was compiled in Microsoft Excel containing called SNPs at 244 loci in 188 RILs and the two parents (Supplementary File 2).
Analysis of KASP genotyping data
Eighty-three monomorphic loci among the parents and the loci containing homozygous alleles in RILs were removed from the genotype data received from LGC Genomics, UK. MACH software (Li 2006) was used for imputation of most likely genotype at missing site (missing rate was around 6% per loci) because it has better efficiency as compared to other imputation software as assessed by (Biernacka et al. 2009). After imputation of missing sites and allele frequency analysis, 145 polymorphic KASP markers remained from which a further 20 KASP markers were removed due to segregation distortion. The remaining 125 polymorphic markers were analyzed in the following steps. GenAELx 6 (Peakall and Smouse 2006) was used to conduct allele frequency analysis for the determination of polymorphic markers percentage. RILs were assessed for heterozygous alleles percentage to determine the heterozygosity in population.
Linkage map construction
A linkage map was calculated using the MAP functionality in QTL IciMapping software and the data generated was then used to draw linkage map using Map Chart v2.32. Binning of redundant markers was also done in QTL IciMapping software v4.2 (Meng et al. 2015) and a chi-square test was used to remove marker loci with segregation distortion from the analysis as also performed by Hirao et al. (2019). Segregation distortion was detected if a locus had a p-value of < 0.05. For grouping and ordering of remaining markers, the regression mapping algorithm RECORD (REcombination Counting and ORDering) based on recombination events between adjacent markers was used and the algorithms for ordering and rippling that gave a shorter linkage map were used. Kosambi mapping function was used to calculate genetic distance (cM) between markers. This linkage map was used for QTL mapping.
QTL analysis
Three software platforms were used for QTL mapping and QTLs that mapped consistently among at least two platforms were considered as stable QTLs.
-
(i)
QTL Ici Mapping was used to predict LOD scores and the phenotypic variance explained (PVE) for each marker using the single marker analysis (SMA) functionality with a LOD threshold of 2.5. Then the inclusive composite interval mapping (ICIM) functionality was used to map putative QTLs related to various traits and additive effect along with PVE percentage also analyzed. The LOD test statistic used was – 2ln (L0/L1), where L0/L1 is the ratio of the likelihood under the null hypothesis (indicating the absence of QTL) and the alternative hypothesis (indicating the presence of QTL). QTLs with PVE ≥ 20% were considered as major-effect QTL (Du et al. 2019). The step length for QTL scanning was set at 1 cM and LOD threshold value required to declare a QTL (p < 0.05) was determined by 1000-permutations test.
-
(ii)
QTL g.CI Mapping (Changrong et al. 2020) was used for genome-wide composite interval mapping using the LOD threshold value previously determined by QTL IciMapping software.
-
(iii)
Windows QTL Cartographer v2.5 (Wang et al. 2012a) was also used to perform CIM.
The naming of the QTLs were as per the standard nomenclatural guidelines published (McCouch 1997). The indica R498 genome positions of left and right KASP markers of the QTLs were converted to japonica reference genome (Nipponbare IRGSP 1.0) positions for reporting herein. QTLs identified commonly from all models were compared with already reported regions in Gramene QTL database (https://archive.gramene.org/qtl/) and QTARO database (https://qtaro.abr.affrc.go.jp/qtab/table#as_table:21:undefined:undefined). QTLs were considered novel if the observed marker interval did not overlap with the marker interval significantly in previous studies (Kulkarni et al. 2020). For the identification of putative gene(s) in the QTL regions, the KASP marker SNP position nearest to QTL peak was selected and 200 kb region (100 kb downstream and upstream of SNP) was searched for candidate genes in the following databases. MSU Rice Genome Annotation Release 7 was obtained used to obtain information about the genes model for all chromosomes as well as functional annotation (http://rice.plantbiology.msu.edu/; Thibaud-Nissen et al. 2007). Q-TARO database was also used to obtain the list of genes and their locations on all chromosomes to find genes with functions that may underly QTLs (Yonemaru et al. 2010). Putative functions of candidate genes in the QTL region were then confirmed from already published knockout studies and association of gene with the trait was determined.
Allelic effect of associated KASP markers with traits
The mean phenotypic data of the traits with respect to each allele of the KASP marker nearest to the QTL peak was compared using the function (t-Test: two-sample assuming equal variances) in analysis ToolPak add-in in MS Excel and allelic effect of the parents on the traits was estimated.
In silico candidate genes analysis
Based on the search window suggested by (Zaw et al. 2019), a 100 kb region upstream and downstream of the KASP marker nearest to the peak LOD was searched for genes in the MSU Rice Genome Annotation Project database and then the resulting list of gene loci for each QTL was evaluated to find candidate genes that could be involved in the expression of respective trait.
Results
Trait variation in RILs
RILs were identified that carried favourable characteristics from one of the parents such as longer and wider grains, semi-dwarf, longer panicle and flag leaf and promising grain weight and grain yield. Transgressive segregation was observed in RILs for panicle length (PL) and hundred grain weight (HGW) and broad distributions was observed for traits such as plant height (PH), tiller number (PTN), flag leaf length (FLL), filled grains number (FG), unfilled grains number (UFG) and grain yield per plant (GYP) (Table 1). RILs showed normal frequency distribution for traits such as PH, FLL and GL while UFG showed significant positive skewness and GYP data was slightly positive skewed. Data from PL,SF% and GW was slightly negative skewed (Fig. 2).

