
Abstract
Microdochium majus and Microdochium nivale cause serious disease problems in grasses and cereal crops in the temperate regions. Both fungi can infect the plants during winter (causing pink snow mould) as well as under cool humid conditions during spring and fall. We conducted a pathogenicity test of 15 M. nivale isolates and two M. majus isolates from Norway at low temperature on four different grass cultivars of Lolium perenne and Festulolium hybrids. Significant differences between M. nivale isolates in the ability to cause pink snow mould were detected. The M. nivale strains originally isolated from grasses were more pathogenic than isolates from cereals. The genetic diversity of M. nivale and M. majus isolates was studied by sequencing four genetic regions; Elongation factor-1 alpha (EF-1α), β-tubulin, RNA polymerase II (RPB2) and the Internal Transcribed Spacer (ITS). Phylogenetic trees based on the sequences of these four genetic regions resolved M. nivale and M. majus isolates into separate clades. Higher genetic diversity was found among M. nivale isolates than among M. majus isolates. M. nivale isolates revealed genetic differences related to different host plants (grasses vs. cereals) and different geographic regions (Norway and UK vs. North America). Sequence results from the RPB2 and β-tubulin genes were more informative than those from ITS and EF-1α. The genetic and phenotypic differences detected between Norwegian M. nivale isolates from cereals and grasses support the assumption that host specialization exist within M. nivale isolates.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Microdochium nivale (Fr.) Samuels and I. C. Hallett is the most prevalent low temperature pathogen in cereals and grasses in Norway (Årsvoll 1973; Ergon et al. 2003; Larsen 1994). The fungus was first described by the Swedish mycologist E.M. Fries (1825) under the name Lanosa nivalis. Fries characterized the fungus by its ability to attack wheat and grass plants under snow cover (Noble and Montgomerie 1956). Due to its similarity with Fusarium species, this fungus was given the name F. nivale Ces. ex Berlese & Voglino (W. Gams and Müller 1980). However, the fungus has been reclassified several times (Booth 1971; Gams 1989; Glynn et al. 2005; Samuels and Hallett 1983). The fungus produces conidia of two different sizes with different numbers of septa, and based on this divided into two varieties, var. majus and var. nivale (Wollenweber 1930). Gams and Müller (1980) reclassified the fungus as Gerlachia nivale due to the absence of conidial foot cells. Later Samuels and Hallett (1983) showed that the fungus rather belong to the genus Microdochium.
Molecular genotyping techniques have been applied to investigate genetic differences between isolates of Microdochium var. majus and Microdochium var. nivale. Parry et al. (1995) were able to differentiate between the two varieties using restriction enzyme analysis of the Internal Transcribed Spacer (ITS) region. Lees et al. (1995) confirmed the distinction between the two varieties by using RAPD (Random Amplified Polymorphic DNA) genotyping, and they found higher levels of diversity among var. nivale isolates than among var. majus isolates. Later Glynn et al. (2005) suggested that var. majus and var. nivale should be classified as two different species, Microdochium majus and Microdochium nivale, based on the elongation factor 1 alpha (EF-1α) gene sequences. Several studies have used constitutive genes such as RNA polymerase II (RPB2) and β-tubulin to study fungal population structure in relation to host or geographic origin (Einax and Voigt 2003; Myllys et al. 2001; Pavlic et al. 2009). Jewell and Hsiang (2013) used β-tubulin, RPB2, and EF-1α gene sequences, in addition to sequences from the ITS region to differentiate between M. nivale and M. majus isolates from different host plants and geographic regions. They found isolates of different geographic origin (Europe vs. North America) and from different host plants (grasses vs. cereals) to be different based on RPB2 and β-tubulin genes sequence. In general RPB2 is considered a very good target for phylogenetic and evolutionary studies in fungi, mainly because it is a large single copy gene and easy to amplify by PCR (Liu et al. 1999). In addition, sequence comparison between RPB2 from fungi, plant and animals showed 12 conserved regions >85% sequence identity, which makes it easier to design PCR primers to amplify these regions from different species (Liu and Hall 2004). Moreover, several studies found that the RPB2 gene has higher taxonomic sensitivity than ITS and β-tubulin (Liu and Hall 2004; Schoch et al. 2012; Větrovský et al. 2016).
