
Introduction
Fusarium ranks as one of the world’s most economically destructive and species-rich groups of mycotoxigenic plant pathogens (Aoki et al. 2014). These ubiquitous molds produce a plethora of toxic secondary metabolites, such as trichothecenes, zearalenone, fumonisins, and enniatins, which pose a significant threat to agricultural biosecurity, food safety, and plant, human and animal health (Marasas et al. 1984). Fusarial-induced diseases of virtually every economically important plant cost the global agricultural economy multi-billion euro losses annually. Moreover, phylogenetically diverse fusaria, including plant pathogens (Short et al. 2011), cause infections in humans, with those involving the cornea and nails being the most common (Chang et al. 2006 and references therein). Because fusaria are broadly resistant to the spectrum of antifungals currently available, disseminated infections in patients who are artificially immunosuppressed or immunocompromised and severely neutropenic are typically fatal (Balajee et al. 2009). The likely reservoir of nosocomial fusarioses is the plumbing system, which has been shown to harbor the most common human opportunistic fusaria (Kuchar 1996; Short et al. 2011). Accurate identification of the etiological and/or toxigenic agent is central to disease management and infection control (Wingfield et al. 2012). Thus, the primary focus of this mini-review is to provide a contemporary guide to the following three web-accessible resources for DNA sequence-based identification of Fusarium: FUSARIUM-ID (http://isolate.fusariumdb.org/; Geiser et al. 2004; Park et al. 2010), Fusarium MLST (http://www.cbs.knaw.nl/fusarium/; O’Donnell et al. 2010), and NCBI GenBank (http://www.ncbi.nlm.nih.gov/). The following brief overview of Fusarium phylogenetic diversity is provided as background information for the sections on DNA sequence-based identification.
Phylogenetic limits, subgeneric clades and species recognition
Fusarium Link (Hypocreales, Nectriaceae) was circumscribed originally in 1809 based on the production of its iconic fusiform multiseptate macroconidia. However, we now know from morphological and molecular phylogenetic studies (Gräfenhan et al. 2011) that this character evolved convergently in different lineages of ascomycetes, and that it has been lost at least once within the F. solani species complex (e.g., F. neocosmosporiellum and related self-fertile species formerly classified in Neocosmospora; O’Donnell et al. 2013). As a result, Fusarium is non-monophyletic in the three mostly widely used morphology-based taxonomic treatments (Nelson et al. 1983; Gerlach and Nirenberg 1982; Leslie and Summerell 2006) that recognize 30, 78 and 70 species, respectively. With the exception of the turf grass pathogen ‘Fusarium’ nivale, which is now recognized as Microdochium nivale within the distantly related order Xylariales (Samuels and Hallet 1983), and ‘Fusarium’ tabacinum, which was reclassified as Monographella cucumerina in the Xylariales (Palm et al. 1995), the other taxa removed from Fusarium and reclassified in four different genera within the Nectriaceae by Gräfenhan et al. (2011) are not known to be pathogenic or mycotoxigenic. Fortunately, the molecular phylogenetic circumscription of a monophyletic Fusarium that includes at least 20 clades referred to as species complexes and nine monotypic lineages (Fig. 1) has received overwhelming support by the Fusarium research community worldwide (Geiser et al. 2013). Given the poor correspondence between the robust RPB1/RPB2 molecular phylogeny and the morphology-based sectional classification adopted by Gerlach and Nirenberg (1982) and Nelson et al. (1983), the nonmonophyletic sections were abandoned in favor of strongly supported, monophyletic species complexes (Geiser et al. 2013; O’Donnell et al. 2013). Following the demise of dual nomenclature 1 January 2013 under the newly named International Code of Nomenclature for Algae, Fungi and Plants (Hawksworth et al. 2011), plant pathologists and other applied biologists are encouraged to only use the Fusarium name (Geiser et al. 2013). Fusarium has priority over the teleomorphs (i.e., Albonectria, Cyanonectria, Gibberella, Haematonectira, Nectria and Neocosmospora), and the anamorphs Bisfusarium and Rectifusarium, which were recently proposed, respectively, for members of the F. dimerum (FDSC) and F. ventricosum (FVSC) species complexes (Lombard et al. 2015). Comparative phylogenomic analyses of low-coverage genomes of the 93 fusaria included in O’Donnell et al. (2013) are in progress to more critically evaluate the circumscription of Fusarium (J. Stajich, pers. commun.).

Diagrammatic representation of Fusarium phylogeny inferred from a combined RPB1 + RPB2 dataset (3383 bp) rooted on sequences of Neonectria and Ilyonectria (modified from Fig. 1 in O’Donnell et al. 2013). GCPSR-based analyses indicate that the genus comprises at least 300 phylogenetically distinct species, 20 species complexes (highlighted in gray), and 9 monotypic lineages. The approximate number of phylogenetically distinct species within each species complex is indicated
Although Fusarium as circumscribed morphologically by Gerlach and Nirenberg (1982) has shrunk in size by approximately 20 species, largely due to the seminal study by Gräfenhan et al. (2011), application of phylogenetic species recognition based on genealogical concordance and non-discordance over the past 20 years (GCPSR sensu Taylor et al. 2000; Dettman et al. 2003) has resulted in its explosive growth. Today at least 300 phylogenetically distinct species have been resolved as genealogically exclusive lineages based on phylogenetic analyses of representative fusaria in the ARS Culture Collection (NRRL), the CBS-KNAW Biodiversity Centre (CBS) and the Fusarium Research Center (FRC). However, the majority of these species are unnamed and many of these are morphologically cryptic (Fig. 1). Four complementary but distinct technological and theoretical advances have been key to the greatly accelerated species discovery within Fusarium over the past two decades. These include the marriage of PCR and automated DNA sequencing in the early 1990s, the acceptance of GCPSR-based studies as the gold standard for species recognition within the Fusarium community, the wealth of fusaria (~40,000 isolates) accessioned in publically accessible culture collections (e.g., CBS, FRC and NRRL), and a highly collaborative global phytopathological community.
