
Abstract
Drought tolerance in maize is a complex and polygenic trait, especially in the seedling stage. In plant breeding, complex genetic traits can be improved by genomic selection (GS), which has become a practical and effective breeding tool. In the present study, a natural maize population named Northeast China core population (NCCP) consisting of 379 inbred lines were genotyped with diversity arrays technology (DArT) and genotyping-by-sequencing (GBS) platforms. Target traits of seedling emergence rate (ER), seedling plant height (SPH), and grain yield (GY) were evaluated under two natural drought stress environments in northeast China. Adequate genetic variations were observed for all the target traits, but they were divergent across environments. Similarly, the heritability of the target trait also varied across years and environments, the heritabilities in 2019 (0.88, 0.82, 0.85 for ER, SPH, GY) were higher than those in 2020 (0.65, 0.53, 0.33) and cross-2-years (0.32, 0.26, 0.33). In total, three marker datasets, 11,865 SilicoDArT markers obtained from the DArT-seq platform, 7837 SNPs obtained from the DArT-seq platform, and 91,003 SNPs obtained from the GBS platform, were used for GS analysis after quality control. The results of phylogenetic trees showed that broad genetic diversity existed in the NCCP population. Genomic prediction results showed that the average prediction accuracies estimated using the DArT SNP dataset under the two-fold cross-validation scheme were 0.27, 0.19, and 0.33, for ER, SPH, and GY, respectively. The result of SilicoDArT is close to the SNPs from DArT-seq, those were 0.26, 0.22, and 0.33. For the trait with lower heritability, the prediction accuracy can be improved using the dataset filtered by linkage disequilibrium. For the same trait, the prediction accuracies estimated with two DArT marker datasets were consistently higher than that estimated with the GBS SNP dataset under the same genotyping cost. The prediction accuracy was improved by controlling population structure and marker quality, even though the marker density was reduced. The prediction accuracies were improved by more than 30% using the significant-associated SNPs. Due to the complexity of drought tolerance under the natural stress environments, multiple years of data need to be accumulated to improve prediction accuracy by reducing genotype-by-environment interaction. Modeling genotype-by-environment interaction into genomic prediction needs to be further developed for improving drought tolerance in maize. The results obtained from the present study provides valuable pathway for improving drought tolerance in maize using GS.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Maize (Zea mays L.) is an important source of food, animal feed, and energy because of its high yield potential (Jiang et al. 2018). Consequently, poor maize production caused by abiotic stresses has a tremendously detrimental impact on global food security. Seed germination and seedling establishments are the initial stages of maize growth, which affect the seedling emergence rate, uniformity, and robustness, and then determine maize yield potential (Tian et al. 2014). However, these initial stages are frequently subjected to severe drought stress worldwide due to global warming, which decreases the emergence rate and retards seedling growth. Therefore, the development of drought-tolerant maize hybrids is essential to address the global temperature rise and end the global food insecurity.
The selection of elite drought-tolerant maize inbred lines is the basis of the development of drought-tolerant maize hybrids. However, traditional methods for selecting inbred lines are time-consuming, labor-intensive, and inefficient. In recent decades, with the development of high-throughput and inexpensive genotyping technologies, genomic selection (GS) has become a practical and effective tool in animal and plant breeding (Michel et al. 2016; Zhang et al. 2017b). The principle of GS is to establish the prediction model based on the genotypic and phenotypic data from a training population (TP), which is used to derive genomic estimated breeding values (GEBVs) for all the individuals in the breeding population (BP) from their genomic profiles (Meuwissen et al. 2001), guiding breeders to select excellent individuals from the population without phenotypic data. GS has been shown to improve the efficiency of recurrent selection of bi-parental populations (Beyene et al. 2015; Massman et al. 2013; Môro et al. 2019; Vivek et al. 2017) and multi-parent populations (Zhang et al. 2017b) to reduce breeding cycle time and accelerate the genetic gain per unit cost and time. At present, GS has been widely used in maize for various traits, including grain yield (Liu et al. 2018), developmental traits (Cui et al. 2020; Zhang et al. 2019), abiotic and biotic stress tolerances (Beyene et al. 2015; Cao et al. 2017; Vivek et al. 2017) and grain nutritional quality (Guo et al. 2020). Compared with the widely used marker-assisted selection (MAS) technology, the GS strategy has the advantage of without detection of quantitative trait loci (QTL) in advance, and it can be used to predict the performance of the breeding population without genetic structure analysis (Bernardo 2016). Moreover, GS utilizes all genes/loci affecting phenotypes, including both major genes/loci and minor genes/loci (Crossa et al. 2017; Desta and Ortiz 2014; Xiao et al. 2017). Therefore, it is a promising genomic tool for improving the complex quantitative traits.
Genomic prediction (GP) can be implemented with statistical models that can estimate the marker effects accurately in a training population. Over the past 2 decades, various statistical models and machine learning methods have been proposed for the implementation of GP. Among the parametric models, ridge regression best linear unbiased predictions (rrBLUP) and genomic-BLUP (GBLUP) are most commonly used in plant breeding (Heslot et al. 2012). Bayes A assumes a prior distribution of effects with a higher probability of moderate to large effects, while Bayes B and Bayes Cπ assume some marker effects to be zero. Reproducing Kernel Hilbert Spaces (RKHS) regression is equivalent to a GBLUP with a linear kernel (de los Campos et al. 2009). Deep Learning (DL), a nonparametric model, is a type of machine learning (ML) approach and a subfield of artificial intelligence (Montesinos-López et al. 2021). For most traits with different genetic architectures, the differences between different models are slight. rrBLUP is a classical method with lower computational requirements and usually performs well across different traits (Kwong et al. 2017; Maulana et al. 2021; Resende et al. 2012).
Prediction accuracy of GS, the correlation between the genomic estimated breeding values (GEBV) and the field observations, is used to evaluate the reliability of GS results (Combs and Bernardo 2013). A variety of factors are known to influence GS prediction accuracy, which includes training population size, relatedness between training and test individuals, DNA marker type, marker quality and density, trait heritability, statistical models, linkage disequilibrium (LD) between markers, and population structure, etc. (Liu et al. 2018; Norman et al. 2018). The quality and density of genomic markers are influenced by genotyping platforms. The ideal sequencing platform should be inexpensive, provide high-throughput and good genomic coverage, as well as be replicable and stable. Genotyping-by-sequencing (GBS), which can simplify complex genomes, has now been used as a high-throughput and cost-effective tool for generating high-density molecular markers in many crop species (Elshire et al. 2011). The Diversity Arrays Technology (DArT) is a DNA hybridization-based molecular marker technique, which can detect variation at numerous genomic loci without sequence information of reference genome. DArT marker has been used for the implementation of MAS in maize (Dos Santos et al. 2016). In addition, DArT can be combined with next-generation sequencing platforms known as DArT-seq, which permits simultaneous detection of several thousands of DNA polymorphisms by scoring the presence or absence of DNA fragments in genomic representations and through a process of reducing the genomic complexity (Kilian et al. 2012).