Variation in pathogenicity between M. majus and M. nivale has been reported in several studies on grasses (Hofgaard et al. 2006; Holmes 1976) and on cereals (Diamond and Cooke 1999; Maurin et al. 1995). Isolates of M. nivale were found to be more pathogenic on winter rye (Simpson et al. 2000), as well as on perennial ryegrass (Hofgaard et al. 2006) compared to M. majus isolates, while M. majus isolates were found to be more pathogenic on winter wheat (Diamond and Cooke 1997; Simpson et al. 2000). Furthermore, differences in pathogenicity between isolates of M. nivale from grasses and those from cereals were found (Årsvoll 1973; Hofgaard et al. 2006; Litschko and Burpee 1987; Smith 1983). Host specialization within the groups of M. nivale isolates has been studied by molecular genotyping techniques such as RAPD and IGS-RFLP (the intergenic spacer restriction fragment length polymorphisms) (Lees et al. 1995; Mahuku et al. 1998; Nicholson et al. 1996), and by DNA sequencing (Glynn et al. 2005; Jewell and Hsiang 2013; Ren et al. 2015). Mahuku et al. (1998) grouped 100 isolates from different grass species into four clusters according to their hosts using IGS-RFLP analysis. Studies by Jewell and Hsiang (2013) and Ren et al. (2015) indicate the existence of host specialization especially within M. nivale isolates.
An effective strategy for disease control should be based on knowledge of population structure of the plant pathogen, and whether the level of genetic variation within pathogen populations reflects the interaction between pathogen and hosts (Allard 1990; McDonald and McDermott 1993). Therefore, the aim of our study was to characterize genetic and phenotypic (pathogenicity) diversity of a selection of Norwegian isolates of M. nivale and M. majus by assessing their pathogenicity on Lolium perenne and Festulolium, and by sequence analyses of four genes or genetic regions; Elongation factor-1 alpha (EF-1α), Beta-tubulin, RNA polymerase II (RPB2), and Internal transcribed spacer (ITS).
Materials and methods
Fungal isolates, identification and cultivation
A majority of the 44 isolates used in this study was collected in Norway in the period 1975–2010. The M. nivale isolates were mainly isolated from leaves of cereals and grasses displaying snow mould symptoms, whereas most of the M. majus isolates were isolated from symptomatic leaves or seeds of wheat (Table 1). M. nivale isolates NG26 and NG42, and M. majus isolates NG3 and NG36 were kindly provided by Simon Edwards, Harper Adams University, UK. One isolate of M. majus (OP2A) was kindly provided by Roy Browne, University College Dublin, Ireland. The isolates were preserved at −80 °C as mycelium on 5 mm PDA (potato dextrose agar) plugs in 1.5-ml microfuge tubes in the fungal culture collection at NIBIO, Plant Health Division, Ås, Norway (Table 1). Fungal isolates were classified into species based on colony and conidial morphology according to Gerlach and Nirenberg (1982). Asporogenic isolates (7 isolates listed in Table 1) were classified using polymerase chain reaction (PCR) according to Glynn et al. (2005).
Pathogenicity test
A pathogenicity test was performed using the Norwegian Lolium perenne cultivars ‘Figgjo’ and ‘Ivar’, and the Festulolium candidate cultivars ‘FuRs9812’ and ‘FuRs0463’. Seeds were germinated in a greenhouse at 18 to 22 °C and 16 h photoperiod. The greenhouse was supplemented with a light source (Constant Color CMH™ Lamps 400 W) of about 250 μmol photosynthetic active radiation (PAR) m−2 s−1. After 2 weeks, the seedlings were transplanted to 10 cm pots containing fertilized soil mixture (Gartnerjord, Tjerbo); 5 plants per pot and 6 pots per cultivar (3 for inoculation and 3 as controls). The plants were fertilized weekly with a mixture of 80 g/L Kristalon™ fertilizer 06–12-36 and 60 g/L of YaraLiva® Calcinit 15.5–0-0 (Yara International ASA, Oslo, Norway).
Fifteen M. nivale and two M. majus isolates were selected for this study (as listed in Table 1). To recover the isolates after storage, they were inoculated onto 9 cm PDA plates and incubated for 10 days at 9 °C in darkness. The isolates were then transferred to new plates and incubated at 20 °C for 7 days under 12-h cycles of near-ultraviolet and white light for sporulation. Conidial suspensions were prepared by washing the agar with 10 ml sterile distilled water containing 0.2% Tween 20, and adjusted to 1 × 106 conidia ml−1.
Each of the 17 isolates was inoculated on each of the four cultivars (three pots per isolate) by spraying 10 ml of the spore suspensions per pot, as described by Hofgaard et al. (2006). Controls were sprayed with distilled water containing 0.2% Tween 20. Inoculated plants were randomly distributed on four trolleys, while non-inoculated plants were placed on two separate trolleys. To maintain high humidity during incubation at 2 °C in darkness, all plants (including controls) were covered with moist cellulose wadding and black plastic sheets immediately after inoculation. The experiment was conducted twice, in the first experiment (A) the plants were incubated for 8 weeks after inoculation while in the second experiment (B), the plants were incubated for 9 weeks.
After the incubation period, the plants were cut to 5 cm above soil surface and allowed to regrow in the greenhouse under the same conditions as described above. After 2 weeks of regrowth, all above ground plant material were harvested and dry weights (g/pot) determined. Pathogenicity was calculated as 1-relative regrowth (as described by Hofgaard et al. 2006).