Based on biodiversity studies that estimate the ~100,000 named and accepted fungi only comprise one-tenth (Hawksworth 2001) to one-fiftieth (Blackwell 2011) of the species in nature, coupled with the fact that most of the fusaria studied to date were isolated from agronomically important plants (i.e., <1% of all vascular plants; Simpson and Ogorzaly 1995) cultivated on a fraction of our planet’s surface, it is reasonable to suggest that the number of novel fusaria in nature could exceed our current estimate of 300 by an order of magnitude. Looking to the future, typing schemes will need to take advantage of next-generation sequencing (NGS) technology (Boers et al. 2012) to rise to the challenge posed by the anticipated seismic shift in species discovery within Fusarium and other agriculturally important plant pathogens. This discovery will be driven in part by metagenomics studies (LeBlanc et al. 2014), surveys of endophytes inhabiting endemics in biogeographically interesting areas (Walsh et al. 2010), and by the inexorable introduction of novel pathogens into nonindigenous areas by the globalization of world trade (Fisher et al. 2012). Much is at stake because an accurate species-level identification is essential for elucidating and communicating all facets of a pathogens’ biology (Wingfield et al. 2012).
Sequence-based identification of fusaria: Some loci reveal while others conceal
Only three of the marker loci tested to date meet three important criteria for phylogenetic species recognition in that they are: 1) applicable across the phylogenetic breadth of Fusarium (Fig. 1), 2) informative at or near the species-level, and 3) orthologous across the genus. These are: translation elongation factor 1-α (TEF1), DNA-directed RNA polymerase II largest (RPB1) and second largest subunit (RPB2). The latter two marker loci were developed as part of the NSF-funded Deep Hyphae and the Assembling the Fungal Tree of Life (AFTOL) projects (Lutzoni et al. 2004; James et al. 2006). In contrast to TEF1, whose highly variable introns can only be aligned reliably across members of a species complex, or several closely related ones, the portions of RPB1 and RPB2 sequenced can easily be aligned across Fusarium (O’Donnell et al. 2013). While all three genes are included in our ongoing GCPSR-based studies that span the breadth of the genus, sequence data from only one of these loci is needed to obtain a reasonably accurate placement of an unknown within a species complex by conducting a nucleotide BLAST query of FUSARIUM-ID and/or Fusarium MLST (Fig. 2A-B), or by phylogenetic analysis (see Bruns et al. 1998 for an example). As discussed below, we also conduct BLASTn queries of GenBank, but these require careful scrutiny of the top ‘hits’ because: 1) many sequences in NCBI are misidentified (Bidartondo et al. 2008; Kang et al. 2010), 2) sequences deposited in NCBI for the majority of newly discovered fusaria employing GCPSR lack binomials, and 3) the taxonomy for many records is out of date (Fig. 3). Because most of the fusaria within the F. solani and F. incarnatum-equiseti species complexes lack binomials, an ad hoc species/multilocus haplotype nomenclature was adopted to allow for accurate communication of information regarding these pathogens within the scientific community (O’Donnell et al. 2008, 2009b).

(A) FUSARIUM-ID (http://isolate.fusariumdb.org) at Pennsylvania State University (Park et al. 2010) and (B) Fusarium MLST at the CBS-KNAW Fungal Biodiversity Centre (http://www.cbs.knaw.nl/Fusarium/) host two complementary web-accessible databases dedicated to the identification of fusaria via nucleotide BLAST queries. Conducting the same BLASTn queries of GenBank (http://blast.ncbi.nlm.nih.gov/Blast.cgi) can be informative, but also challenging given the large number of misidentified sequences in NCBI (see Fig. 3)

Twenty-one of the 100 BLASTn ‘hits’ that were recovered from GenBank using accession JN014954.1 as the query. Black boxes were added to the GenBank query results to identify sequences deposited under 11 different names. The black star inserted by the record for Fusarium sp. NRRL 22244 indicates the accession was identified in the notes/comments as phylogenetic species FIESC 25 (O’Donnell et al. 2009b)
At the suggestion of Christopher L. Schardl, University of Kentucky (Tsai et al. 1994), β-tubulin was the first protein-encoding gene that we used for molecular phylogenetics in Fusarium (O’Donnell and Cigelnik 1997; O’Donnell et al. 1998a). However, we discovered that its utility is limited due to the presence of divergent paralogs within the F. solani, F. incarnatum-equiseti, and F. chlamydosporum species complexes. After Stephen A. Rehner (ARS-USDA, Beltsville, MD) called our attention to the utility of TEF1 for species-level studies in insects (Cho et al. 1995), we developed degenerate PCR primers that amplify the intron-rich 5` end of this ortholog in all fusaria (O’Donnell et al. 1998b), and we have used this sequence data to populate FUSARIUM-ID (Geiser et al. 2004; Park et al. 2010) and Fusarium MLST (O’Donnell et al. 2010). Although the internal transcribed spacer (ITS) region of the nuclear ribosomal DNA was selected as the official ‘barcode’ locus for the Fungi (Schoch et al. 2012), its utility within Fusarium, and many other groups of fungi (Bruns 2001; Du et al. 2012; Gazis et al. 2011), is limited by the fact that it is often uninformative at the species-level, and like TEF1, it can only be aligned reliably across members of a species complex or several closely related ones. In recently evolved phytopathologically-important lineages such as the F. graminearum (Sarver et al. 2011), F. oxysporum (O’Donnell et al. 2009a) and F. fujikuroi species complexes (O’Donnell et al. 1998a), many of the species share identical or nearly ITS rDNA alleles. Using the ITS rDNA for species identification and phylogenetic inference in Fusarium is further complicated by the presence of highly divergent ITS2 rDNA paralogs or xenologs within every strain tested within six closely related species complexes (i.e., concolor-to-fujikuroi in Fig. 1; O’Donnell et al. 1998a). In addition, we recently detected this phenomenon within the F. buharicum species complex, which suggests that the gene duplication or horizontal gene transfer event took place at least 49 million years ago (O’Donnell et al. 2013).