The maize growth from seed germination stage to seedling establishment stage involves a variety of complicated metabolic transformations, physiological activities, and cellular and tissue differentiation, which are regulated by many genomic loci, including both major and minor effect loci expressed under drought stress. These abundant genetic variations exist in maize germplasms with different genetic backgrounds. In this study, three marker datasets of a natural maize population obtained from the DArT and GBS genotyping platforms, as well as the phenotypic data of emergence rate (ER) and plant height (SPH) in the seedling stage, and grain yield (GY) in the mature stage evaluated under two drought stress environments, were used to perform GS analysis. The main objectives of the present study were to: (1) evaluate the phenotypic variation of several target traits in the NCCP in natural drought stress environments; (2) compare the genomic prediction accuracies of the target traits of ER, SPH, and GY estimated from the three marker datasets obtained from the DArT and GBS genotyping platforms; (3) assess the prediction accuracies of three traits by improving the marker quality; and (4) improve the prediction accuracies by controlling population structure and incorporating trait-marker associations.
Materials and methods
Plant materials, phenotype evaluation, and heritability estimation
A collection of 391 inbred lines, designated as the northeast China core population (NCCP), were used for genomic prediction analyses in the current study. All these inbred lines were selected from China, Mexico, and America, and they adapted to the spring maize area of Northeast China. The drought stress tolerance of this population was determined in the Fuxin Mongolian autonomous county, Liaoning Province, China (42°06′ N, 122°55′ E) in 2019 and 2020, respectively, where drought stress occurs frequently during the spring season. The drought trial was planted on 12 May and harvested on 7 October in 2019, and planted on 11 May and harvested on 9 October in 2020. During the maize growing season, the average temperature was 21 °C both in 2019 and 2020. The average rainfall was 4 mm and 3 mm in 2019 and 2020, respectively. The highest temperature was above 35 °C in both years, and the precipitation was less than 20 mm per month, causing serious drought stress in the seedling stage (Fig. 1). Irrigation was not performed before planting, and planting was scheduled before precipitation according to the weather forecast for getting good germination. A completely randomized block design with three replications was applied in each environment. The inbred lines were sown using one seed per hole in a single plot, 3 m long, with plant spacing of 10 cm and row spacing of 60 cm. Target traits of emergence rate (ER), seedling plant height (SPH) and grain yield (GY) were evaluated to represent the drought tolerance of the tested plant materials. The target traits of ER and SPH were measured at the seedling stage, i.e. 20 days after planting. While GY was measured at the maturity stage, the moisture content was measured by LDS-1G Grain Moisture Meter. ER was measured as the proportion of the number of surviving plants to the total number of seeds planted. SPH was measured as the distance from the plant base to the highest of the seedling plant. The average dry grain weight from five plants was used to represent the GY for each entry.

Distribution of temperature and rainfall in maize planting plots of Fuxin Mongolian Autonomous County in 2019 and 2020. The horizontal axis shows the date from sowing to harvest. The panels of a and c are temperature (°C) data, including maximum temperature (tem_max), minimum temperature (tem_min), and average temperature (tem_avg). The panels of b and d are rainfall data, and the unit is mm
The best linear unbiased estimator (BLUE) values and broad-sense heritability (H2) of ER, SPH, and GY were calculated within and across years using the META-R software version 6.04 (Alvarado et al. 2020) (http://hdl.handle.net/11529/10201). The linear mixed model used in META-R was implemented in the LME4 R-package, functions of lmer and REML were used to estimate the variance components.
where \({Y}_{ijk}\) is the trait of interest, μ is the overall mean, \(Ge{n}_{i}\), \(En{v}_{j}\), and \(Ge{n}_{i}\times En{v}_{j}\) are the effects of the i-th genotype, j-th year, and i-th genotype by j-th year interaction, respectively. All the effects are considered random, which is assumed to be normally and independently distributed, with mean zero and homoscedastic variance σ2. Genotype and environment is a fixed effect when calculating BLUEs, the covariate is always a fixed effect. \({Rep}_{k}\) is the effect of k-th replication. \({\varepsilon }_{ijk}\) is the random error term of the i-th genotype, j-th year, k-th replication. Traits with heritability below 0.05 within the individual environment were excluded from the across-location analysis.
Broad-sense heritability (H2) based on the entry means within the trial was estimated as follows:
where \({\sigma }_{g}^{2}\), \({\sigma }^{2}\), and \({\sigma }_{ge}^{2}\) are the genotypic variance, error variance, and genotype-by-environment interaction variance, respectively, and nreps and nEnvs are the numbers of replications and environments, respectively (Carena et al. 2010).
Genotyping and quality control
Leaf samples of three individual plants were collected for each inbred line during the seeding stage for DNA extraction with a CTAB procedure, and genotyping was carried out by DArT (https://www.diversityarrays.com/) and GBS (Elshire et al. 2011) platforms.
Genotyping of all inbred lines with the GBS method was performed at Wuhan medical laboratory, BGI Co., Ltd, following a protocol widely used by the maize research community (Elshire et al. 2011). The genomic DNA was digested with the ApeKI restriction enzyme, and a DNA library was constructed in 96-plex and sequenced on Illumina-hiseq XTen sequencing system. Each sample obtained an average of about seven hundred and fifty thousand reads. The B73 RefGen_v4 was the reference genome, and BWA software was applied to match all sequencing data to the reference genome. Details in single nucleotide polymorphism (SNP) calling and imputation have been previously described (Cao et al. 2017). Each line has an average of approximately 77,000 SNPs distributed on each chromosome. In total, 776,285 loci were aggregated to cover all SNPs, and 768,558 of them were mapped to the ten maize chromosomes (Table S1), while 7727 could not be anchored to any chromosome (data not displayed).
Genotyping of all the inbred lines with DArT-seq method was carried out at the SAGA (https://seedsofdiscovery.org/about/genotyping-platform/) sequencing lab, jointly established by the DArT company and CIMMYT. Two enzymes of PstI (CTGCAG) and HpaII (CCGG) were used to digest the DNA samples to reduce the complexity of the genome. For each 96 wells plate, 16% of the samples were replicated to assess reproducibility (Pereira et al. 2020). After enzyme digestion, DNA from different samples was linked with barcodes of different base combinations of sequences to construct a simplified sequencing DNA library. Equimolar amounts of amplification products from each sample were pooled by plate and amplified by c-Bot (Illumina) bridge PCR, followed by fragment sequencing on Illumina Hiseq 2500 (www.illumina.com). The short fragment sequencing technology (150 bp) and the simplified sequencing DNA library of mixed samples was sequenced on a single lane (Kilian et al. 2012) (https://www.diversityarrays.com/). SNPs were called using the DArTsoft analytical pipeline (http://www.diversityarrays.com/software.html) (Chen et al. 2016). DArT's SNP development is different from GBS, which does not rely on the reference genome information, but mainly depends on the sequencing library data from the DArT company. All reads were aligned by sequence analysis based on maize tags of metagenome representation. Two types of marker datasets were generated by DArT-seq: SilicoDArT and SNP. The SilicoDArT dataset is the presence/absence variation of the tag sequences, while the SNP dataset was always used for genetics analysis and molecular marker-assisted selection in previous studies. The SNP dataset was obtained by mapping the SNPs to the B73 RefGen_v4 reference genome using the BLAST tool, and some of the fragments in SilicoDArT were not able to be aligned to the reference genome. The number of markers in SilicoDArT dataset was 62,794, while the number of markers in the SNP dataset was 39,659, and 547 of them were not able to be anchored to any ten maize chromosomes (Table S1).