A–O Frequency distributions of phenotype data of 188 RILs and parents. Blue down arrow indicates Super Basmati and red down arrow indicates IR55419-04 (where data available). (Color figure online)
There was significant variation between the parents for most traits (Table 1). Super Basmati was about 22.5 cm taller than IR55419-04 and it also showed significantly higher values for the traits i.e., tiller number, flag leaf length, panicle length and grain length. However, IR55419-04 had on average 98 more filled grains/panicle (p < 0.0001) than Super Basmati (53 grains/panicle) and its average paddy grain width was 0.33 mm wider than Super Basmati (2.6 mm).
Correlation analysis of yield-related traits
Correlations of GYP, FG and PH with all agronomic traits are summarized in histograms (Fig. 3). GYP was positively correlated with PH, PTN, PL, HGW, FLL, FG, GW and SF% (Table 2). PL was also found to be positively correlated with PH, FLL and FG. HGW was positively correlated with GL and GW.

Histogram of Pearson’s correlation (r) between yield related traits plant height (PH), number of tillers per plant (PTN), flag leaf length (FLL), panicle length (PL), number of filled grains per panicle (FG), number of unfilled grains (UFG), spikelet fertility percentage (SF%), hundred grains weight per plant (HGW), grain yield per plant (GYP), grain length (GL) and grain width (GW)
UFG negatively correlated with PH, PTN, FG, HGW, GYP, GL, GW and most significant with SF%. FLL had negative correlation with HGW, GL, GW and SF% ranging from −0.111 (SF%) to −0.059 (GL). GL was found to be negatively correlated with GYP, FG and UFG with a value of −0.015, −0.23 and −0.109 respectively. PL had slightly negative correlation only with HGW with a score of −0.015.
Genotyping and linkage analysis
Of 188 RILs, 152 RILs were 85% homozygous so only those lines were further used in linkage analysis. The total genetic distance of the genetic map was found to be 1369.4 cM with an average interval of 10.59 cM (Table 3; Fig. 4) which is within the recommended range (Collard et al. 2005). There were only two markers on chromosome 10; otherwise, 6–18 markers were located on each chromosome with an average interval of 11 cM. The order of the 125 genetically mapped markers was identical to the physical map order (Supplementary File 1).

Linkage map of 125 KASP markers on all 12 chromosomes drawn using Map Chart v2.32
QTL analysis
A total of 31 QTLs were detected collectively among all platforms with QTLs detected for all traits except spikelet fertility percentage (SF%). For SMA results see Supplementary File 1, Fig. S1). Of these 31 QTLs, WinQTL Cartographer detected 10 additional QTLs including two QTLs for PH on Chr 6, one QTL for PTN on Chr 3, four QTLs for FLL on Chr 5, 11 and 3, two QTLs for HGW on Chr 3 and a QTL for GYP on Chr 3 that were not included because they are identified in only one platform.) The remaining 21 QTLs were consistent in at least two platforms and thus considered stable QTLs (Supplementary File 1, Fig. S2). These stable QTLs included twelve novel QTLs and four well-known major effect QTLs related to height, hundred grain weight and grain width. The detailed summary of statistics related to these QTLs is present in Table 4 (including citations for QTLs previously reported) and the direction of QTLs based on positive or negative additive effect is present in Table 5. The QTLs with positive additive effect are inherited from Super Basmati while the ones with negative additive effect are inherited from IR55419-04. The increasing or decreasing effect on grain traits for alleles of KASP markers nearest to the seven QTLs could be observed visually (Supplementary File 1 Fig. S3). The allelic effects on traits at the KASP marker nearest to the QTL position are shown in Fig. 5.

Box and whiskers plots of allelic class means and variations for plant height (PH), hundred grains weight per plant (HGW), panicle length (PL), grain length (GL) number of filled grains per panicle (FG), number of unfilled grains (UFG), grain width (GW), flag leaf length (FLL) and grain yield per plant (GYP). Means are indicated with crosses, boxes show interquartile range, whiskers show 95% confidence interval while dots represent outliers
In-silico analysis of putative candidate genes identification
The KASP markers nearest to the detected QTLs were selected for the identification of putative (candidate) genes. A total of 814 genes were found within ± 100 kb region for all 21 QTLs collectively, with an average of 38 genes per QTL (Supplementary File 3).
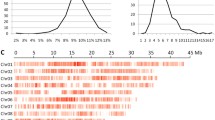
In-silico analysis detected putative cloned genes for 18 out of 21 QTLs (Fig. 6; Table 6). No putative gene associated with respective traits were found for qGL2.2, qGL5.1 and qGL6.1.

Graphical representation of QTL peak positions (shown in red) mapped using QTL IciMapping on respective chromosomes and the location of putative candidate genes (shown in black) associated with the trait on both physical (Mb = positions in Os-Nipponbare-Reference-IRGSP-1.0) and genetic map (cM). (Color figure online)
Comparative analysis of QTL clusters and phenotype correlation
Two QTLs on chromosome 2 (Fig. 7) comprising of qHGW2.1 (peak position 30.58 Mb) and qGL2.1 (peak position 31.29 Mb) were 0.71 Mb apart. Positive additive effect of these QTLs reflected the role of Super Basmati in enhancing yield and grain length. A positive phenotypic correlation (r = 0.329) was found between HGW and GL (Table 2). The RILs carrying the Super Basmati haplotype-set in the 0.71 Mb region (between KASP markers gs_id391_ff and gs_id395_ff) all had greater grain length and grain weight.