Analysis of variance of pathogenicity was performed using PROC GLM in SAS version 9.2 (SAS Institute Inc., Cary, NC, USA). Significant differences between isolates (P < 0.05) in pathogenicity were calculated by the Ryan-Einot-Gabriel-Welsch (REGWQ) multiple range test in SAS.
DNA extraction
Thirty M. nivale and fourteen M. majus isolates were selected for DNA sequencing. The isolates were a part of the same collection as listed in Table 1. Fungal isolates were inoculated on PDA plates and incubated for 1 week at 20 °C in darkness. Mycelium was harvested by carefully scraping it off the agar surface using a clean razor blade, frozen quickly in liquid nitrogen, and ground using a mortar and pestle. DNA was extracted from 100 mg ground mycelium using DNeasy Plant Mini Kit (Qiagen Inc., Germany), according to the manufacturers’ protocol. DNA quality was verified by agarose gel electrophoresis. DNA was stored at −20 °C prior to PCR amplification.
PCR amplification and sequencing
PCR amplification was performed in a 25 μl volume containing 2.5 μl of Taq polymerase buffer (10 x GeneAmp PCR buffer contain 15 mM MgCl2), 10 pmol each of forward and reverse primer (Invitrogen Ltd., UK), 200 μM dNTP, 1.0 unit Taq DNA polymerase (AmpliTaq - Applied Biosystems, Foster City, CA) and 2 μl of fungal DNA. PCR amplification of the ITS region was performed according to White et al. (1990), amplification of EF-1α gene according to Glynn et al. (2005), and amplification of the RPB2 gene and the β-tubulin gene followed the protocols described by Jewell and Hsiang (2013). The PCR products were visualized using gel electrophoresis and a UV-transilluminator (GelDoc 1000 gel documentation system, BioRad). The PCR products were purified and sequenced in both directions at GATC Biotech (Germany).
Sequence data analysis
Good quality sequences were obtained from 30 isolates of the EF-1α gene (21 M. nivale and 9 M. majus), 36 isolates of the β-tubulin gene (22 M. nivale and 14 M. majus), 32 isolates of the RPB2 gene (23 M. nivale and 9 M. majus), and 40 isolates of the ITS region (28 M. nivale and 12 M. majus). The DNA sequences were assembled and analysed using the Lasergene Seqman software (DNAStar Inc.). DNA sequences used in further analyses have been deposited in the NCBI GenBank® database (accession numbers: KT736151 - KT736180 for EF-1α, KT736181 - KT736220 for ITS, KT736221 - KT736256 for β-tubulin and KT736257 - KT736288 for RBP2). Fragments of 723, 647, 404, and 399 bp were used to produce multiple sequence alignments for RBP2, β-tubulin, EF-1α and ITS, respectively. Sequences of the RPB2 and β-tubulin genes in the North American isolates were obtained from the NCBI GenBank® database (Supplementary Table 1). The sequence alignments were performed using the CLC Main Workbench version 6.9.1 (CLC Inc. Aarhus, Denmark) with default parameters. Neighbour-joining (NJ) phylogenetic trees were constructed for each gene by the CLC software with 1000 bootstrap replicates. Pairwise comparisons were computed based on aligned sequences using the CLC Genomic Workbench version 6.9.1 (CLC Inc. Aarhus, Denmark) to estimate the percentage of identity (percentage of identical residues in alignment position to overlapping alignment between sequences), as well as the pairwise distance based on the Jukes-Cantor distance. The sequences of Microdochium bolleyi were used as out-group for RPB2 and β-tubulin genes. The branch tips of each tree were labelled with isolate number, species (M or N for M. majus or M. nivale, respectively), host origin (C or G for cereals or grasses, respectively), and geographic origin (NA for North America, NO for Norway, UK for United Kingdom and IR for Ireland).
Results
Pathogenicity and host specialization
Significant and large differences in pathogenicity were found between M. nivale and M. majus isolates (Fig. 1), between M. nivale isolates from cereals and grasses (P ≤ 0.0001 (exp A) and 0.003 (exp B), Table 2), and among M. nivale isolates (P ≤ 0.0001 in both experiments, Table 2). M. nivale isolates 200,231, 201,050, 200,272 and 201,053 were highly aggressive, while isolates 200,114 and 200,136 had a poor ability to cause snow mould in these experiments (Fig. 1). The two M. majus isolates showed very low pathogenicity on Lolium and Festulolium (Fig. 1). M. nivale isolates from grasses were more pathogenic (mean value of the relative regrowth was 0.4) than isolates from cereals (mean value of the relative regrowth was 0.6), when inoculated on the grasses (Fig. 2). The cultivars displayed significantly different disease tolerances in both experiments, and this was due to a differential reaction of the two L. perenne cultivars in the 8-week experiment (P ≤ 0.001), and between cultivars of both species in the 9-weeks experiment (P ≤ 0.016 and P ≤ 0.005, Table 2). On average, there were no differences in disease tolerance between the two grass species. There was a small but significant cultivar x isolate interaction (P ≤ 0.026, Table 2) only in the experiment with the shortest incubation time, indicating that the pathogenicity of the isolates differed among the four grass cultivars.