In both DArT and GBS datasets, TASSEL 5 (Bradbury et al. 2007) was used to filter out markers with minor allele frequency (MAF) < 0.05 and a missing rate > 20%. The samples with a missing rate of more than 20% were discarded. The number of lines in DArT and GBS datasets is 379 and 378, respectively. In total, the number of markers is 11,865 in the SilicoDArT dataset, 7837 in the DArT SNP dataset, and 91,003 in the GBS SNP dataset (Table 1). Imputation was performed using the method of Cao et al. (2017).
SNP distribution across the whole maize genome
The distribution of SNPs in the genome is one of the indicators of the quality of the genotyping platforms. A package named RIdeogram (Hao et al. 2020), written in R programming (R Core Team, 2021), was used to assess the distribution of SNPs obtained from both the DArT-seq and GBS platforms. This analysis was not applied to the SilicoDArT dataset, due to a lack of information on physical positions on the reference genome. The plot of the SNP distribution was drawn using the ideogram function. The customized R scripts to plot the heatmap of the SNP distribution on maize chromosomes using the HapMap format input dataset were provided (https://aozhangchina.github.io/R/chromesomeheatmapTool/ChromesomeHeatmap.html).
Genomic prediction model
The rrBLUP model assumes homogenous variance of all markers and shrinks all marker effects equally to zero. rrBLUP is equivalent to BLUP and uses the realized relationship matrix estimated from the markers. The genomic prediction analysis was performed with the ridge regression BLUP (rrBLUP) package (Endelman, 2011). The mixed model is described as:
where y is the vector (n × 1) of BLUPs from the phenotypic data analysis, X is a design matrix for fixed effects; ε is the vector (n × 1) of independently random errors with assumed distribution N (0, Iσε2), Z is the design matrix (n × n) for random effects and u is the vector of random effects with u ∼ N (0, Kσu2) and K was an a realized (additive) relationship matrix computed from marker genotypes proposed by VanRaden (2008). In addition, n is the number of individuals (Endelman 2011; Liu et al. 2018). Variance components are estimated by REML using the spectral decomposition algorithm of Kang et al. (2008). The mixed.solve function, a linear mixed-model equation estimates marker effects and GEBVs. GEBVs are derived from the realized (additive) relationship matrix of individuals calculated from marker genotypes (Tecle et al. 2014).
Effect of training population size (TPS) on the estimation of genomic prediction accuracy
To evaluate the effect of TPS on the estimation of genomic prediction accuracy, the cross-validation scheme was used to randomly generate different training and prediction sets and assess the prediction accuracy for different target traits in prediction sets. The training populations were extracted from the whole population The TPS ranged from 10 to 90%, with an interval of 10%. Each cross-validation scheme was repeated 100 times. For the same trait, the prediction accuracies estimated from the DArT and GBS molecular marker datasets were compared. A gradient random cross-validation analysis was implemented for all traits. For prediction, regression models were evaluated by the prediction accuracy, a Pearson's correlation between the GEBVs and the estimated breeding values in the testing population (Thavamanikumar et al. 2015). All predictions were repeated 100 times, and the mean value was taken as the average prediction accuracy.
Effect of marker quality on the estimation of genomic prediction accuracy
For the same target trait, genomic prediction accuracy estimated by the data from the drought stress condition is always relatively low than that estimated by the data from the optimal condition. Higher marker quality was proved to improve prediction accuracy. As mentioned in in materials and methods section, different levels of MAF and missing rates were filtered in the DArT and GBS marker datasets to control the marker quality and quantity.
For evaluating the effect of linkage disequilibrium (LD) among markers on the estimation of the genomic prediction accuracy, neighbor SNPs not in a strong LD phase, i.e., LD values lower than a specific threshold (0.1 and 0.2) within an LD block, were excluded from the prediction analyses. TASSEL 5.0 (Bradbury et al. 2007) was used to perform LD analysis, and LD decay plots were developed using the customized R scripts (https://aozhangchina.github.io/R/LDdecay/LDdecayPlotTool.html). Genomic prediction accuracy was calculated from 100 times repetition using twofold cross-validation. A twofold cross-validation analysis was implemented for all traits. For prediction, regression models were evaluated by the prediction accuracy. All predictions were repeated 100 times, and the mean value was taken as the average prediction accuracy.
Effect of genetic structure on the estimation of genomic prediction accuracy
It was shown that the relationship between training and prediction sets had a significant impact on the estimation of prediction accuracy (Zhang et al. 2017a). In the current study, NCCP consists of multiple heterotic groups, such as the Reid, Reid_Chinese, Lancaster, Tangsipingtou, Lvda Red Cob, Longdan, Jidan, CML, Mixed, etc. Some elite breeding lines from Northeast China were also included in this population, including Shen118, Shen3336, Liao6049, Dan340, Ji818, etc.
A phylogenetic tree was used to demonstrate the genetic relationship among all the inbred lines used in NCCP. The genetic distances among all the inbred lines were calculated in Bio-R (Biodiversity Analysis with R) software developed by CIMMYT using the filtered DArT and GBS datasets' (https://hdl.handlenet/11529/10820). The formula to compute the modified Rogers distance (MRD) used in the present study is as follows:
where \({p}_{lax}\) is the estimated frequency of allele a, which is located in locus l, and genotype x; l is the number of loci, \({n}_{l}\) is the number of alleles in the locus l. Markers from both platforms were used to create the phylogenetic tree. Based on the genetic distance matrix, the Neighbor-Joining (NJ) method of MEGA X (https://www.megasoftware.net/) and FigTree (http://tree.bio.ed.ac.uk/software/figtree/) software were used to create the phylogenetic trees (Saitou and Nei 1987).
The principal component analysis (PCA) was performed to assess the effects of genetic structure on the estimation of prediction accuracy. All the inbred lines in NCCP were divided into two subgroups. The prcomp function from the stats package in R and the plot function from the base package in R were applied to generate the PCA plot. Genomic prediction accuracy was estimated within each subgroup from 100 times repetitions using twofold cross-validation.
Genomic prediction analysis with the significant trait associated markers
For each trait, all SNPs were divided into the significant marker and non-significant marker groups according to the GWAS results. The GWAS was conducted by GAPIT (version 3) (Wang and Zhang 2021) using the MLM model to obtain the association markers from GBS and DArT platforms. This model simultaneously incorporates population structure (Yu et al. 2006). The first three PCs were used to estimate the Q value in this study. The MLM model is described as follows:
where \({\varvec{X}}\) is the SNP marker matrix, \({\varvec{Q}}\) and \({\varvec{Z}}\) represent the subpopulation membership matrix and kinship matrix respectively, \({\varvec{\beta}}\) and \({\varvec{v}}\) are the coefficient vectors of SNP markers and subpopulation membership respectively, \({\varvec{u}}\) is the random genetic effect vector, and \({\varvec{\varepsilon}}\) is the random error vector, which is Var[\(\varepsilon ]=I{\sigma }_{e}^{2}\) (Yu et al. 2006; Fan et al. 2016).
The thresholds of significant markers were set as P < 0.5, 0.1, 0.01, 0.001. The prediction accuracies were estimated using significant markers, equal amount of non-significant markers, and the rest of non-significant markers. Genomic prediction accuracy was calculated from 100 times repetitions using twofold cross-validation.