Linked QTLs and the haplotype-sets associated with their phenotypes. For chromosome 2 RILs with Super Basmati (SB) haplotype-set for KASP markers (gs_id391_ff, gs_id392_ff, gs_id393_ff, gs_id394, gs_id395_ff) had longer grains and higher grain weight. For chromosome 8 RILs with IR55419-04 haplotype-set (gs_id1421_fn, xa13_SNP_nn_2, gs_id1422_ff, xa13_SNP_nn_5) had wider grains
Two QTLs for grain weight on chromosome 8 (Fig. 7), qHGW8.1 (peak position 26.44 Mb) and qGW8.1 (peak position 26.95 Mb), were 0.51 Mb apart. Both QTLs showed negative additive effect and positive correlation of 0.561 (Table 2) as supported by previous studies (Zhang et al. 2020). RILs carrying the IR55419-04 haplotype-set in the 0.51 Mb region (the region between KASP markers gs_id1421_fn and xa13_SNP_nn_5) had greater grain width and weight.
Two QTLs, qUFG3.1 and qGW3.1, were found on chromosome 3 with a distance of 0.28 Mb between them. Both these QTLs had negative additive effect and negative correlation of −0.103 (Table 2).
Discussion
Validity of KASP markers for QTL mapping
KASP have been available for identification of genetic variation at nucleotide level for around a decade (He et al. 2014), yet their suitability for QTL mapping is yet to be widely appreciated. This study has shown that KASP-based SNP genotyping is efficient for mapping of QTLs in a segregating bi-parental rice population. In a single genotyping screen targeting 250 SNPs which had not previously been tested for polymorphism in the parents, half of them amplified polymorphic markers, providing sufficient coverage to identify QTLs on 10 of the 12 chromosomes. This compares favorably to the outcome of SSR-based QTL analysis in RILs derived from Basmati 370 and Pusa Basmati 1121, which also detected no QTLs on Chromosomes 9 and 10 (Sharma et al. 2021). Eight out of 21 QTLs mapped in this study were co-localized with already reported QTL regions, thus validating the accuracy of KASP markers for QTL mapping. With more than a million KASP markers available (Steele et al. 2018), another round of genotyping with < 50 selected markers should give additional map coverage, if needed for future analysis or fine mapping. However, in-silico analysis of rice gene databases permitted to detection of putative genes underpinning the majority of the QTLs identified, without further marker analysis.
RILs performance
The mean plant height of RILs was closer to the height of the semi-dwarf parent (IR55419-04) (Table 1). The mean tiller number (PTN) of RILs was slightly less than both parents while mean flag leaf length and panicle length of RILs was higher than both parents. There were more filled grains (FG) in IR55419-04 than Super Basmati but the mean FG in RILs was lower than IR55419-04. However, grain weight and width were higher in RILs than both parents. Grain length mean data of RILs was closer to the grain length of Super Basmati suggesting some dominance in the allelic contribution from Super Basmati.
Grain yield (GYP) was positively correlated with all other traits except UFG and GL, although its negative correlation with GL was not significant (Table 2); this contrasts with the results of Li et al. (2021) who found both GL and GW to be positively correlated with grain yield in RILs derived from two indica varieties. FG correlation and PH were also significantly positively correlated with all traits except UFG and GL (Table 2). Decoupling of grain size and yield has been observed in a study in which knocking out GS3 gene (which is a negative regulator of grain size) increased the grain length but decreased the grain yield (Yuyu et al. 2020).
Candidate genes for QTLs
A total of 814 genes were found within ± 100 kb region of peak LOD for the 21 QTLs identified (Supplementary file 3). The closest KASP markers to the peak LOD scores were considered to be useful for selection for that QTL in this mapping population. At least one candidate gene was identified based on the functions of genes previously published for 18 of the QTLs identified in this study (Table 6).
Plant height
qPH1.1 corresponds to frequently reported QTLs for plant height (Changrong et al. 2020; Kulkarni et al. 2020; Li et al. 2021; Sabar et al. 2019; Yadav et al. 2019). Of 34 genes within the ± 100 kb region flanking the QTL peak at 38.4 Mb, only Os01g0883800 at 38.38 Mb position had biological function related to plant height (Spielmeyer et al. 2002). This is the semi-dwarf 1 (sd-1) locus responsible for semi-dwarf phenotype because of a defective gibberellin (GA) 20-oxidase that fails to provide enough gibberellin (GA) plant growth hormones in elongating stem. IR55419-04 contributed the height reducing allele for this QTL (Table 6) since sd-1 is a major-effect QTL, this might lead to the mean PH of RILs being below 120 cm. This evidence validates the association of the qPH1.1 with a KASP marker RM1068_SNP_nn_1.
A novel QTL for plant height (qPH3.1) was found on chromosome 3 (peak position 32.3 Mb). There were 58 genes within the ± 100 kb region of QTL peak and only Os03g0782500 was found to be associated with plant height at 32.43 Mb. This gene encodes a phytochrome-interacting factor-like protein (OSPIL1/OsPIL13) and it is a key regulator of internode elongation. Its expression is inhibited during drought stress conditions. This locus also suggests that the short stature of IR55419-04 during drought stress conditions could be a result of regulatory pathway that involves OsPIL1/OsPIL13 (Todaka et al. 2012). qPH12.1 (peak position 9.18 Mb) is adjacent to already reported QTLs for plant height (Kulkarni et al. 2020). Of 32 genes found ± 100 kb from the QTL peak, only Os12g0272800 at 9.98 Mb was related to plant height. This locus is the site of a semi-dominant mutant necrotic root tip 1 (nrtp1-D) that encodes a typical coiled-coil nucleotide binding leucine rich repeat (CC-NB-LRR) type protein, identified in a mutagenized population of Kasalath (Yu et al. 2018). Homozygous mutants were rootless with very poor shoot growth and overall development and heterozygotes had poor development as compared with wild type. This is evidence for an overall effect of this gene on plant height and proper development of plant.