ITS rDNA and domains D1 and D2 at the 5` end of the ribosomal large subunit (LSU rDNA) were used in GCPSR-based MLST schemes for the F. solani (O’Donnell et al. 2008), F. dimerum (Schroers et al. 2009), F. incarnatum-equiseti (FIESC) and F. chlamydosporum species complexes (O’Donnell et al. 2009b), but this locus consistently contributed the least number of phylogenetically informative characters among the loci sampled. For example, in the FIESC study, 162/717 (23%) of the aligned nucleotide positions in TEF1 and 220/1766 (12%) in RPB2 were parsimony informative; however, only 11/1125 (1%) were synapomorphic within the ITS+LSU rDNA (O’Donnell et al. 2009b). In hindsight, inclusion of the fungal ‘barcode’ locus in this typing scheme was a mistake. However, we should note that in the numerous instances where ITS+LSU rDNA sequence data did not resolve at the species level, it does have utility for placing unidentified fusaria within a species complex (Balajee et al. 2009).
Currently we are generating low-coverage genome sequences for the 93 fusaria included in our published molecular phylogeny of Fusarium (O’Donnell et al. 2013) to mine them for additional phylogenetically informative loci for species-level studies (López-Giráldez and Townsend 2011) and for comparative phylogenomics (Stajich, unpubl.). This and other whole genome sequence data will be critical for developing NGS typing schemes necessary to characterize the deluge of novel species that will be discovered by phytopathological, biogeographical and metagenomic studies in the future. The newly developed marker loci should prove to be invaluable in expanding the ad hoc species-haplotype nomenclature (O’Donnell et al. 2008, 2009a, b) to all agriculturally and medically important fusaria so that information concerning the unnamed, morphologically cryptic mycotoxigenic plant and human pathogens can be accurately communicated within the scientific community. This informal naming system is also useful for identifying agriculturally and medically important species that should be formally described with Latin binomials.
Sequence-based identification of fusaria: A primer for conducting BLASTn queries via the Internet
Querying one of the web-accessible databases using partial DNA sequence data from TEF1, RPB1 and/or RPB2 to identify an unknown ordinarily is the easy part; however, interpreting the results is often challenging. As previously discussed in detail (Geiser et al. 2004; O’Donnell et al. 2010), the advantage of conducting nucleotide BLAST queries of FUSARIUM-ID or Fusarium MLST first, rather than NCBI GenBank, is that they house broadly sampled, well-characterized phylogenetically informative sequences from isolates that can be obtained from FRC (http://plantpath.psu.edu/facilities/fusarium-research-center), the ARS Culture Collection (NRRL, http://nrrl.ncaur.usda.gov/cgi-bin/usda) or the CBS-KNAW Biodiversity Centre (http://www.cbs.knaw.nl/Fusarium/).
When conducting BLASTn queries of the CBS-KNAW’s Fusarium MLST database, we recommend only searching the reference files for Fusarium, which is the default setting, and setting the ‘Minimum similarity to keep results’ to 50% (Fig. 2B). Most of the sequences housed in FUSARIUM-ID are also present in Fusarium MLST, so a query of only one of these databases may be needed. Once the results of a BLASTn query of Fusarium MLST are returned, with the top ‘hits’ displayed in tabular form, click on Expand Alignments to see the alignment of your query sequence with each of the reference sequences retrieved from the database. Results obtained from BLASTn queries of GenBank and FUSARIUM-ID differ slightly in that horizontal red lines are used to identify sequence length of the top ‘hits’, which span the length of the region if the sequences are full-length (see % query cover in Fig. 3). GenBank differs from FUSARIUM-ID and Fusarium MLST in that all of the top BLASTn hits are displayed together in tabular form immediately below the horizontal red lines, followed by the alignment of your query sequence with each of the reference sequences that were recovered (Fig. 4). All three databases provide a hyperlink to each accession record, a description that includes the taxon name that the accession was deposited under and the locus. Also included are several statistical measures, with the two most important ones being percentage identity and query coverage. Using the loci mentioned herein, the E-value for each ‘hit’ should be zero (Fig. 3), which means the match is not due to chance.