Results
Phenotypic data and correlation analysis
The phenotypic variations and standard deviations of the three traits in 2019, 2020, and across 2 years were shown in Table 2. Sufficient genetic variations for genomic selection were found under the drought environment. The phenotypic values of ER ranged from 0.16 to 1.00, with an average value of 0.81 in 2019; ranged from 0.32 to 0.90, with an average value of 0.70 in 2020; and ranged from 0.60 to 0.84 with an average value of 0.76 across 2 years. For SPH, the phenotypic values ranged from 9.14 to 20.64, with an average value of 14.96 in 2019; ranged from 8.61 to 15.06 with an average value of 11.67 in 2020; and ranged from 12.00 to 14.48, with an average value of 13.36 across 2 years. The GY values ranged from 12.22 to 93.96, from 7.59 to 23.32, and from 18.32 to 38.19 in 2019, 2020, and across 2 years, respectively. The average value of GY was 40.00, 10.97, and 25.04 in 2019, 2020, and across 2 years, respectively. The coefficient of variation (CV) in 2020 was greater than that in 2019 for each trait (Table 2). Correspondingly, the variance of the environment component was great, low to moderate heritabilities for all the three traits were obtained in 2020 and across 2 years, and relative high heritabilities were observed in 2019 for all the three traits. The H2 of ER, SPH, and GY in 2019 was 0.88, 0.82, and 0.85, respectively. In 2020, the H2 of ER, SPH, and GY was 0.65, 0.53, and 0.33, respectively. Across 2 years, the H2 of ER, SPH, and GY was 0.32, 0.26, and 0.33, respectively (Table 2). The phenotypic data analysis based on BLUEs revealed that ER, SPH, and GY had normal distributions or skewed normal distributions (Fig. 2). The phenotypic correlations among all the three traits were positive and significant, and the correlation coefficients were 0.36, 0.35, and 0.25 (P < 0.01) between ER and SPH, between ER and GY, and between SPH and GY, respectively.

Frequency distributions and correlations of BLUE (best unbiased linear estimator) as phenotype values were calculated from the maize seedling emergence rate (ER), seedling plant height (SPH), and grain yield (GY) of maize measured in 19FX (2019 Fuxin) and 20FX (2020 Fuxin) under drought environment. The plots on the diagonal represent the phenotypic distribution frequency of ER, SPH, GY. The values above the diagonal line are the Pearson’s correlation coefficients between every two traits. The values below the diagonal line are scattered plots for every two traits. *represents a significant difference at the 0.05 level; **represents a significant difference at the 0.01 level
Quality control on genotypic data and marker distribution
Two kinds of marker sets were obtained in the current research. The SilicoDArT markers were from DArT-seq, and SNPs were from the GBS and DArT-seq. The plots of MAF and missing rate distribution before filtering for three marker sets were shown in Fig. 3. The SilicoDArT markers had an average MAF of 0.10, and the DArT-seq SNPs set had an average MAF of 0.11 before filtering. For the GBS SNP set, the average MAF before filtering was 0.11. The highest peak before filtering was observed at the MAF interval of 0 to 0.05 for all the three marker sets. The MAF distributions in other intervals from 0.05 to 0.50 were relatively uniform. Average MAF after filtering was 0.24, 0.24, and 0.23 for SilicoDArT, SNPs from DArT-seq, and SNPs from GBS, respectively (Table 1). Consistent MAF distributions between the two genotyping platforms potentially indicated that the quality of sequencing and molecular marker datasets generated by both genotyping platforms were high and reliable (Fig. 3a–c).

Distribution of MAF and missing rate before filtering in three marker datasets: a MAF distribution of the SilicoDArT marker dataset; b MAF distribution of the DArT marker dataset; c MAF distribution of the GBS marker dataset; d missing rate distribution of the SilicoDArT marker dataset; e missing rate distribution of the DArT marker dataset; f missing rate distribution of the GBS marker dataset
Both in the SilicoDArT and DArT-seq SNP datasets, distributions of missing rates showed a decreasing trend from the interval of 0–10% to the interval of 90–100%, and close to 40% of molecular markers had the missing rates less than 10%. In contrast, close to 40% of molecular markers in the GBS SNP dataset had the missing rates more than 90%, and 10% of the GBS SNPs appeared in each interval of missing rate of 0–10%, 10–20%, 20–30%, 30–40%, and 40–50% (Fig. 3d–f). The irregular distribution of missing rates in the GBS SNP dataset indicated that the quality of this SNP dataset is not high.
High-quality marker sets were obtained after filtering, and the average missing rate was 0.070, 0.073, and 0.068 in the SilicoDArT dataset, DArT-seq SNP dataset, and GBS SNP dataset, respectively (Table 1). Heterozygosity is usually a critical index for marker filtering, especially for inbred lines. In this research, the average heterozygosity rates were very low in all the three marker datasets, i.e., less than 0.01 before filtering, and less than 0.03 after filtering. It indicated that the breeding lines in the NCCP are homozygous (Table S1 and Table 1).
After filtering, the number of markers decreased by 81%, 80%, and 88% in the SilicoDArT dataset, DArT-seq SNP dataset, and GBS SNP dataset, respectively (Table S1 and Table 1). The missing rate was mainly responsible for the reduction of markers, the missing rate before filtering was 25%, 24%, and 56% in the SilicoDArT dataset, DArT-seq SNP dataset, and GBS SNP dataset, respectively (Table S1).
The markers of DArT-seq SNP dataset and GBS SNP dataset were distributed across the entire maize genome (Fig. 4). Generally, the density of markers at both ends of the chromosomes was high and decreased toward the centromere regions. The SNPs in both datasets were enriched at the ends of chromosomes 5 and 8. More uniform distribution was observed in the GBS SNP dataset, whereas the DArT-seq SNP dataset had higher marker density on the long arms of chromosomes 8 and 9. Uneven distributions may potentially lead to a decrease in prediction accuracy.

The markers density thermogram of DArT and GBS datasets. a The markers density thermogram of DArT’s SNP; b the markers density thermogram of GBS
The phylogenetic tree of NCCP
The phylogenetic trees were generated with the DArT-seq SNPs and GBS SNPs (Fig. 5). In total, the NCCP was divided into 13 groups, corresponding to the heterotic groups of NSS, SS, Huanglvxi, Lvxi, Reid, Mixed, France, PA, Jidan, Longdan, Lvdahonggu, Tangsipingtou and Unknown. The inbred lines from CIMMYT are tropical material, only two of them were regenerated in northeast China and classified into the Mixed group. Some small groups and inbred lines selected from the multiple parental lines also were classified as the unknown group. DArT SNPs produced a better phylogenetic tree, and heterotic groups of Reid, Lvxi, and Huanglvxi were separated clearly. However, the phylogenetic tree produced with the GBS SNPs did not separate the heterotic groups clearly in the present study. Some inbred lines from local breeders lost their source information, so they were classified into the unknown group. Some materials were collected from third parties with inaccurate source information, it is not easy to classify them into heterotic groups properly. For some inbred lines, pedigree information conflicted with the phylogenetic tree results generated with molecular markers, it is hard to generate a perfect phylogenetic tree in a practical breeding program.