Flag leaf length
A novel QTL for flag leaf length (FLL) (qFLL6.1) was found on chromosome 6 (peak position 21.5 Mb). About 31 genes were located ± 100 kb region of QTL peak. Among these, only Os06g0572400 at 22.19 Mb was found to be associated with flag leaf length. This gene encodes a tetraspanin protein (OsTET9) which accumulates in young flag leaves and its expression is suppressed during abiotic stress conditions (Mani et al. 2015).
Panicle length
A QTL for panicle length (PL) (qPL2.1) was found on chromosome 2 (peak position 25.93 Mb). A similar QTL was found between 24.42 and 25.87 Mb by Vemireddy et al., (2015) where Basmati 370 contributed a similar effect as Super Basmati in this study. Among the 32 genes that were present within ± 100 kb region of QTL peak, Os02g0673100 present at 27.39 Mb was found related with panicle length and development. This locus is aluminum-activated malate transporter (OsALMT7) that transports malate to apical portion of developing panicle which is required for normal panicle development (Heng et al. 2018).
Grain filling
Two QTLs for grain filling (qFG1.1 and qFG7.1) were found on chromosome 1 and 7 at peak positions 4.24 and 2.4 Mb respectively. Although qFG1.1 is considered novel in this population, we note that Waheed et al. (2021) reported a QTL designated qFG1 with Super Basmati alleles negatively affecting this trait at 4.5 MB. About 30 genes were found within ± 100 kb region of QTL peak of qFG1.1. A locus at 2.2 Mb on chromosome 1 (Os01g0142800) was found related to grain filling. This locus is peptide transporter 8.1/protein transporter 7 (OsPTR7) which is involved in transport of dimethylarsenate (DMA) from roots to shoots (Tang et al. 2017). About 33 genes were found ± 100 kb region of QTL peak of qFG7.1 and a locus Os07g0182000 at 4.3 Mb on chromosome 7 was found related to grain filling. This locus is basic leucine zipper transcription factor 58 (OsbZIP58) gene which is responsible for promoting expression of many seed storage proteins and starch synthesis genes. High temperature affects alternative splicing of this gene that results in modified form of OsbZIP58 which shows reduced activity (Xu et al. 2020).
Unfilled grains
A novel QTL for number of unfilled grains (qUFG3.1) was found on chromosome 3 (peak position 10.18 Mb). QTL analysis of spikelet sterility percentage identified a QTL at the same location with a LOD threshold > 2 but < 2.5. About 43 genes were present within ± 100 kb of the QTL peak of qUFG3.1 and a locus Os03g0308200 at 10.97 Mb on chromosome 3 was found associated with unfilled grain number. This locus is Narrow and rolled leaf 2 (NRL2) which regulates male fertility and pollen development. The same gene was also found within the ± 100 kb region of a QTL peak related to grain width (qGW3.1) which was found at 9.9 Mb on the same chromosome and already reported by (Bai et al. 2010). A mutant form of this gene (nrl2-1) carrying a base-pair deletion and another (nrl2-2) carrying a base-pair substitution both caused male sterility and wider seeds as compared to wild-type NRL2 as reported by (Zhao et al. 2016).
Grain weight
A novel QTL for hundred grain weight (qHGW1.1) was found on chromosome 1 (peak position 14.11 Mb). Thirty-two genes were found within the ± 100 kb region of QTL peak of qHGW1.1 and a locus Os01g0367100 at 14.99 Mb was found related to HGW. This locus is photoassimilate defective 1 (PHD1) which encodes chloroplast-localized UDP-glucose epimerase (UGE). PHD1 is involved in chloroplast biogenesis by supplying galactolipids to thylakoid membranes (Li et al. 2011).
Another QTL for HGW (qHGW2.1) was found on chromosome 2 (peak position 30.58 Mb) corresponding to already reported QTL for HGW by Donde et al. (2020), and a similar QTL for single plant yield was identified by Vemireddy et al. (2015). Thirty-six genes were found in 100 kb upstream and downstream region of QTL peak LOD and a locus Os02g0735200 at 30.67 Mb was found related to HGW. This locus is cytosolic glutamine synthetase 1;1 (OsGS1;1) which is responsible for normal growth and grain filling in rice (Tabuchi et al. 2005).
A major-effect QTL for HGW was found on chromosome 8 (peak position 26.44 Mb) corresponding to already reported QTL for HGW on chromosome 8 (Li et al. 2021). Thirty-six genes were present in ± 100 kb region of QTL peak LOD of qHGW8.1 and a locus Os08g0535200 at 26.7 Mb was found associated with HGW (Ma et al. 2017). This locus is OsSWEET11 which is found to express significantly during grain development.
Yield
A novel QTL for grain yield per plant (qGYP4.1) was found on chromosome 4 (peak position 34.44 Mb. Forty genes were found in the QTL peak ± 100 kb region and a locus Os04g0674700 at 34.44 Mb was found related with GYP. This locus is BTH-induced AMP binding protein 1 (OSBIABP1) which is found to express in stem, leaves and flowers and regulates plant growth and development during environmental stress (Zhang et al. 2009).