Pathogenicity (0 = min; 1 = max) of seventeen Microdochium sp. isolates (fifteen M. nivale and two M. majus) on four different grass cultivars (Figgjo, Ivar, FuRs9812 and FuRs0463). Pathogenicity was measured as 1- relative regrowth (dry weight of inoculated plants divided by dry weight of non-inoculated plants after incubation for eight and nine weeks (experiments A and B respectively) under artificial snow cover, followed by 2 weeks of regrowth (average value of four cultivars). Isolates ID is labelled with host origin (Lp for L. perenne, Fe for Festulolium, Fp for F. pratensis, Ag for A. stolonifera, Sc for S. cereal, Tr for Triticum sp., Fr for F. rubra and Dg for D. glomerata). Error bars indicate standard errors of the mean of the two experiments. Bars with the same letter are not significantly different (P value <0.05)

Pathogenicity (0 = min; 1 = max) of M. nivale isolates from cereals (average value of five isolates) and grasses (average value of ten isolates) on four different grass cultivars (Figgjo, Ivar, FuRs9812 and FuRs0463). Pathogenicity was measured as 1- relative regrowth (dry weight of inoculated plants divided by dry weight of non-inoculated plants at 8 weeks (experiment A) and 9 weeks (experiment B) after incubation under artificial snow cover followed by 2 weeks of regrowth. Error bars indicate standard errors of the mean of the two experiments
Sequence variation between isolates
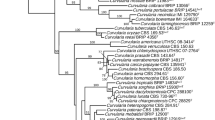
The sequences of the genomic regions of RPB2, ITS, EF-1α and β-tubulin revealed slightly different phylogenetic structures. The RPB2 and the β-tubulin sequences separated M. nivale and M. majus isolates more clearly than the other two. For the β-tubulin gene, all isolates of M. majus formed a single clade with a bootstrap value of 99% and 99.6% sequence identity, while the M. nivale isolates formed two clades with bootstrap values of 100% and pairwise distance of 0.05 (Fig. 3a). The first M. nivale clade represents 17 isolates from grasses from Norway and North America with 99.6% sequence identity, and one isolate (200108) from wheat in Norway. The second M. nivale clade (10 isolates), represents only isolates from cereals originating from Norway, United Kingdom and North America with 99.4% sequence identity. For the RPB2, all M. majus isolates grouped together in one cluster, which was divided into subclades, all with bootstrap values of 100% and a pairwise distance of 0.07 (Fig. 3b). One clade represents isolates from Norway and United Kingdom (8 from Norway and one from UK) with 99.4% sequence identity; the other represents three isolates from North America with 97.2% sequence identity. All M. nivale isolates from Norway (21) and United Kingdom (2) grouped in one sub-cluster with a bootstrap value of 100% and 96.4% sequence identity. Six isolates from North America formed one cluster, which was divided into two sub-clusters corresponding to the host origin, i.e. cereals and grasses.

Neighbour-Joining phylogenetic trees obtained with 1000 bootstrap replicates. The phylogenetic trees were constructed based on a the β-tubulin gene sequence; b the RNA polymerase II gene sequence (RPB2) from the different fungal isolates included. The tips of the tree are labelled with isolate number, species (M or N for M. majus or M. nivale, respectively), host origin (C or G for cereals or grasses, respectively), and geographic origin (NA for North America, NO for Norway, UK for United Kingdom and IR for Ireland)
The phylogenetic analysis based on the EF-1α gene sequences (Fig. 4a) only showed two major clusters with pairwise distance of 0.08, one represents 20 isolates of M. nivale with 99.7% sequence identity, and the other 10 isolates of M. majus with 96.1% sequence identity. No clear sub-clusters were formed and we found no clear difference between isolates based on host origin. The ITS sequence analysis (Fig. 4b) revealed low genetic diversity, but still two major clusters were formed for 28 isolates of M. nivale and 12 isolates of M. majus with a pairwise distance of 0.02 and 99.8% sequence identity.

Neighbour-Joining phylogenetic trees obtained with 1000 bootstrap replicates. The phylogenetic tree was constructed based on a the elongation factor 1-alpha (EF-1α) gene sequence and b the internal transcribed spacer (ITS) genetic region from the different fungal isolates included. The tips of the trees are labelled with isolate number, species (M or N for M. majus or M. nivale, respectively), host origin (C or G for cereals or grasses, respectively), and geographic origin (NA for North America, NO for Norway, UK for United Kingdom and IR for Ireland)
Discussion
The main aim of this study was to investigate pathogenicity of M. nivale isolates from different host plants and the genetic variation among isolates of M. nivale and M. majus. Therefore, a pathogenicity tests was conducted with 15 isolates of M. nivale and two isolates of M. majus, all from Norway, on two L. perenne and two Festulolium cultivars. In addition, the nucleotide sequences of four different genomic regions were studied in approximately 40 M. nivale and M. majus isolates from Norway, England and Ireland to investigate genetic diversity within and among isolates of the two species and the phylogeographic relationships among isolates.