One of the 100 BLASTn nucleotide alignments that followed the tabular display shown in Fig. 3. Alignment of ITS rDNA GenBank accession JN014954.1 used as the query and GQ505685.1 NRRL 22244 FIESC 25-a strongly suggests that the query sequence contains three sequencing errors (indicated by arrows). Alignments such as this are ideal for identifying nucleotide positions within chromatograms that should be checked for errors before sequences are analyzed and deposited in NCBI
After conducting a query of Fusarium MLST or FUSARIUM-ID, we frequently use the same nucleotide sequence to query GenBank, bearing in mind that many sequences in NCBI are misidentified, others are deposited as Fusarium sp. without further annotation, and the taxonomy especially for older records may be out of date (e.g., GenBank accession AF178356.1 was deposited as F. solani f. sp. glycines in 1999, a year before this soybean pathogen was formally described as F. virguliforme). For those who opt to query GenBank, we recommend the use of TEF1, RPB1 and/or RPB2 sequences, rather than sequences from the ITS or LSU rDNA, and look for sequences obtained from NRRL strains among the top ‘hits’. The majority of the sequences we deposited in these databases were listed as Fusarium sp., because they represent unnamed species based on the results of several GCPSR-based studies (O’Donnell et al. 2010, 2014 and references therein). However, notes or comments were often included in the accession records to help identify the phylogenetic species and/or multilocus haplotype (Fig. 5; i.e., Haplotype=”FSSC 11-b” from O’Donnell et al. 2008). Another tip for correctly interpreting BLASTn results is to check the taxon names. Identical or nearly identical sequences deposited under multiple names, as illustrated in Figs. 3 and 6, should raise suspicions that some sequences are misidentified. In addition, sorting through the display of BLASTn ‘hits’ in some searches is complicated by the legacy of dual nomenclature (e.g., Gibberella zeae and F. graminearum for the same species).


Seven of the 100 BLASTn ‘hits’ from a search of NCBI using a partial TEF1 sequence of GenBank accession FJ939721.1 (deposited as F. venenatum) as the query. However, the relatively low 97% similarity to accession GQ915515.1, which was obtained from an authentic isolate of F. venenatum (FRC R-9186), likely indicates the query sequence is from a novel species within the F. sambucinum species complex (see Fig. 1). The latter appears to be conspecific with DAOM 167768 accession DQ842081.1, which was incorrectly deposited in GenBank as F. equiseti. This finding suggests that if an emerencia-like system (Nilsson et al. 2005) was constructed to mine Fusarium TEF1, RPB1 and RPB2 sequences in GenBank, it could prove to be an invaluable tool for discovering many novel phylogenetically distinct species
We don’t recommend using sequences from the nuclear ITS rDNA and/or domains D1+D2 of the LSU rDNA from an unknown to query GenBank because 50% or more of the Fusarium sequences from this locus are misidentified in NCBI. Besides, as previously mentioned, ITS+LSU rDNA sequences are too conserved to resolve species limits of most fusaria. The 10 steps outlined below are recommended to increase the likelihood of obtaining an accurate species- or species complex-level identification of an unknown Fusarium (also see Fig. 7). Here, we used a study by Suthar and Bhatt (2011) to illustrate common mistakes/errors associated with species identification based on BLASTn queries and how to avoid them. They deposited their ITS rDNA sequence of a putative cumin wilt pathogen (NFCCI 2157) in GenBank incorrectly as F. equiseti (accession JN014954.1). Of the 100 ITS rDNA sequences that were recovered from GenBank, using their accession JN014954.1 as the BLASTn query, we determined that all 48 sequences deposited with binomials were incorrectly identified to the species level. This finding highlights the importance of carefully inspecting the name attached to each of the records (Fig. 3) and consulting the primary literature (O’Donnell et al. 2009b). This simple but critical step would have revealed to these authors that their queries of GenBank and FUSARIUM-ID yielded contradictory results. A cursory inspection of the 48 named accessions retrieved by the BLASTn query of GenBank revealed that they were incorrectly deposited under seven different species names (Fig. 3), representing fusaria that are nested within six phylogenetically divergent species complexes (Fig. 1; i.e., incarnatum-equiseti, chlamydosporum, sambucinum, tricinctum, fujikuroi and oxysporum), and as an unrelated fungus, Septogloeum mori.

Flowchart outlining 10 steps that are recommended to increase the likelihood of obtaining an accurate DNA sequence-based species- or species complex-level identification of an unknown Fusarium
Sequence-based identification of fusaria: 10 simple steps to increase your odds of obtaining an accurate species-level identification
-
1)
Carefully check sequence chromatograms for errors before conducting a BLASTn query (also check problematic nucleotide positions at step 4 if necessary). Based on a detailed GCPSR-based study of the F. incarnatum-equiseti species complex (O’Donnell et al. 2009b), it seems likely that GenBank accession JN014954.1 contains three sequencing errors (Fig. 4); errors are most commonly found at either end of the sequence and within homopolymers.
-
2)
Avoid using ITS+LSU rDNA sequences to identify unknown fusaria because, compared with TEF1, RPB1 and RPB2, they are frequently uninformative at the species-level. However, we do plan to deposit ITS+LSU rDNA sequences of the 93 fusaria included in a robust phylogeny of the genus (O’Donnell et al. 2013) in FUSARIUM-ID, Fusarium MLST and GenBank to facilitate identification to species complex, and in some instances, to several phylogenetically divergent species (Fig. 1; Balajee et al. 2009).