Grain length
Two novel QTLs related to grain length (qGL2.1 and qGL2.2) were found on chromosome 2 at peak positions 31.29 Mb and 35.1Mb respectively. About 35 genes were found in ± 100 kb region of qGL2.1 peak and one locus, a little downstream of searched range, Os02g0747900 at 31.42 Mb was found related with grain length. This locus is positive regulator of grain length 2 (PGL2) that interacts with a negative regulator of grain length i.e., basic helix-loop-helix (bHLH) proteins, to positively regulate grain length (Heang and Sassa 2012).
Another QTL for grain length was located on chromosome 3 at peak position 25.05 Mb co-localized with already reported QTLs for GL (Bai et al. 2010; Changrong et al. 2020; Li et al. 2021; Zaw et al. 2019). Thirty-seven genes were found in the QTL peak interval of ± 100 kb and a locus Os03g0646900 at 25.04 Mb was found associated with GL. This locus is phosphatase with Kelch-like repeat domain (OsPPKL1) also known as qGL3, a major QTL for grain length (Zhang et al. 2012).
A novel QTL for grain length was found on chromosome 11 at peak position of 21.1 Mb. Thirty-one genes were found within ± 100 kb of QTL peak region and a locus Os11g0528700 at 19.1 Mb was found associated with grain length. This locus is OsGH3.13 that encodes an indole-3-acetic acid (IAA)-amido synthetase. OsGH3.13 is also identified as a candidate gene for grain length QTL (qGL11) found in the interval of 19–20.1 Mb by (Wang et al. 2021).
Grain width
A major-effect QTL for grain width (qGW6.1) was found on chromosome 6 at peak position of 4.62 Mb co-localized with already reported QTL for GW (Li et al. 2021). Forty-three genes were found within the QTL peak 100kb up and downstream region and a locus Os06g0154500 present at 2.8 Mb was found related with grain width. This locus is mitogen activated protein kinase phosphatase1 (OsMPK1) also known as Dwarf and small grain 1 that negatively regulates grain size. Its overexpression results in smaller grains (Xu et al. 2018).
Another major-effect QTL for GW (qGW8.1) was found on chromosome 8 at peak position of 26.95 Mb adjacent to already reported QTL for GW by (Li et al. 2021). Forty-six genes were found within ± 100 kb region of QTL peak and a locus Os08g0537800 at 26.88 Mb was found to be related with grain width. This locus is Wide and thick grain 1 (WTG1) which encodes an otubain-like protease (a deubiquitinating enzyme). The mutant form of WTG1 produces wide, short and thick grains (Huang et al. 2017). Another locus Os08g0531600 also a well-known QTL for Grain width (GW8) (Wang et al. 2012b) was present a little upstream of searched range at 26.5 Mb.
Conclusion
KASP genotyping in a RIL population succeed in locating 21 QTLs. Of these 12 were novel, 4 major-effect QTLs and the remaining QTLs were co-localized with previously identified well-known QTLs for yield-related traits. From candidate genes found at 18 of the QTLs, we identified 6 previously cloned candidate genes related to yield within 100 kb region flanking the QTL peaks of qPH1.1, qHGW2.1, qGL3.1, qPH3.1, qGYP4.1 and qGW8.1. Further candidate cloned genes were found within 130 kb (qGL2.1) to less than 2 Mb region (qFG7.1) from QTL peak positions. The KASP markers RM1068_SNP_nn_1 (qPH1.1) on chromosome 1, gs_id1421_fn (qHGW8.1) on chromosome 8, gs_id395_ff (qGL2.1) on chromosome 2 and RM15585_SNP_nn_2 (qGL3.1) on chromosome 3 can be used with confidence for selection of shorter plant height, increased grain weight and increased grain length, respectively.
Change history
09 August 2023
A Correction to this paper has been published: https://doi.org/10.1007/s10681-023-03217-x
References
Abbas S, Mayo ZA (2021) Impact of temperature and rainfall on rice production in Punjab, Pakistan. Environ Dev Sustain 23(2):1706–1728. https://doi.org/10.1007/s10668-020-00647-8
Bai X, Luo L, Yan W, Kovi MR, Zhan W, Xing Y (2010) Genetic dissection of rice grain shape using a recombinant inbred line population derived from two contrasting parents and fine mapping a pleiotropic quantitative trait locus qGL7. BMC Genet 11(1):1–11. https://doi.org/10.1186/1471-2156-11-16
Biernacka JM, Tang R, Li J, McDonnell SK, Rabe KG, Sinnwell JP, Rider DN, De Andrade M, Goode EL, Fridley BL (2009) Assessment of genotype imputation methods. BMC Proc. https://doi.org/10.1186/1753-6561-3-S7-S5
Changrong Y, Hengming L, Wei D, Jinwen Z, Yuran X, Anyu G, Yonggang L, Wei N, Shengli S, Hua A (2020) Genome-wide association study on agronomic traits of temperate japonica rice (Oryza sativa L.). Crop Breed Appl Biotechnol. https://doi.org/10.1590/1984-70332020v20n1a1