Isolates of M. nivale from grasses were significantly more pathogenic than isolates from cereals when tested on the four grass cultivars. The low pathogenicity of M. majus isolates observed in our study, confirm previous studies demonstrating that M. majus are relatively less pathogenic than M. nivale on grasses (Hofgaard et al. 2006; Holmes 1976). However, only two M. majus isolates were used in our experiments. We identified M. nivale isolate 200,231 (host origin L. perenne) as the most pathogenic isolate. Thus, this isolate was chosen for further screening of 23 grass populations for snow mould resistance under controlled and field conditions (Abdelhalim et al. 2016) and was used as inoculum source for the transcriptome analysis following snow mould infection in controlled conditions (Kovi et al. 2016). According to Hofgaard et al. (2006), highly pathogenic isolates had faster growth on PDA at 2 °C, as well as higher activity of the cell wall-degrading enzyme β-glucosidase than isolates with low pathogenicity. This may indicate that pathogenicity and host specialization of an isolate may be related to cell wall composition of the host and the ability of the isolate to produce specific cell wall degrading enzymes.
The significant isolate x cultivar interaction found in experiment A (8 week incubation) but not in experiment B (9 weeks incubation) indicate that the relative ranking of an isolates’ pathogenicity depend on the host plant and the incubation conditions. A significant isolate x cultivar interaction may be a result of an incubation period which is too short for the isolates to infect the tolerant cultivars (thus, no differences in pathogenicity between isolates are detected), whereas clear differences in pathogenicity may be detected on the more susceptible cultivars. Others have also found that the developmental stage of the plants at the time of inoculation, growth conditions and the duration of incubation can affect the results of this screening method (Hofgaard et al. 2006; Abdelhalim et al. 2016).
The wide host range and high level of genetic diversity possessed by M. nivale relative to M. majus (Mahuku et al. 1998) makes it interesting to explore the relationship between the genetic diversity and the host range of M. nivale isolates, and also to study the relation between genetic diversity and geographic origin. In general fungal specices show high levels of intragenomic heterogeneity and using only one genomic region for phylogenetic studies is not a proper choice (Hibbett et al. 2016; O’Donnell et al. 2015). Thus we used different genomic regions to investigate the sequence diversity among M. nivale and M. majus isolates. The four genic regions used in this study represent the most common gene sequences used to study fungal taxonomy (Hibbett et al. 2016; James et al. 2006; Větrovský et al. 2016). ITS is the most common followed by β-tubulin, EF-1α and RPB2 (Hibbett et al. 2016).
Clear genetic differences between the two snow mould species were detected for all four genomic regions. This result agrees with previous studies by Glynn et al. (2005) and Jewell and Hsiang (2013). Our results do support the proposition by Glynn et al. (2005) that M. nivale and M. majus should be considered as two separate species. However, Jewell and Hsiang (2013) were not able to differentiate between these two fungal species using the ITS sequences. Similarly, in our study the ITS sequences were the least informative of all. The multi-copy nature of the ITS sequence makes it easy to amplify from low-quality DNA, however, it also limits the ability of this genomic region to distinguish between interspecific and intraspecific variation (Hibbett et al. 2016; James et al. 2006; O’Donnell et al. 2015)
To obtain a better understanding of the phylogeographic relationship among M. nivale and M. majus isolates we utilized sequence variation within the protein coding genes RPB2, EF-1α and β-tubulin. Based on the sequences of these genes larger genetic variation was found among M. nivale isolates than among M. majus isolates. This result agrees with previous studies by Maurin et al. (1995), Glynn et al. (2005) and Mahuku et al. (1998). Fungal populations with high levels of genetic diversity, such as M. nivale, are more likely to have a wider host range, and to develop resistance to fungicides faster than species with low genetic diversity (McDonald and McDermott 1993; Walker et al. 2009). Therefore understanding the population structure of this pathogen may have a large impact on snow mould management and grass production.
In order to study the association between geographic origin of the isolates and sequence variation, the gene sequences of RPB2 and β-tubulin from six North American isolates were obtained from NCBI GenBank®. Only RPB2 clustered M. nivale isolates according to their geographic origins; North-American isolates were placed in one cluster with a bootstrap value of 100%, while isolates from Norway and UK formed another cluster. The β-tubulin gene sequences revealed less clear relationship with geographic origin. These results agrees with a study by Jewell and Hsiang (2013) who found that isolates of different geographic origin (Europe vs. North America) were genetically different based on the RPB2 gene sequence, whereas the β-tubulin gene sequence did not reveal the same difference. It will be of great interest to understand when this separation took place and what kind of events caused this separation such as population size, environmental factors or gene fitness.