-
3)
Avoid querying GenBank with ITS+LSU rDNA sequences because the majority of named sequences are misidentified. When GenBank ITS rDNA accession JN014954.1 was used to query FUSARIUM-ID, it showed 99.78% identity to sequences of four different species within the F. incarnatum-equiseti species complex (i.e., FIESC 15, 17, 23 and 25). Instead of depositing the sequence as Fusarium sp., which would have been the only correct identification based on the ITS rDNA sequence data, it was deposited under the name attached to the majority of the named sequences retrieved in their BLASTn query of GenBank (i.e., F. equiseti). Our search of the NCBI GenBank database, using accession JN014954.1 as the query, found that the 48 accessions with binomials were all misidentified, including 28 deposited as F. equiseti. The real F. equiseti, however, corresponds to phylogenetic species FIESC 14 (O’Donnell et al. 2009b).
-
4)
Nucleotide polymorphisms or gaps in the alignments displayed after a BLASTn query should always be confirmed by rechecking the chromatograms. The available evidence suggests that the three mismatches in the ITS rDNA alignment of GenBank accessions JN014954.1 and GQ505685.1 NRRL 22244 FIESC 25 (Fig. 4) are due to sequencing errors in the former.
-
5)
Carefully check the taxon names associated with the top hits (i.e., % identity and sequence coverage), who deposited the sequence, and look for notes/comments included in the accession record (Fig. 5). Be prepared to scrutinize multiple names among the top ‘hits’ in BLASTn queries of GenBank. The example provided by GenBank accession JN014954.1 might seem extreme (i.e., all 48 ITS rDNA sequences with binomials were misidentified); however, it serves to highlight an intractable problem created by open deposit of uncurated sequences without allowing third party annotation (Bidartondo et al. 2008). By contrast, sequences deposited in FUSARIUM-ID and Fusarium MLST were generated primarily as part of GCPSR-based studies of Fusarium. When conducting queries of GenBank, it is good practice to look for NRRL strains among the top hits, and if they were deposited as Fusarium sp., then look for notes/comments in the accession records.
-
6)
It is prudent to compare BLASTn results from multiple loci where possible; the identification tools built into Fusarium MLST allow for queries using single or multiple sequences. Generating a partial TEF1 sequence is an excellent place to start because both FUSARIUM-ID and Fusarium MLST are well-populated with data from this locus and because it frequently resolves at the species level. Where possible, it is highly desirable to try to identify unknowns by conducting molecular phylogenetic analyses of published MLST datasets to which the unknowns have been added; MLST datasets can be downloaded from FUSARIUM-ID or Fusarium MLST. Bootstrap analyses of the individual and combined dataset often yield the most reliable identification of unknowns, when the criteria of genealogical exclusivity and non-discordance under GCPSR are employed (Taylor et al. 2000; Dettman et al. 2003).
-
7)
The results of prior GCPSR-based studies provide invaluable guidance in interpreting when a given % similarity equates with conspecificity. For the following interpretations to hold true, we assume that carefully edited TEF1, RPB1 and/or RPB2 sequences are being used as a query against FUSARIUM-ID and/or Fusarium MLST. As discussed previously (Geiser et al. 2004; O’Donnell et al. 2012), an identical match with 99-100% sequence coverage (Fig. 3), in most but not all cases can be interpreted as a definitive species identification. However, it is important to note that our GCPSR-based studies have shown that sequences from these genes sometime fail to distinguish recently evolved sister species (Kasson et al. 2013; Sarver et al. 2011; Schroers et al. 2009). Queries that show similarity at or below 99.4% (i.e., ≥4/680 nucleotide differences in TEF1) should be subjected to a GCPSR analysis, given the likelihood the unknown represents a novel phylogenetic species (Fig. 6). To increase confidence in TEF1 queries that show 1-3 base pair differences from the top ‘hit’ in FUSARIUM-ID and Fusarium MLST, it is advisable to conduct similar queries using RPB1 and/or RPB2 together with bootstrapped phylogenetic analyses as outlined in step 6. To increase the taxonomic representation of fusaria in agronomically important species complexes that haven’t been subjected to GCPSR, and to promote phylogenetic species recognition, we plan to update FUSARIUM-ID and Fusarium MLST by depositing several hundred TEF1, RPB1 and RPB2 unpublished sequences from the F. sambucinum, F. tricinctum and F. lateritium species complexes (Fig. 1).
-
8)
The primary literature should be consulted to take advantage of the ad hoc species and/or species-haplotype nomenclature developed for the F. incarnatum-equiseti and F. chlamydosporum (O’Donnell et al. 2009b), F. solani (O’Donnell et al. 2008), F. oxysporum (O’Donnell et al. 2009a) species complexes and Ambrosia Fusarium Clade (AFC; Kasson et al. 2013; O’Donnell et al. 2014). The AFC represents a monophyletic lineage within clade 3 of the F. solani species complex that comprises at least 12 phylogenetically distinct mutualistic symbionts of the fungus-farming ambrosia beetle Euwallacea (Fig. 8).