Cheon K-S, Jeong Y-M, Lee Y-Y, Oh J, Kang D-Y, Oh H, Kim SL, Kim N, Lee E, Baek J (2019) Kompetitive allele-specific PCR marker development and quantitative trait locus mapping for bakanae disease resistance in Korean japonica rice varieties. Plant Breed Biotechnol 7(3):208–219. https://doi.org/10.9787/PBB.2019.7.3.208
Collard BC, Jahufer M, Brouwer J, Pang E (2005) An introduction to markers, quantitative trait loci (QTL) mapping and marker-assisted selection for crop improvement: the basic concepts. Euphytica 142(1):169–196. https://doi.org/10.1007/s10681-005-1681-5
Donde R, Mohapatra S, Baksh SY, Padhy B, Mukherjee M, Roy S, Chattopadhyay K, Anandan A, Swain P, Sahoo KK (2020) Identification of QTLs for high grain yield and component traits in new plant types of rice. PLoS ONE 15(7):e0227785. https://doi.org/10.1371/journal.pone.0227785
Du H, Yu Y, Ma Y, Gao Q, Cao Y (2017) Sequencing and de novo assembly of a near complete indica rice genome. Nat Commun 8:15324. https://doi.org/10.1038/ncomms15324
Du B, Wang Q, Sun G, Ren X, Cheng Y, Wang Y, Gao S, Li C, Sun D (2019) Mapping dynamic QTL dissects the genetic architecture of grain size and grain filling rate at different grain-filling stages in barley. Sci Rep 9(1):1–16. https://doi.org/10.1038/s41598-019-53620-5
Hammer Ø, Harper DA, Ryan PD (2001) PAST: Paleontological statistics software package for education and data analysis. Palaeontol Electron 4(1):9
He C, Holme J, Anthony J (2014) SNP genotyping: the KASP assay. In: Delphine F, Whitford R (eds) Crop breeding. Springer, pp 75–86. https://doi.org/10.1007/978-1-4939-0446-4_7
Heang D, Sassa H (2012) An atypical bHLH protein encoded by POSITIVE REGULATOR OF GRAIN LENGTH 2 is involved in controlling grain length and weight of rice through interaction with a typical bHLH protein APG. Breed Sci 62(2):133–141. https://doi.org/10.1270/jsbbs.62.133
Heng Y, Wu C, Long Y, Luo S, Ma J, Chen J, Liu J, Zhang H, Ren Y, Wang M (2018) OsALMT7 maintains panicle size and grain yield in rice by mediating malate transport. Plant Cell 30(4):889–906. https://doi.org/10.1105/tpc.17.00998
Hirao T, Matsunaga K, Hirakawa H, Shirasawa K, Isoda K, Mishima K, Tamura M, Watanabe A (2019) Construction of genetic linkage map and identification of a novel major locus for resistance to pine wood nematode in Japanese black pine (Pinus thunbergii). BMC Plant Biol 19(1):1–13. https://doi.org/10.1186/s12870-019-2045-y
Huang K, Wang D, Duan P, Zhang B, Xu R, Li N, Li Y (2017) WIDE AND THICK GRAIN 1, which encodes an otubain-like protease with deubiquitination activity, influences grain size and shape in rice. Plant J 91(5):849–860. https://doi.org/10.1111/tpj.13613
Hussain S, Fujii T, McGoey S, Yamada M, Ramzan M, Akmal M (2014) Evaluation of different rice varieties for growth and yield characteristics. J Anim Plant Sci 24(5):1504–1510
Kang J-W, Lee S-B, Lee J-Y, Kwon Y-H, Lee S-M, Kabange NR, Shin D, Cha J-G, Park D-S, Ko J-M (2020) Development and validation of KASP markers for Stv-bi, a rice stripe virus resistance gene in rice (Oryza sativa L.). Plant Breed Biotechnol 8(2):196–201. https://doi.org/10.9787/PBB.2020.8.2.196
Khan S (2015) QTL mapping: a tool for improvement in crop plants. Res J Recent Sci 2277:2502
Kulkarni SR, Balachandran S, Ulaganathan K, Balakrishnan D, Praveen M, Prasad AH, Fiyaz R, Senguttuvel P, Sinha P, Kale RR (2020) Molecular mapping of QTLs for yield related traits in recombinant inbred line (RIL) population derived from the popular rice hybrid KRH-2 and their validation through SNP genotyping. Sci Rep 10(1):1–21. https://doi.org/10.1038/s41598-020-70637-3
Lee J-W, Chin JH, Yoo S-C (2021) Development of Kompetitive allele specific PCR markers for anaerobic germination 1 locus in Rice. Plant Breed Biotechnol 9(1):20–31. https://doi.org/10.9787/PBB.2021.9.1.20
Li Y (2006) Mach 1.0: rapid haplotype reconstruction and missing genotype inference. Am J Hum Genet 79:2290
Li C, Wang Y, Liu L, Hu Y, Zhang F, Mergen S, Wang G, Schläppi MR, Chu C (2011) A rice plastidial nucleotide sugar epimerase is involved in galactolipid biosynthesis and improves photosynthetic efficiency. PLoS Genet 7(7):e1002196. https://doi.org/10.1371/journal.pgen.1002196
Li G, Yang Q, Li D, Zhang T, Yang L, Qin J, Tang B, Guo X, Cao Y, You S (2021) Genome-wide SNP discovery and QTL mapping for economic traits in a recombinant inbred line of Oryza sativa. Food Energy Secur 10(2):313–328. https://doi.org/10.1002/fes3.274
Ma L, Zhang D, Miao Q, Yang J, Xuan Y, Hu Y (2017) Essential role of sugar transporter OsSWEET11 during the early stage of rice grain filling. Plant Cell Physiol 58(5):863–873. https://doi.org/10.1093/pcp/pcx040
Mani B, Agarwal M, Katiyar-Agarwal S (2015) Comprehensive expression profiling of rice tetraspanin genes reveals diverse roles during development and abiotic stress. Front Plant Sci 6:1088. https://doi.org/10.3389/fpls.2015.01088