Phylogenetic Trees for SNP markers from DArT-seq and GBS platforms. The letters a and b represent NCCP’s phylogenetic trees from DArT’s SNPs and GBS’s SNPs
The genomic prediction accuracy estimated from the DArT and GBS markers
Genomic prediction accuracies were estimated for all the three target traits with the BLUE values and the SNP datasets filtered with different parameters (Fig. 6). Using DArT SNP datasets, the prediction accuracy increased continuously as the TPS increased across all three traits evaluated under a natural drought stress environment (Fig. 6). The prediction accuracy slightly increased across all three traits when the TPS increased from 50 to 90%. The smallest standard error was observed in prediction accuracy when 50% to 60% of the training population size was used to predict the target traits of ER. For the target traits of SPH and GY, 40% of the training population size gained the smallest standard error, which indicated that 40 to 60% of the total genotypes assigned as the training set could achieve good prediction accuracy in the DArT SNP markers. For the GBS SNP markers, a similar trend was observed for GY. With the increase of the TPS, the average and median prediction accuracies showed slightly different for the target traits of ER and SPH (Fig. 6). The prediction accuracies estimated in a single year were similar to those using BLUEs across 2 years (Figs. S1, S2). The heritabilities of the three traits were higher in 2019, slightly improving the prediction accuracy. Prediction accuracies estimated with SilicoDArT markers were consistent with those estimated with DArT SNP markers (Fig. S3). Therefore, 50% of TPS was selected for further analysis.

Genomic prediction accuracies of ER, SPH, and GY in NCCP across two natural drought conditions, when the training population size was set from 10 to 90% of total genotypes, with an interval of 10%. Panel a ER estimated with DArT markers; b ER estimated with GBS markers; c SPH estimated with DArT markers; d SPH estimated with GBS markers; e GY estimated with DArT markers; f GY estimated with GBS markers
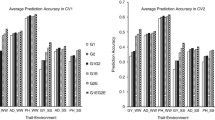
Genomic prediction accuracies estimated using twofold cross-validation (50% of TPS) for all three traits were shown in Fig. 7. The prediction accuracies estimated with the markers from different genotypic platforms showed significant differences for three interesting traits. For traits of SPH, the prediction accuracies estimated with the SilicoDArT dataset and DArT-seq SNP dataset showed significant differences, which means alignment of the markers to the reference genome resulting in a loss of prediction accuracy. For three traits, the prediction accuracies estimated from the DArT-seq SNPs were significantly higher than those estimated from the GBS SNPs (P < 0.01). Among the three traits, the prediction accuracy of SPH was the lowest in both the DArT and GBS markers, which was consistent with the lowest heritability of SPH. The average prediction accuracies using the SNPs from the DArT-seq were 0.27, 0.19, and 0.33 for ER, SPH, and GY traits, respectively. The prediction result estimated with SilicoDArT markers was similar to that estimated with DArT-seq SNPs, which were 0.26, 0.22, and 0.33 for ER, SPH, and GY traits, respectively. While, the prediction accuracies estimated from the GBS SNP markers were − 0.02, − 0.06, and 0.20 for the traits of ER, SPH, and GY, respectively.

Genomic prediction accuracies of ER, SPH, and GY estimated with the SilicoDArT, DArT and GBS marker datasets under twofold cross-validation. The significant difference was analyzed by one-way ANOVA of SPSS
Different prediction accuracies affected by marker quality
For all the three traits, the results of prediction accuracies estimated from the three molecular marker datasets filtered with the different combinations of MAF and missing rate were presented in Table 3, Tables S2, and S3. For the ER trait, the average prediction accuracies range from 0.19 to 0.29, and the highest prediction accuracy was observed using 2,762 SilicoDArT markers. Interestingly, increasing the filtering of missing rates improved the prediction accuracy, while MAF had little effect on the estimation of the prediction accuracy. Similar trends were also observed for ER and GY. In most cases, the prediction accuracies did not show significant differences under different combinations of MAF and missing rate. The LD decay distances estimated with the DArT SNPs and GBS SNPs were 6.16 kb and 3.83 kb, respectively (Fig. S4). The result of prediction accuracy estimated with the different DArT/GBS marker datasets filtered by various LD values was presented in Table 4. Filtering with different LD values caused different marker densities, and the number of markers decreased as the values increased. SPH obtained the highest average prediction accuracy in the LD0.1 of DArT with 800 SNPs, followed by the accuracy in the DArT-LD0.2 with 2681 markers and without filtering with 7,837 SNPs. In contrast, the DArT-LD0.1 in ER and GY traits were found to have the highest accuracies. Using the GBS markers, the prediction accuracies for traits of ER and GY estimated from 2333 SNPs filtered with LD value (r2 = 0.1) were higher than those estimated from all the 91,003 SNPs without filtering. Slight improvements were observed, when a smaller number of high-quality markers filtered with different parameters was used for prediction, but the best filtering parameters for improving prediction accuracy varied in different traits and genotyping platforms.
Prediction accuracy estimated within subgroups
In general, the population with close relationships is more likely to obtain high prediction accuracy. Effects of controlling the population structure on improving the prediction accuracy were assessed. For keeping enough lines in each subgroup, NCCP was divided into two subgroups, according to the population structure result. The PCA plots show the distribution of two subgroups using PC1 and PC2 estimated with the DArT and GBS markers, and the first two PC explained 35% and 31% of the genetic variation, respectively (Fig. 8). The PCA results estimated with the markers from the two platforms were very similar. Finally, 180 inbred lines weres classified into Subgroup 1, and 199 inbred lines were classified into Subgroup 2.

The first two principal components of DArT and GBS datasets. a The first two principal components of DArT. b The first two principal components of GBS. Subgroup 1 is marked with black and subgroup 2 is marked with red
For all the traits, the prediction accuracies estimated within each subgroup were shown in Table 5. Using the DArT SNP markers, the prediction accuracies in Subgroup 1 with 50% of TPS were 0.17, 0.11, and 0.11 for ER, SPH, and GY traits, respectively. For Subgroup 2, the prediction accuracies were 0.29, 0.20, and 0.33 for ER, SPH and GY traits, respectively. The prediction accuracies in Subgroup 1 were significantly lower than those using the whole population for all three traits, whereas the prediction accuracies in Subgroup 2 were higher than those using the entire population for all three traits. For the GBS markers, no differences were observed in the prediction accuracy between the two subgroups.
Prediction accuracy estimated with the trait-marker associations
To improve the prediction accuracy under the condition of natural drought in the maize seedling stage, the trait-marker associations detected in GWAS were selected for predictions using different significance levels (P < 0.5, 0.1, 0.01, 0.001). The same amount of randomly selected markers and all non-significant markers excluding significant markers were used as controls for comparison analysis. The results showed that the prediction accuracies estimated with significant markers were significantly higher than those estimated from the same amount of random markers for all the traits evaluated under drought tolerance environment at the seedling stage. When SNPs are from GBS, the prediction accuracies estimated with 46,168, 9074, 872 and 72 significant SNPs in P < 0.5, 0.1, 0.01 and 0.01were 0. 36, 0. 59, 0. 61 and 0. 57 for ER trait, respectively. For SPH and GY traits, both had the same trend as ER, and the maximum prediction accuracy was obtained when P < 0.01, which were 0.62 and 0.63 respectively (Fig. 9). When SNPs were from DArT-seq, 3858, 775, 90 and 11 significant SNPs were selected for prediction for ER trait in P < 0.5, 0.1, 0.01 and 0.001, and their prediction accuracies reached to 0.54, 0.71, 0.66 and 0.53, respectively. The same trends were also observed for SPH and GY traits, and the prediction accuracies were the highest when P < 0.1, i.e. 0.75 for SPH, and 0.60 for GY (Fig S5).