Fig. 8 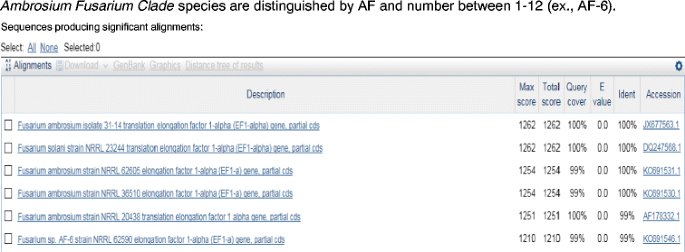
Because binomials for 11/12 species farmed by Euwallacea ambrosia beetles were lacking, an informal nomenclature was adopted for the 12 phylogenetically distinct species within the Ambrosia Fusarium Clade (AFC). The latter is nested within clade 3 of the F. solani species complex (Kasson et al. 2013; O’Donnell et al. 2014). AFC species are distinguished by AF followed by a unique number between 1 and 12 (see AF-6 below). Although a partial TEF1 sequence can be used to identify most of the species within the AFC, multilocus sequence data is recommended for a definitive identification because several species share an identical TEF1 allele, and because hybrid introgression involving this locus was detected
-
9)
When questions concerning the metadata or availability of strains arise, contact Kerry O’Donnell at kerry.odonnell@ars.usda.gov and the curators of FUSARIUM-ID (David M. Geiser, dmg17@psu.edu) or Fusarium MLST (Vincent Robert, v.robert@cbs.knaw.nl) for clarification.
-
10)
Publish in journals where submissions are more likely to be reviewed by those knowledgeable in the pitfalls associated with sequence-based identification. Here, it is worth reiterating the point made in Wingfield et al. (2012) that an accurate species identification is crucial for communicating findings to the scientific community.

Although Fusarium molecular phylogenetics and systematics has been characterized as a model (Kang et al. 2010), species limits and evolutionary relationships within several of the most important plant pathogenic lineages (i.e., F. fujikuroi, F. oxysporum, F. solani and F. sambucinum species complexes) are not fully resolved, especially within the most recently evolved clades (O’Donnell et al. 2013). As mentioned earlier, one of the primary objectives for generating genome-scale data across the breadth of Fusarium is to mine it for additional phylogenetically informative loci for NGS-based species-level studies. In the not too distant future, our goal is to identify additional genes that are as informative as TEF1, RPB1 and RPB2 to further Fusarium identification, molecular diagnostics and robust phylogenetic inference.
References
Aoki, T., O’Donnell, K., & Geiser, D. M. (2014). Systematics of key phytopathogenic Fusarium species: current status and future challenges. Journal of General Plant Pathology, 80, 189–201.
Balajee, S. A., Borman, A. M., Brandt, M. E., Cano, J., Cuenca-Estrella, M., Dannaoui, E., et al. (2009). Sequence-based identification of Aspergillus, Fusarium, and Mucorales species in the clinical mycology laboratory: Where are we and where should we go from here? Journal of Clinical Microbiology, 47, 877–884.
Bidartondo, M. I., Bruns, T. D., Blackwell, M., Edwards, I., Taylor, A. F. S., Horton, T., et al. (2008). Preserving accuracy in GenBank. Science, 319, 1616.
Blackwell, M. (2011). The Fungi: 1, 2, 3 … 5.1 million species? American Journal of Botany, 98, 926–938.
Boers, S. A., van der Reijden, W. A., & Jansen, R. (2012). High-throughput multilocus sequence typing: bringing molecular typing to the next level. PLoS ONE, 7, e39630.
Bruns, T. D. (2001). ITS reality. Inoculum, 52, 2–3.
Bruns, T. D., Szaro, T. M., Gardes, M., Cullings, K. W., Pan, J. J., Taylor, D. L., et al. (1998). A sequence database for the identification of ectomycorrhizal basidiomycetes by phylogenetic analysis. Molecular Ecology, 7, 257–272.
Chang, D. C., Grant, G. B., O’Donnell, K., Wannemuehler, K. A., Noble-Wang, J., Rao, C. Y., et al. (2006). Multistate outbreak of Fusarium keratitis associated with use of a contact lens solution. Journal of the American Medical Association, 296, 953–963.
Cho, S., Mitchell, A., Regier, J. C., Mitter, C., Poole, R. W., Friedlander, T. P., et al. (1995). A highly conserved nuclear gene for low-level phylogenetics: elongation factor-1α recovers morphology-based tree for heliothine moths. Molecular Biology and Evolution, 12, 650–656.
Dettman, J. R., Jacobson, D. J., & Taylor, J. W. (2003). A multilocus genealogical approach to phylogenetic species recognition in the model eukaryote Neurospora. Evolution, 57, 2703–2720.
Du, X.-H., Zhao, Q., Yang, Z. L., Hansen, K., Taşkın, H., Büyükalaca, S., et al. (2012). How well do ITS rDNA sequences differentiate species of true morels (Morchella)? Mycologia, 104, 1351–1368.
Fisher, M. C., Henk, D. A., Briggs, C. J., Brownstein, J. S., Madoff, L. C., McCraw, S. L., et al. (2012). Emerging fungal threats to animal, plant and ecosystem health. Nature, 484, 186–194.
Gazis, R., Rehner, S., & Chaverri, P. (2011). Species delimitation of fungal endophyte diversity studies and its implications in ecological and biogeographic inferences. Molecular Ecology, 20, 3001–3013.
Geiser, D. M., Aoki, T., Bacon, C. W., Baker, S. E., Bhattacharyya, M. K., Brandt, M. E., et al. (2013). LETTER TO THE EDITOR: One fungus, one name: Defining the genus Fusarium in a scientifically robust way that preserves longstanding use. Phytopathology, 103, 400–408.
Geiser, D. M., del Mar Jiménez-Gasco, M., Kang, S., Makalowska, I., Veeraraghavan, N., Ward, T. J., et al. (2004). FUSARIUM-ID v. 1.0: a DNA sequence database for identifying Fusarium. European Journal of Plant Pathology, 110, 473–479.