McCouch S (1997) Report on QTL nomenclature. Rice Genet Newsl 14:11–13
Meng L, Li H, Zhang L, Wang J (2015) QTL IciMapping: integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J 3(3):269–283. https://doi.org/10.1016/j.cj.2015.01.001
Mideros SX, Warburton ML, Jamann TM, Windham GL, Williams WP, Nelson RJ (2014) Quantitative trait loci influencing mycotoxin contamination of maize: analysis by linkage mapping, characterization of near-isogenic lines, and meta-analysis. Crop Sci 54(1):127–142. https://doi.org/10.2135/cropsci2013.04.0249
Nagaraju J, Kathirvel M, Kumar RR, Siddiq E, Hasnain SE (2002) Genetic analysis of traditional and evolved Basmati and non-Basmati rice varieties by using fluorescence-based ISSR-PCR and SSR markers. Proc Natl Acad Sci 99(9):5836–5841
Pariasca-Tanaka J, Lorieux M, He C, McCouch S, Thomson MJ, Wissuwa M (2015) Development of a SNP genotyping panel for detecting polymorphisms in Oryza glaberrima/O. sativa interspecific crosses. Euphytica 201(1):67–78. https://doi.org/10.1007/s10681-014-1183-4
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6(1):288–295. https://doi.org/10.1111/j.1471-8286.2005.01155.x
Pollard DA (2012) Design and construction of recombinant inbred lines. Quantitative trait loci (QTL). Springer, pp 31–39. https://doi.org/10.1007/978-1-61779-785-9_3
Sabar M, Arif M (2014) Phenotypic response of rice (Oryza sativa) genotypes to variable moisture stress regimes. Int J Agric Biol 16:32–40
Sabar M, Shabir G, Shah SM, Aslam K, Naveed SA, Arif M (2019) Identification and mapping of QTLs associated with drought tolerance traits in rice by a cross between Super Basmati and IR55419-04. Breed Sci 69(1):169–178. https://doi.org/10.1270/jsbbs.18068
Sharma M, Gangurde SS, Salgotra RK et al (2021) Genetic mapping for grain quality and yield-attributed traits in Basmati rice using SSR-based genetic map. J Biosci 46:50. https://doi.org/10.1007/s12038-021-00169-z
Spielmeyer W, Ellis MH, Chandler PM (2002) Semidwarf (sd-1), “green revolution” rice, contains a defective gibberellin 20-oxidase gene. Proc Natl Acad Sci 99(13):9043–9048. https://doi.org/10.1073/pnas.132266399
Steele KA, Quinton-Tulloch MJ, Amgai RB, Dhakal R, Khatiwada SP, Vyas D, Heine M, Witcombe JR (2018) Accelerating public sector rice breeding with high-density KASP markers derived from whole genome sequencing of indica rice. Mol Breed 38(4):1–13. https://doi.org/10.1007/s11032-018-0777-2
Steele K, Tulloch MQ, Burns M, Nader W (2021) Developing KASP markers for identification of basmati rice varieties. Food Anal Methods 14(4):663–673. https://doi.org/10.1007/s12161-020-01892-3
Tabuchi M, Sugiyama K, Ishiyama K, Inoue E, Sato T, Takahashi H, Yamaya T (2005) Severe reduction in growth rate and grain filling of rice mutants lacking OsGS1; 1, a cytosolic glutamine synthetase1; 1. Plant J 42(5):641–651. https://doi.org/10.1111/j.1365-313x.2005.02406.x
Tanabata T, Shibaya T, Hori K, Ebana K, Yano M (2012) SmartGrain: high-throughput phenotyping software for measuring seed shape through image analysis. Plant Physiol 160(4):1871–1880. https://doi.org/10.1104/pp.112.205120
Tang Z, Chen Y, Chen F, Ji Y, Zhao F-J (2017) OsPTR7 (OsNPF8. 1), a putative peptide transporter in rice, is involved in dimethylarsenate accumulation in rice grain. Plant Cell Physiol 58(5):904–913. https://doi.org/10.1093/pcp/pcx029
Thibaud-Nissen F, Malek R, Lee Y, Zheng L, Orvis J, Haas B, Wortman J, Buell C (2007) The TIGR rice genome annotation resource: improvements and new features. Nucleic Acids Res 35:D883–D887. https://doi.org/10.1093/nar/gkl976
Todaka D, Nakashima K, Maruyama K, Kidokoro S, Osakabe Y, Ito Y, Matsukura S, Fujita Y, Yoshiwara K, Ohme-Takagi M (2012) Rice phytochrome-interacting factor-like protein OsPIL1 functions as a key regulator of internode elongation and induces a morphological response to drought stress. Proc Natl Acad Sci 109(39):15947–15952. https://doi.org/10.1073/pnas.1207324109
USDA (2023) Country summary: Pakistan: rice https://ipad.fas.usda.gov/countrysummary/?id=PK&crop=Rice. Accessed 31 May 2023
Usman B, Nawaz G, Zhao N, Liao S, Qin B, Liu F, Liu Y, Li R (2021) Programmed editing of rice (Oryza sativa L.) OsSPL16 gene using CRISPR/Cas9 improves grain yield by modulating the expression of pyruvate enzymes and cell cycle proteins. Int J Mol Sci 22(1):249. https://doi.org/10.3390/ijms22010249
Vemireddy LR, Noor S, Satyavathi V et al (2015) Discovery and mapping of genomic regions governing economically important traits of Basmati rice. BMC Plant Biol 15:207. https://doi.org/10.1186/s12870-015-0575-5
Waheed R, Ignacio JC, Arbelaez JD, Juanillas VM, Asif M, Henry A, Kretzschmar T, Arif M (2021) Drought response QTLs in a Super Basmati× Azucena population by high-density GBS-based SNP linkage mapping. Plant Breed 140(5):758–774. https://doi.org/10.1111/pbr.12961