The Neighbour-Joining phylogenetic tree constructed for the β-tubulin gene sequences, clustered M. nivale isolates according to the host plants they were isolated from (cereals vs. grasses). M. nivale isolates from cereals (except isolate 200,108) formed one cluster with a bootstrap value of 54%, while isolates from grasses (including the cereal isolate 200,108) formed another cluster with a bootstrap value of 50%. M. nivale has a wide host range (Matsumoto and Hsiang 2016), but different isolates show different host preferences (Hofgaard et al. 2006; Matsumoto and Hsiang 2016; Tronsmo et al. 2001). M. nivale isolates from different host plants may constitute specialized strains with restricted gene flow between them. Therefore, further studies are needed to understand the relationship between host preference and genetic variation in M. nivale and M. majus. The good resolution provided by the sequences of the single copy genes RPB2 and β-tubulin can be utilized for detection and quantification of M. nivale and M. majus isolates. For example, Elbelt et al. (2018) used β-tubulin gene sequences from M. nivale and M. majus for detecting and quantification of these species involved in wheat head blight infections.
In conclusion, the result from this study supports the classification of M. nivale and M. majus as two different fungal species. The genotypic and phenotypic (pathogenicity) differences detected between Norwegian M. nivale isolates from cereals and grasses, support the existence of host specialization within M. nivale isolates. Further studies may reveal that the “pathotypes” of this species should be regarded as separate species or subspecies. In conducting screening tests for snow mould resistant cultivars of different grass species, it is therefore important to select isolates of the right “pathotype” of the fungus.
References
Abdelhalim, M., Rognli, O. A., Hofgaard, I. S., Østrem, L., & Tronsmo, A. M. (2016). Snow mould resistance under controlled conditions and winter survival in the field in populations of perennial ryegrass, meadow fescue, and Festulolium are partly dependent on ploidy level and degree of northern adaptation. Canadian Journal of Plant Science, 96(4), 579–589.
Allard, R. W. (1990). The Genetics of Host-Pathogen Coevolution: Implications for Genetic Resource Conservation. J Hered 81:1–6.
Årsvoll, K. (1973). Winter damage in Norwegian grasslands, 1968-1971. Meldinger fra Norges Landbrukshoegskole, 52(3).
Booth, C. (1971). The genus Fusarium. Kew: Commonwealth Mycological Inst.
Diamond, H., & Cooke, B. M. (1997). Host specialisation in Microdochium nivale on cereals. Cereal Research Communications, 25(3), 533–538.
Diamond, H., & Cooke, B. M. (1999). Towards the development of a novel in vitro strategy for early screening of Fusarium ear blight resistance in adult winter wheat plants. European Journal of Plant Pathology, 105(4), 363–372.
Einax, E., & Voigt, K. (2003). Oligonucleotide primers for the universal amplification of β-tubulin genes facilitate phylogenetic analyses in the regnum fungi. Organisms Diversity & Evolution, 3(3), 185–194.
Elbelt, S., Siou, D., Gelisse, S., Cruaud, C., Lannou, C., Lebrun, M.-H., et al. (2018). Optimized real-time qPCR assays for detecting and quantifying the Fusarium and Microdochium species responsible for wheat head blight, as defined by MIQE guidelines. bioRxiv, 272534.
Ergon, Å., Skinnes, H., & Tronsmo, A. M. (2003). Testing snow Mould resistance of winter wheat: Inoculation experiments with Microdochium nivale in the field. Acta Agriculturae Scandinavica, Section B - Soil & Plant Science, 53(3), 110–117.
Fries, E. M. (1825). Systema orbis vegetabilis (Vol. 1): e Typographia academica.
Gams, W. (1989). Chapter 11 - Taxonomy and nomenclature of Microdochium nivale (Fusarium nivale). In: Chełkowski, J. (ed) Fusarium. Elsevier, Amsterdam, 195-198.
Gams, W., & Müller, E. (1980). Conidiogenesis of Fusarium nivale and Rhynchosporium oryzae and its taxonomic implications. Netherlands Journal of Plant Pathology, 86(1), 45–53.
Gerlach, W., & Nirenberg, H. (1982). The genus Fusarium--a pictorial atlas. Mitteilungen aus der Biologischen Bundesanstalt fur Land-und Forstwirtschaft Berlin-Dahlem, 209.
Glynn, N. C., Hare, M. C., Parry, D. W., & Edwards, S. G. (2005). Phylogenetic analysis of EF-1 alpha gene sequences from isolates of Microdochium nivale leads to elevation of varieties majus and nivale to species status. Mycological Research, 109(8), 872–880.