Gerlach, W., & Nirenberg, H. (1982). The genus Fusarium – a pictorial atlas. Mitteilungen aus der Biologischen Bundesanstalt für Land- und Forstwirtschaft Berlin-Dahlem, 209, 1–406.
Gräfenhan, T., Schroers, H.-J., Nirenberg, H. I., & Seifert, K. A. (2011). An overview of the taxonomy, phylogeny and typification of nectriaceous fungi in Cosmospora, Acremonium, Fusarium, Stilbella and Volutella. Studies in Mycology, 68, 79–113.
Hawksworth, D. L. (2001). The magnitude of fungal diversity: the 1.5 million species estimate revisited. Mycological Research, 105, 1422–1432.
Hawksworth, D. L., Crous, P. W., Redhead, S. A., Reynolds, D. R., Samson, R. A., Seifert, K. A., et al. (2011). The Amsterdam Declaration on fungal nomenclature. IMA Fungus, 2, 105–112.
James, T. Y., Kauff, F., Schoch, C. L., Matheny, P. B., Hofstetter, V., Cox, C. J., et al. (2006). Reconstructing the early evolution of Fungi using a six-gene phylogeny. Nature, 443, 818–822.
Kang, S., Mansfield, M. A., Park, B., Geiser, D. M., Ivors, K. L., Coffey, M. D., et al. (2010). The promise and pitfalls of sequence-based identification of plant-pathogenic fungi and oomycetes. Phytopathology, 100, 732–737.
Kasson, M. T., O’Donnell, K., Rooney, A. P., Sink, S., Ploetz, R. C., Ploetz, J. N., et al. (2013). An inordinate fondness for Fusarium: Phylogenetic diversity of fusaria cultivated by ambrosia beetles in the genus Euwallacea on avocado and other plant hosts. Fungal Genetics and Biology, 56, 147–157.
Kuchar, R. T. (1996). Isolation of Fusarium from hospital plumbing fixtures: implications for environmental health and patient care. M.S. thesis. The University of Texas Health Science Center, Houston, TX.
LeBlanc, N., Kinkel, L. L., & Kistler, H. C. (2014). Soil fungal communities respond to grassland plant community richness and soil edaphics. Microbial Ecology, 70, 188--95. doi:10.1007/s00248-014-0531-1.
Leslie, J. F., & Summerell, B. A. (2006). The Fusarium Laboratory Manual. Ames, Iowa, USA: Blackwell Publishing.
Lombard, L., van der Merwe, N. A., Groenewald, J. Z., & Crous, P. W. (2015). Generic concepts in Nectriaceae. Studies in Mycology, 80, 189–245.
López-Giráldez, F., & Townsend, J. P. (2011). PhyDesign: an online application for profiling phylogenetic informativeness. BMC Evolutionary Biology, 11, 152.
Lutzoni, F., Kauff, F., Cox, C. J., McLaughlin, D., Celio, G., Dentinger, B., et al. (2004). Assembling the fungal tree of life: progress, classification, and evolution of subcellular traits. American Journal of Botany, 91, 1446–1480.
Marasas, W. F. O., Nelson, P. E., & Toussoun, T. A. (1984). Toxigenic Fusarium Species. Identity and Mycotoxicology. University Park, Pennsylvania, USA: The Pennsylvania State University Press.
Nelson, P. E., Tousson, T. A., & Marasas, W. F. O. (1983). Fusarium species: An illustrated manual for identification. University Park, Pennsylvania, USA: The Pennsylvania State University Press.
Nilsson, R. H., Kristiansson, E., Ryberg, M., & Larsson, K.-H. (2005). Approaching the taxonomic affiliation of unidentified sequences in public databases – an example from the mycorrhizal fungi. BMC Bioinformatics, 6, 178.
O’Donnell, K., & Cigelnik, E. (1997). Two divergent intragenomic rDNA ITS2 types within a monophyletic lineage of the fungus Fusarium are nonorthologous. Molecular Phylogenetics and Evolution, 7, 103–116.
O’Donnell, K., Cigelnik, E., & Nirenberg, H. I. (1998a). Molecular systematics and phylogeography of the Gibberella fujikuroi species complex. Mycologia, 90, 465–493.
O’Donnell, K., Gueidan, C., Sink, S., Johnston, P. R., Crous, P. W., Glenn, A., et al. (2009a). A two-locus DNA sequence database for typing plant and human pathogens within the Fusarium oxysporum species complex. Fungal Genetics and Biology, 46, 936–948.
O’Donnell, K., Humber, R. A., Geiser, D. M., Kang, S., Park, B., Robert, V. A. R. G., et al. (2012). Phylogenetic diversity of insecticolous fusaria inferred from multilocus DNA sequence data and their molecular identification via FUSARIUM-ID and Fusarium MLST. Mycologia, 104, 427–445.
O’Donnell, K., Kistler, H. C., Cigelnik, E., & Ploetz, R. C. (1998b). Multiple evolutionary origins of the fungus causing Panama disease of banana: Concordant evidence from nuclear and mitochondrial gene genealogies. Proceedings of the National Academy of Sciences USA, 95, 2044–2049.
O’Donnell, K., Rooney, A. P., Proctor, R. H., Brown, D. W., McCormick, S. P., Ward, T. J., et al. (2013). Phylogenetic analyses of RPB1 and RPB2 support a middle Cretaceous origin for a clade comprising all agriculturally and medically important fusaria. Fungal Genetics and Biology, 52, 20–31.