Prediction accuracy of GS under different situations and number of markers at different significance levels using the SNP of GBS. “SM” represents the significance markers, “ENSM” represents the randomly selected same amount of non-significant markers with the significance markers, “NSM” represents all non-significant markers excluding significant markers from genotype datasets and “ALL” represents the prediction of all markers under 60% of the training population. a and b respectively represent the prediction accuracy obtained by GS and number of markers at different significance levels by GWAS of ER trait. c and d respectively represent the prediction accuracy obtained by GS and number of markers at different significance levels by GWAS of SPH trait. e and f respectively represent the prediction accuracy obtained by GS and number of markers at different significance levels by GWAS of GY trait
Discussion
Drought tolerance, especially in the seedling stage, is a complex and inherent trait of maize (Wang et al. 2016). The global climate has changed recently, resulting in drastic fluctuations in rainfall patterns and increasing temperature. Sudden climate changes can cause significant economic losses worldwide. There are signs of grain yield stagnation in maize, especially in drought-stressed and semi-arid regions. Fuxin County is a region providing a natural test site located in the main maize producing area of Northeast China that has suffered years of drought. In this region, we can deploy more large-scale experiments without using greenhouses. In the current study, the three traits have found enough phenotypic variation in our NCCP for ER, SPH, and GY traits. We transformed the data to the BLUEs (best unbiased linear estimator) as phenotype values for the rrBLUP model assumptions (Fig. 2). The results of correlation analysis show that ER and SPH potentially impact GY under a drought environment. The phylogenetic tree shows our population has a rich genetic variation for improving drought resistance, although the two phylogenetic trees are not the same.
GS is an effective breeding tool for improving complex traits in maize (Crossa et al. 2014). The GS can accelerate the genetic gain per unit time and unit cost by reducing the selection cycle time and the phenotyping cost when the prediction accuracy is high. In the present study, a natural maize population genotyped with GBS and DArT markers were used to estimate the genomic prediction accuracies of ER, SPH, and GY under drought stress environments. Results indicated that the prediction for the inbred line permance under a drought environment is tough through both GBS and DArT genotyping platforms. The average prediction accuracies of ER, SPH and GY in the panel estimated using SNPs of DArT were 0.27, 0.19 and 0.33, respectively, SilicoDArT markers were slightly higher than the DArT SNPs; while the prediction accuracies with GBS markers were 0.14, and 0.20 (Fig. 7). Low prediction accuracy may be mainly affected by the quality of markers and heritabilities. Our heritabilities and prediction accuracies for the GY trait were within the range of the previous study that used 22 populations under water stress environments (Zhang et al. 2017a). Interestingly, unlike under normal conditions, the heritabilities of the three traits were more than 0.8 in 2019, but the average prediction accuracies were only slightly higher than that in 2020 and across 2 years. This implied that the prediction of drought resistance needs to accumulate more year data to improve the heritability across years and achieve stable phenotype.
In the same trait, the prediction accuracy estimated from the DArT markers was higher than that estimated from the GBS marker dataset, and the difference was significant. Both DArT and GBS are cost efficient and high-throughput genotyping platforms that can be used to implement GS. The GBS markers were implemented successfully in maize to improve multiple traits with different levels of genetic complexity (Crossa et al. 2013; Wang et al. 2020). Comparatively, only a few studies were reported using DArT markers to perform GS in wheat (Crossa et al. 2016; Liu et al. 2020). We spent a similar amount of money on two genotyping platforms, about $30 per sample, to control the budget within an acceptable range for breeders. This price can obtain DArT markers with normal quality, but a little bit lower quality for GBS markers. That means the genome coverage from GBS has not reached 1×, which is probably the reason why the prediction accuracies are not suitable when using GBS markers in this research. Our research indicated that DArT genotyping platform is more suitable than GBS when working with a lower budget. GS using DArT markers is also being implemented in CIMMYT maize breeding programs for improving grain yield and the other major agronomic traits.
This study showed that the average prediction accuracy was improved along with the TPS increasing in all the traits in both genotyping platforms. In contrast, the standard error was at first raised and then decreased as the TPS increased (Fig. 6). Relatively high prediction accuracies with the slightest standard error were observed in all traits when 50% to 60% of the total genotypes were used as a training set. Results of this study are consistent with a previous report (Guo et al. 2020) that can be applied in the practical breeding program. The marker density is an essential effect on prediction accuracy if population sets and heritabilities are the same. Our research found that the prediction accuracy of about 8000 markers from DArT is significantly higher than that of 90,000 markers from GBS, which emphasized the importance of marking quality and indicated 8000 markers is enough to implement genomic prediction in maize. Low-density marker populations appeal to GS because they decrease multicollinearity and computational time consumption and allow more individuals to be genotyped for the same cost. However, this prediction was made in a breeding program's initial stage, and higher marker density could play a key role in the following breeding cycle for the long term. As a mature genotyping platform, GBS markers from the sufficient sequencing depth have a superb performance on genomic prediction (Liu et al. 2021). The higher marker density of GBS also has a high application prospect in the multi-cycle breeding process.
We tried to increase the prediction accuracy by improving the marker quality and controlling the population structure, but at the same time, the marker density was also reduced. Our results show that the prediction accuracy was improved in some cases (Tables 3, 4, 5), even if the density of the marker was reduced. However, this strategy doesn't always work, and may be due to the fact that some complex traits require markers with specific effects. Also, there is a tradeoff between the number of markers and marker quality or population structure because marker quality decreases as the number of markers increase in a specific marker dataset. This study suggests that genomic prediction can also be performed using high-quality and low-density markers in the initial breeding stage under the drought environment, especially when heritability is low.