Wang D, Sun W, Yuan Z, Sun Q, Fan K, Zhang C, Yu S (2021) Identification of a novel QTL and candidate gene associated with grain size using chromosome segment substitution lines in rice. Sci Rep 11(1):1–11. https://doi.org/10.1038/s41598-020-80667-6
Wang S, Basten C, Zeng Z (2012a) Windows QTL cartographer 2.5. Department of Statistics, North Carolina State University, Raleigh, NC
Wang S, Wu K, Yuan Q, Liu X, Liu Z, Lin X, Zeng R, Zhu H, Dong G, Qian Q (2012b) Control of grain size, shape and quality by OsSPL16 in rice. Nat Genet 44(8):950–954
Xu R, Yu H, Wang J, Duan P, Zhang B, Li J, Li Y, Xu J, Lyu J, Li N (2018) A mitogen-activated protein kinase phosphatase influences grain size and weight in rice. Plant J 95(6):937–946. https://doi.org/10.1111/tpj.13971
Xu H, Li X, Zhang H, Wang L, Zhu Z, Gao J, Li C, Zhu Y (2020) High temperature inhibits the accumulation of storage materials by inducing alternative splicing of OsbZIP58 during filling stage in rice. Plant Cell Environ 43(8):1879–1896. https://doi.org/10.1111/pce.13779
Yadav S, Sandhu N, Singh VK, Catolos M, Kumar A (2019) Genotyping-by-sequencing based QTL mapping for rice grain yield under reproductive stage drought stress tolerance. Sci Rep 9(1):1–12. https://doi.org/10.1038/s41598-019-50880-z
Yang H, Li C, Lam H-M, Clements J, Yan G, Zhao S (2015) Sequencing consolidates molecular markers with plant breeding practice. Theor Appl Genet 128(5):779–795. https://doi.org/10.1007/s00122-015-2499-8
Yonemaru J, Yamamoto T, Fukuoka S, Uga Y, Hori K, Yano M (2010) Q-TARO: QTL annotation rice online database. Rice 3:194–203. https://doi.org/10.1007/s12284-010-9041-z
Yu Z, Dong L, Jiang Z, Yi K, Zhang J, Zhang Z, Zhu Z, Wu Y, Xu M, Ni J (2018) A semi-dominant mutation in a CC-NB-LRR-type protein leads to a short–root phenotype in rice. Rice 11(1):1–11. https://doi.org/10.1186/s12284-018-0250-1
Yuyu C, Aike Z, Pao X, Xiaoxia W, Yongrun C, Beifang W, Yue Z, Liaqat S, Shihua C, Liyong C (2020) Effects of GS3 and GL3. 1 for grain size editing by CRISPR/Cas9 in Rice. Rice Sci 27(5):405–413
Zaw H, Raghavan C, Pocsedio A, Swamy BM, Jubay ML, Singh RK, Bonifacio J, Mauleon R, Hernandez JE, Mendioro MS (2019) Exploring genetic architecture of grain yield and quality traits in a 16-way indica by japonica rice MAGIC global population. Sci Rep 9(1):1–11
Zhang X-c, Yu X, Zhang H-j, Song F-m (2009) Molecular characterization of a defense-related AMP-binding protein gene, OsBIABP1, from rice. J Zhejiang Univ SCI B 10(10):731–739. https://doi.org/10.1631/jzus.B0920042
Zhang X, Wang J, Huang J, Lan H, Wang C, Yin C, Wu Y, Tang H, Qian Q, Li J (2012) Rare allele of OsPPKL1 associated with grain length causes extra-large grain and a significant yield increase in rice. Proc Natl Acad Sci 109(52):21534–21539. https://doi.org/10.1073/pnas.1219776110
Zhang J, Guo T, Yang J, Hu M, Wang H, Sun K, Chen Z, Wang H (2020) QTL mapping and haplotype analysis revealed candidate genes for grain thickness in rice (Oryza sativa L.). Mol Breed 40:1–12. https://doi.org/10.1007/s11032-020-01128-6
Zhao S, Zhao L, Liu F, Wu Y, Zhu Z, Sun C, Tan L (2016) NARROW AND ROLLED LEAF 2 regulates leaf shape, male fertility, and seed size in rice. J Integr Plant Biol 58(12):983–996. https://doi.org/10.1111/jipb.12503
Acknowledgements
The authors duly acknowledge that funding was received from Innovate UK (Project 103,711) to develop KASP including bioinformatics by Mark Quinton-Tulloch and technical support by LGC Biosearch Technologies UK for the genotyping conducted in this study.
Author information
Authors and Affiliations
Contributions
Phenotyping, QTL mapping analysis, preparation of figures and initial draft writing of the manuscript were done by HA. In-silico analyses were performed by RR and ADB. NP and UM helped with genotype data handling. Plant material was provided by MA. Co-ordination and funding acquisition for KASP designs and genotyping were managed by KS, who also reviewed and edited all versions of the manuscript. Study conceptualization and finalization of the manuscript were overseen by MA
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest (financial or non-financial).
Ethical approval
This research was screened under Bangor University Research Ethics Framework, no issues were identified.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ashfaq, H., Rani, R., Perveen, N. et al. KASP mapping of QTLs for yield components using a RIL population in Basmati rice (Oryza sativa L.). Euphytica 219, 79 (2023). https://doi.org/10.1007/s10681-023-03206-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10681-023-03206-0