Hibbett, D., Abarenkov, K., Kõljalg, U., Öpik, M., Chai, B., Cole, J., Wang, Q., Crous, P., Robert, V., Helgason, T., Herr, J. R., Kirk, P., Lueschow, S., O'Donnell, K., Nilsson, R. H., Oono, R., Schoch, C., Smyth, C., Walker, D. M., Porras-Alfaro, A., Taylor, J. W., & Geiser, D. M. (2016). Sequence-based classification and identification of Fungi. Mycologia, 108(6), 1049–1068.
Hofgaard, I., Wanner, L., Hageskal, G., Henriksen, B., Klemsdal, S., & Tronsmo, A. (2006). Isolates of Microdochium nivale and M. majus differentiated by pathogenicity on perennial ryegrass (Lolium perenne L.) and in vitro growth at low temperature. Journal of Phytopathology, 154(5), 267–274.
Holmes, S. (1976). A comparative study of the infection of perennial ryegrass by Fusarium nivale and F. culmorum in sterilized soil. Annals of Applied Biology, 84(1), 13–19.
James, T. Y., Kauff, F., Schoch, C. L., Matheny, P. B., Hofstetter, V., Cox, C. J., Celio, G., Gueidan, C., Fraker, E., Miadlikowska, J., Lumbsch, H. T., Rauhut, A., Reeb, V., Arnold, A. E., Amtoft, A., Stajich, J. E., Hosaka, K., Sung, G. H., Johnson, D., O'Rourke, B., Crockett, M., Binder, M., Curtis, J. M., Slot, J. C., Wang, Z., Wilson, A. W., Schüssler, A., Longcore, J. E., O'Donnell, K., Mozley-Standridge, S., Porter, D., Letcher, P. M., Powell, M. J., Taylor, J. W., White, M. M., Griffith, G. W., Davies, D. R., Humber, R. A., Morton, J. B., Sugiyama, J., Rossman, A. Y., Rogers, J. D., Pfister, D. H., Hewitt, D., Hansen, K., Hambleton, S., Shoemaker, R. A., Kohlmeyer, J., Volkmann-Kohlmeyer, B., Spotts, R. A., Serdani, M., Crous, P. W., Hughes, K. W., Matsuura, K., Langer, E., Langer, G., Untereiner, W. A., Lücking, R., Büdel, B., Geiser, D. M., Aptroot, A., Diederich, P., Schmitt, I., Schultz, M., Yahr, R., Hibbett, D. S., Lutzoni, F., McLaughlin, D., Spatafora, J. W., & Vilgalys, R. (2006). Reconstructing the early evolution of Fungi using a six-gene phylogeny. Nature, 443(7113), 818–822.
Jewell, L. E., & Hsiang, T. (2013). Multigene differences between Microdochium nivale and Microdochium majus. Botany, 91(2), 99–106.
Kovi, M. R., Abdelhalim, M., Kunapareddy, A., Ergon, Å., Tronsmo, A. M., Brurberg, M. B., et al. (2016). Global transcriptome changes in perennial ryegrass during early infection by pink snow mould. Scientific Reports, 6.
Larsen, A. (1994). Breeding winter hardy grasses. Euphytica, 77(3), 231–237.
Lees, A., Nicholson, P., Rezanoor, H., & Parry, D. (1995). Analysis of variation within Microdochium nivale from wheat: evidence for a distinct sub-group. Mycological Research, 99(1), 103–109.
Litschko, L., & Burpee, L. (1987). Variation among isolates of Microdochium nivale collected from wheat and turfgrasses. Transactions of the British Mycological Society, 89(2), 252–256.
Liu, Y. J., & Hall, B. D. (2004). Body plan evolution of ascomycetes, as inferred from an RNA polymerase II phylogeny. Proceedings of the National Academy of Sciences of the United States of America, 101(13), 4507–4512.
Liu, Y. J., Whelen, S., & Hall, B. D. (1999). Phylogenetic relationships among ascomycetes: evidence from an RNA polymerse II subunit. Molecular Biology and Evolution, 16(12), 1799–1808.
Mahuku, G. S., Hsiang, T., & Yang, L. (1998). Genetic diversity of Microdochium nivale isolates from turfgrass. Mycological Research, 102(5), 559–567.
Matsumoto, N., & Hsiang, T. (2016). Snow Mold Fungi. In Snow Mold: The Battle under snow between fungal pathogens and their plant hosts (pp. 55–94). Singapore: Springer Singapore.
Maurin, N., Rezanoor, H., Lamkadmi, Z., Somé, A., & Nicholson, P. (1995). A comparison of biological, molecular and enzymatic markers to investigate variability within Microdochium nivale (Fries) Samuels and Hallett. Agronomie, 15(1), 39–47.
McDonald, B. A., & McDermott, J. M. (1993). Population genetics of plant pathogenic fungi. Bioscience, 43(5), 311–319.