The low-cost DArT platform increases the possibility of integrating genomic selection strategies into practical breeding programs for small breeding companies and the private sector. The SilicoDArT markers are possible to capture lost effects using a single reference genome for a diversity-rich population, which is essential for challenging drought tolerance prediction. The key to the genomic prediction of maize drought resistance is to improve heritability and stabilize the phenotype. Natural drought experimental sites can increase the heritability of planting materials by accumulating data for many years without a greenhouse. Fuxin county provides a chance to study genomic prediction for drought tolerance. The SNPs significantly associated with traits can remarkably improve the prediction accuracy to drought tolerance by removing low effect or invalid markers. This result is consistent with a previous study (Cerrudo et al. 2018). The GP accuracies obtained from the marker-trait associated SNPs were relatively higher than those obtained from the genome-wide SNPs for most of the target traits, by about 5–30% (Yuan et al. 2019).New models for integrating genotype-by-environment interaction into genomic prediction needs to be further developed to improve the selection of maize drought tolerance, although the genomic prediction using significant markers has dramatically promoted the prediction accuracy.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ANOVA:
-
Analysis of variance
- BLUE:
-
Best linear unbiased estimate
- BP:
-
Breeding population
- DArT:
-
Diversity arrays technology
- DL:
-
Deep learning
- ER:
-
Emergence rate
- GBS:
-
Genotyping-by-sequencing
- GEBV:
-
Genomic estimated breeding values
- GS:
-
Genomic selection
- GY:
-
Grain yield
- H2 :
-
Broad-sense heritability
- LD:
-
Linkage disequilibrium
- MAF:
-
Minor allele frequency
- MAS:
-
Marker-assisted selection
- MRD:
-
Modified Rogers' distance
- NCCP:
-
Northeast China core population
- NJ:
-
Neighbor-Joining
- PCA:
-
Principal component analysis
- QTL:
-
Quantitative trait loci
- rrBLUP:
-
Ridge regression BLUP
- SNP:
-
Single nucleotide polymorphism
- SPH:
-
Seedling plant height
- TP:
-
Training population
- TPS:
-
Training population size
References
Alvarado G, Rodríguez FM, Pacheco A, Burgueño J, Crossa J, Vargas M, Pérez-Rodríguez P, Lopez-Cruz MA (2020) META-R: a software to analyze data from multi-environment plant breeding trials. Crop J 8:745–756
Bernardo R (2016) Bandwagons I, too, have known. Theor Appl Genet 129:2323–2332
Beyene Y, Semagn K, Mugo S, Tarekegne A, Babu R, Meisel B, Sehabiague P, Makumbi D, Magorokosho C, Oikeh S, Gakunga J, Vargas M, Olsen M, Prasanna BM, Banziger M, Crossa J (2015) Genetic gains in grain yield through genomic selection in eight bi-parental maize populations under drought stress. Crop Sci 55:154–163
Bradbury PJ, Zhang ZW, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Cao SL, Loladze A, Yuan Y, Wu YS, Zhang A, Chen JF, Huestis G, Cao JS, Chaikam V, Olsen M, Prasanna BM, San Vicente F, Zhang XC (2017) Genome-wide analysis of tar spot complex resistance in maize using genotyping-by-sequencing SNPs and whole-genome prediction. Plant Genome 10:plantgenome2016-10
Carena MJ, Hallauer AR, Filho JM (2010) Quantitative genetics in maize breeding. Springer, New York
Cerrudo D, Cao S, Yuan Y, Martinez C, Suarez EA, Babu R, Zhang TS (2018) Genomic selection outperforms marker assisted selection for grain yield and physiological traits in a maize doubled haploid population across water treatments. Front Plant Sci 9:366
Chen J, Zavala C, Ortega N, Petroli C, Franco J, Burgueño J, Costich DE, Hearne SJ (2016) The development of quality control genotyping approaches: a case study using elite maize lines. PLoS ONE 11:e0157236
Combs E, Bernardo R (2013) Accuracy of genomewide selection for different traits with constant population size, heritability, and number of markers. Plant Genome 6:plantgenome2012-11
Crossa J, Beyene Y, Kassa S, Pérez P, Hickey JM, Chen C, de los Campos G, Burgueño J, Windhausen VS, Buckler E, Jannink J-L, Lopez Cruz MA, Babu R (2013) Genomic prediction in maize breeding populations with genotyping-by-sequencing. G3 Genes Genomes Genet 3:1903
Crossa J, Pérez P, Hickey J, Burgueño J, Ornella L, Cerón-Rojas J, Zhang X, Dreisigacker S, Babu R, Li Y, Bonnett D, Mathews K (2014) Genomic prediction in CIMMYT maize and wheat breeding programs. Heredity (edinb) 112:48–60
Crossa J, Jarquín D, Franco J, Pérez-Rodríguez P, Burgueño J, Saint-Pierre C, Vikram P, Sansaloni C, Petroli C, Akdemir D, Sneller C, Reynolds M, Tattaris M, Payne T, Guzman C, Peña RJ, Wenzl P, Singh S (2016) Genomic prediction of gene bank wheat landraces. G3 Genes Genomes Genet 6:1819
Crossa J, Pérez-Rodríguez P, Cuevas J, Montesinos-López O, Jarquín D, de los Campos G, Burgueño J, González-Camacho JM, Pérez-Elizalde S, Beyene Y, Dreisigacker S, Singh R, Zhang XC, Gowda M, Roorkiwal M, Rutkoski J, Varshney RK (2017) Genomic selection in plant breeding: methods, models, and perspectives. Trends Plant Sci 22:961–975
Cui ZH, Dong HX, Zhang A, Ruan YY, He Y, Zhang ZW (2020) Assessment of the potential for genomic selection to improve husk traits in maize. G3-Genes Genomes Genet 10:g3.401600.402020
de los Campos G, Gianola D, Rosa GJM (2009) Reproducing Kernel Hilbert spaces regression: a general framework for genetic evaluation. J Anim Sci 87:1883–1887
Desta ZA, Ortiz R (2014) Genomic selection: genome-wide prediction in plant improvement. Trends Plant Sci 19:592–601
Dos Santos JPR, Pires LPM, de Castro Vasconcellos RC, Pereira GS, Von Pinho RG, Balestre M (2016) Genomic selection to resistance to stenocarpella maydis in maize lines using DArTseq markers. BMC Genet 17:86
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6:e19379
Endelman JB (2011) Ridge regression and other kernels for genomic selection with R package rrBLUP. Plant Genome 4:250–255
Fan Y, Zhou G, Sergey S, Chen Z, Cai S, Li C, Zhou M (2016) Genome-wide association study reveals a new qtl for salinity tolerance in barley (Hordeum vulgare L.). Front Plant Sci 7:946
Guo R, Dhliwayo T, Mageto EK, Palacios-Rojas N, Lee M, Yu D, Ruan Y, Zhang A, San Vicente F, Olsen M, Crossa J, Prasanna BM, Zhang LJ, Zhang XC (2020) Genomic prediction of kernel zinc concentration in multiple maize populations using genotyping-by-sequencing and repeat amplification sequencing markers. Front Plant Sci 11:534
Hao Z, Lv D, Ge Y, Shi J, Weijers D, Yu G, Chen J (2020) RIdeogram: drawing SVG graphics to visualize and map genome-wide data on the idiograms. PeerJ Comput Sci 6:e251
Heslot N, Yang H-P, Sorrells ME, Jannink J-L (2012) Genomic selection in plant breeding: a comparison of models. Crop Sci 52:146–160
Jiang B, Wang P, Zhuang S, Li MS, Li Z, Gong ZH (2018) Detection of maize drought based on texture and morphological geatures. Comput Electron Agric 151:50–60
Kang HM, Zaitlen NA, Wade CM, Kirby A, Heckerman D, Daly MJ, Eskin E (2008) Efficient control of population structure in model organism association mapping. Genetics 178:1709–1723
Kilian A, Wenzl P, Huttner E, Carling J, Xia L, Blois H, Caig V, Heller-Uszynska K, Jaccoud D, Hopper C, Aschenbrenner-Kilian M, Evers M, Peng K, Cayla C, Hok P, Uszynski G (2012) Diversity arrays technology: S generic genome profiling technology on open platforms. Methods Mol Biol (clifton, NJ) 888:67–89
Kwong QB, Ong AL, Teh CK, Chew FT, Tammi M, Mayes S, Kulaveerasingam H, Yeoh SH, Harikrishna JA, Appleton DR (2017) Genomic selection in commercial perennial crops: applicability and improvement in oil palm (Elaeis guineensis Jacq.). Sci Rep 7:2872
Liu XG, Wang HW, Wang H, Guo ZF, Xu XJ, Liu JC, Wang SH, Li WX, Zou C, Prasanna BM, Olsen MS, Huang CL, Xu YB (2018) Factors affecting genomic selection revealed by empirical evidence in maize. Crop J 6:341–352
Liu C, Sukumaran S, Jarquin D, Crossa J, Dreisigacker S, Sansaloni C, Reynolds M (2020) Comparison of array- and sequencing-based markers for genome-wide association mapping and genomic prediction in spring wheat. Crop Sci 60:211–225
Liu Y, Hu G, Zhang A, Loladze A, Hu Y, Wang H, Qu J, Zhang X, Olsen M, San Vicente F, Crossa J, Lin F, Prasanna BM (2021) Genome-wide association study and genomic prediction of fusarium ear rot resistance in tropical maize germplasm. Crop J 9:325–341
Massman JM, Gordillo A, Lorenzana RE, Bernardo R (2013) Genomewide predictions from maize single-cross data. Theor Appl Genet 126:13–22
Maulana F, Kim K-S, Anderson JD, Sorrells ME, Butler TJ, Liu S, Baenziger PS, Byrne PF, Ma X-F (2021) Genomic selection of forage agronomic traits in winter wheat. Crop Sci 61:410–421
Meuwissen THE, Hayes BJ, Goddard ME (2001) Prediction of total genetic value using genome-wide dense marker maps. Genetics 157:1819–1829
Michel S, Ametz C, Gungor H, Epure D, Grausgruber H, Löschenberger F, Buerstmayr H (2016) Genomic selection across multiple breeding cycles in applied bread wheat breeding. Theor Appl Genet 129:1179–1189
Montesinos-López OA, Montesinos-López A, Pérez-Rodríguez P, Barrón-López JA, Martini JWR, Fajardo-Flores SB, Gaytan-Lugo LS, Santana-Mancilla PC, Crossa J (2021) A review of deep learning applications for genomic selection. BMC Genom 22:19
Môro GV, Santos MF, de Souza Júnior CL (2019) Comparison of genome-wide and phenotypic selection indices in maize. Euphytica 215:76
Norman A, Taylor J, Edwards J, Kuchel H (2018) Optimising genomic selection in wheat: effect of marker density, population size and population structure on prediction accuracy. G3 Genes Genomes Genet 8:2889–2899
Pereira WJ, de CastroRodriguesPappas M, Grattapaglia D, Pappas GJ (2020) A cost-effective approach to DNA methylation detection by methyl sensitive DArT sequencing. PLoS ONE 15:e0233800
R Core Team (2021) R: a language and environment for statistical computing, Vienna. https://www.R-project.org/.
Resende MFR Jr, Muñoz P, Resende MDV, Garrick DJ, Fernando RL, Davis JM, Jokela EJ, Martin TA, Peter GF, Kirst M (2012) Accuracy of genomic selection methods in a standard data set of loblolly pine (Pinus taeda L.). Genetics 190:1503–1510
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Tecle IY, Edwards JD, Menda N, Egesi C, Rabbi IY, Kulakow P, Kawuki R, Jannink J-L, Mueller LA (2014) solGS: a web-based tool for genomic selection. BMC Bioinform 15:398–398
Thavamanikumar S, Dolferus R, Thumma BR (2015) Comparison of genomic selection models to predict flowering time and spike grain number in two hexaploid wheat doubled haploid populations. G3 (bethesda) 5:1991–1998
Tian Y, Guan B, Zhou DW, Yu JB, Li GD, Lou YJ (2014) Responses of seed germination, seedling growth, and seed yield traits to seed pretreatment in maize (Zea mays L.). Sci World J 2014:834630
VanRaden PM (2008) Efficient methods to compute genomic predictions. J Dairy Sci 91:4414–4423
Vivek BS, Krishna GK, Vengadessan V, Babu R, Zaidi PH, Kha LQ, Mandal SS, Grudloyma P, Takalkar S, Krothapalli K, Singh IS, Ocampo ETM, Xingming F, Burgueño J, Azrai M, Singh RP, Crossa J (2017) Use of genomic estimated breeding values results in rapid genetic gains for drought tolerance in maize. Plant Genome 10:plantgenome2016.2007.0070
Wang J, Zhang Z (2021) GAPIT version 3: boosting power and accuracy for genomic association and prediction. Genom Proteomics Bioinform 19:1–12
Wang X, Wang H, Liu S, Ferjani A, Li J, Yan J, Yang X, Qin F (2016) Genetic variation in ZmVPP1 contributes to drought tolerance in maize seedlings. Nat Genet 48:1233–1241
Wang N, Yuan Y, Wang H, Yu D, Liu Y, Zhang A, Gowda M, Nair SK, Hao Z, Lu Y, San Vicente F, Prasanna BM, Li X, Zhang X (2020) Applications of genotyping-by-sequencing (GBS) in maize genetics and breeding. Sci Rep 10:16308
Xiao YJ, Liu HJ, Wu LJ, Warburton M, Yan JB (2017) Genome-wide association studies in maize: praise and stargaze. Mol Plant 10:359–374
Yu J, Pressoir G, Briggs WH, Vroh Bi I, Yamasaki M, Doebley JF, McMullen MD, Gaut BS, Nielsen DM, Holland JB, Kresovich S, Buckler ES (2006) A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38:203–208
Yuan Y, Cairns JE, Babu R, Gowda M, Makumbi D, Magorokosho C, Zhang A, Liu Y, Wang N, Hao Z, San Vicente F, Olsen MS, Prasanna BM, Lu Y, Zhang X (2019) Genome-wide association mapping and genomic prediction analyses reveal the genetic architecture of grain yield and flowering time under drought and heat stress conditions in maize. Front Plant Sci 9:1919
Zhang A, Wang H, Beyene Y, Semagn K, Liu Y, Cao S, Cui Z, Ruan Y, Burgueno J, San Vicente F, Olsen M, Prasanna BM, Crossa J, Yu H, Zhang X (2017a) Effect of trait heritability, training population size and marker density on genomic prediction accuracy estimation in 22 bi-parental tropical maize populations. Front Plant Sci 8:1916
Zhang XC, Pérez-Rodríguez P, Burgueño J, Olsen M, Buckler E, Atlin G, Prasanna BM, Vargas M, San Vicente F, Crossa J (2017b) Rapid cycling genomic selection in a multiparental tropical maize population. G3 (bethesda) 7:2315–2326
Zhang HH, Yin LL, Wang MY, Yuan XH, Liu XL (2019) Factors affecting the accuracy of genomic selection for agricultural economic traits in maize, cattle, and pig populations. Front Genet 10:189
Funding
This work was supported by Shanghai Agriculture Applied Technology Development Program, China (Z20190101), the National Science Foundation for Young Scientists of China (31801442), the CIMMYT-China Specialty Maize Research Center (KF201802), the Natural Sciences Foundation of Liaoning Provincial Department of Education (LJKZ0657).
Author information
Authors and Affiliations
Contributions
AZ, SY, and ZC collected materials; AZ, SC, DD analyzed data; AZ, SC, CL, JN, XD, YG, YZ and JF carried out the field experiments; HZ and YR designed the study; SC, AZ and YL wrote the manuscript; XZ, JC, LZ, YR, XD and JF revised the manuscript. All authors have read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, A., Chen, S., Cui, Z. et al. Genomic prediction of drought tolerance during seedling stage in maize using low-cost molecular markers. Euphytica 218, 154 (2022). https://doi.org/10.1007/s10681-022-03103-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10681-022-03103-y