Myllys, L., Lohtander, K., & Tehler, A. (2001). β-tubulin, ITS and group I intron sequences challenge the species pair concept in Physcia aipolia and P. caesia. Mycologia, 335–343.
Nicholson, P., Lees, A. K., Maurin, N., Parry, D. W., & Rezanoor, H. N. (1996). Development of a PCR assay to identify and quantify Microdochium nivale var.nivale and Microdochium nivale var.majus in wheat. Physiological and Molecular Plant Pathology, 48(4), 257–271.
Noble, M., & Montgomerie, I. G. (1956). Griphosphaeria nivalis (Schaffnit) Müller & von Arx and Leptosphaeria avenaria weber on oats. Transactions of the British Mycological Society, 39(4), 449–445.
O’Donnell, K., Ward, T. J., Robert, V. A., Crous, P. W., Geiser, D. M., & Kang, S. (2015). DNA sequence-based identification of Fusarium: current status and future directions. Phytoparasitica, 43(5), 583–595.
Parry, D. W., Rezanoor, H. N., Pettitt, T. R., Hare, M. C., & Nicholson, P. (1995). Analysis of Microdochium nivale isolates from wheat in the UK during 1993. Annals of Applied Biology, 126(3), 449–455.
Pavlic, D., Slippers, B., Coutinho, T. A., & Wingfield, M. J. (2009). Molecular and phenotypic characterization of three phylogenetic species discovered within the Neofusicoccum parvum/N. ribis complex. Mycologia, 101(5), 636–647.
Ren, R., Yang, X., & Ray, R. V. (2015). Comparative aggressiveness of Microdochium nivale and M. majus and evaluation of screening methods for Fusarium seedling blight resistance in wheat cultivars. European Journal of Plant Pathology, 141(2), 281–294.
Samuels, G. J., & Hallett, I. C. (1983). Microdochium stoveri and Monographella stoveri, new combinations for Fusarium stoveri and Micronectriella stoveri. Transactions of the British Mycological Society, 81(3), 473–483.
Schoch, C. L., Seifert, K. A., Huhndorf, S., Robert, V., Spouge, J. L., Levesque, C. A., et al. (2012). Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proceedings of the National Academy of Sciences, 109(16), 6241–6246.
Simpson, D., Rezanoor, H., Parry, D., & Nicholson, P. (2000). Evidence for differential host preference in Microdochium nivale var. majus and Microdochium nivale var. nivale. Plant Pathology, 49(2), 261–268.
Smith, J. D. (1983). Fusarium nivale(Gerlachia nivalis) from cereals and grasses: Is it the same fungus? Canadian Plant Disease Survey, 63(1), 25–26.
Tronsmo, A. M., Hsiang, T., Okuyama, H., & Nakajima, T. (2001). Low temperature diseases caused by Microdochium nivale (pp. 75–86). Sapporo: Low temperature plant microbe interactions under snow. Hokkaido National Agricultural Experiment Station.
Větrovský, T., Kolařík, M., Žifčáková, L., Zelenka, T., & Baldrian, P. (2016). The rpb2 gene represents a viable alternative molecular marker for the analysis of environmental fungal communities. Molecular Ecology Resources, 16(2), 388–401.
Walker, A. S., Auclair, C., Gredt, M., & Leroux, P. (2009). First occurrence of resistance to strobilurin fungicides in Microdochium nivale and Microdochium majus from French naturally infected wheat grains. Pest Management Science, 65(8), 906–915.
White, T. J., Bruns, T., Lee, S., & Taylor, J. (1990). Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protocols: a Guide to Methods and Applications, 18, 315–322.
Wollenweber, H. W. (1930). Fusaria autographice delineata. Selbstverlag, Berlin. Tafel Nr. 969.
Acknowledgements
Elisa Gauslaa, Øyvind Jørgensen and Monica Skogen are acknowledged for their excellent technical assistance.
Funding
Open Access funding provided by Norwegian University of Life Sciences. This work was funded by the Research Council of Norway project ‘VARCLIM: Understanding the genetic and physiological basis for adaptation of Norwegian perennial forage crops to future climates’ (project no. 199664). Mohamed Abdelhalim was funded by a PhD scholarship from the Norwegian University of Life Sciences.
Author information
Authors and Affiliations
Contributions
MBB, ISH and AMT conceived and designed the experiments. MA performed the experiments in collaboration with MBB, ISH and AMT. OAR helped with the statistical analyses. MA drafted the manuscript with inputs from all co-authors. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
ESM 1
(DOCX 13 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abdelhalim, M., Brurberg, M.B., Hofgaard, I.S. et al. Pathogenicity, host specificity and genetic diversity in Norwegian isolates of Microdochium nivale and Microdochium majus. Eur J Plant Pathol 156, 885–895 (2020). https://doi.org/10.1007/s10658-020-01939-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10658-020-01939-5