O’Donnell, K., Sink, S., Libeskind-Hadas, R., Hulcr, J., Kasson, M. T., Ploetz, R. C., et al. (2014). Discordant phylogenies suggest repeated host shifts in the Fusarium – Euwallacea ambrosia beetle mutualism. Fungal Genetics and Biology. doi:10.1016/j.fgb.2014.10.014.
O’Donnell, K., Sutton, D. A., Fothergill, A., McCarthy, D., Rinaldi, M. G., Brandt, M. E., et al. (2008). Molecular phylogenetic diversity, multilocus haplotype nomenclature, and in vitro antifungal resistance within the Fusarium solani species complex. Journal of Clinical Microbiology, 46, 2477–2490.
O’Donnell, K., Sutton, D. A., Rinaldi, M. G., Gueidan, C., Crous, P. W., & Geiser, D. M. (2009b). Novel multilocus sequence typing scheme reveals high genetic diversity of human pathogenic members of the Fusarium incarnatum-F. equiseti and F. chlamydosporum species complexes within the United States. Journal of Clinical Microbiology, 47, 3851–3861.
O’Donnell, K., Sutton, D. A., Rinaldi, M. G., Sarver, B. A. J., Balajee, S. A., et al. (2010). Internet-accessible DNA sequence database for identifying fusaria from human and animal infections. Journal of Clinical Microbiology, 48, 3708–3718.
Palm, M. E., Gams, W., & Nirenberg, H. I. (1995). Plectosporium, a new genus for Fusarium tabacinum, the anamorph of Plectosphaerella cucumerina. Mycologia, 87, 397–406.
Park, B., Park, J., Cheong, K.-C., Choi, J., Jung, K., et al. (2010). Cyber infrastructure for Fusarium: three integrated platforms supporting strain identification, phylogenetics, comparative genomics, and knowledge sharing. Nucleic Acids Research, 39, D640–D646.
Samuels, G. J., & Hallet, I. C. (1983). Microdochium stoveri and Monographella stoveri, new combinations for Fusarium stoveri and Micronectriella stoveri. Transactions of the British Mycological Society, 81, 473–483.
Sarver, B. A. J., Ward, T. J., Gale, L. R., Broz, K., Kistler, H. C., Aoki, T., et al. (2011). Novel Fusarium head blight pathogens from Nepal and Louisiana revealed by multilocus genealogical concordance. Fungal Genetics and Biology, 48, 1096–1107.
Schoch, C. L., Seifert, K. A., Huhndorf, S., Robert, V., Spouge, J. L., Levesque, C. A., et al. (2012). Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proceedings of the National Academy of Sciences USA, 109, 6241–6246.
Schroers, H.-J., O’Donnell, K., Lamprecht, S. C., Kammeyer, P. L., Johnson, S., Sutton, D. A., et al. (2009). Taxonomy and phylogeny of the Fusarium dimerum species group. Mycologia, 101, 44–70.
Short, D. P. G., O’Donnell, K., Zhang, N., Juba, J. H., & Geiser, D. M. (2011). Widespread occurrence of diverse human pathogenic types of the fungus Fusarium detected in plumbing drains. Journal of Clinical Microbiology, 49, 4264–4272.
Simpson, B. B., & Ogorzaly, M. C. (1995). Economic Botany: Plants in Our World. New York, New York, USA: McGraw-Hill, Inc.
Suthar, R. S., & Bhatt, P. N. (2011). In silico identification of Fusarium strain NFCCI 2157 isolated from cumin wilt. International Journal of Plant, Animal and Environmental Sciences, 1, 51–54.
Taylor, J. W., Jacobson, D. J., Kroken, S., Kasuga, T., Geiser, D. M., Hibbett, D. S., et al. (2000). Phylogenetic species recognition and species concepts in fungi. Fungal Genetics and Biology, 31, 21–32.
Tsai, H.-F., Liu, J.-S., Staben, C., Christensen, M. J., Latch, G. C. M., Siegel, M. R., et al. (1994). Evolutionary diversification of fungal endophytes of tall fescue grass by hybridization with Epichloë species. Proceedings of the National Academy of Sciences USA, 91, 2542–2546.
Walsh, J. L., Laurence, M. H., Liew, E. C. Y., Sangalang, A. E., Burgess, L. W., Summerell, B. A., et al. (2010). Fusarium: two endophytic novel species from tropical grasses of northern Australia. Fungal Diversity, 44, 149–159.
Wingfield, M. J., de Beer, Z. W., Slippers, B., Wingfield, B. D., Groenewald, J. Z., Lombard, L., et al. (2012). One fungus, one name promotes progressive plant pathology. Molecular Plant Pathology, 13, 604–613.
Acknowledgments
Special thanks are due to Stacy Sink for assistance with the DNA sequence data, Nathane Orwig for running DNA sequences in the NCAUR DNA core facility, and Bongsoo Park for maintaining FUSARIUM-ID. The mention of firm names or trade products does not imply that they are endorsed or recommended by the US Department of Agriculture over other firms or similar products not mentioned. The USDA is an equal opportunity provider and employer.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
O’Donnell, K., Ward, T.J., Robert, V.A.R.G. et al. DNA sequence-based identification of Fusarium: Current status and future directions. Phytoparasitica 43, 583–595 (2015). https://doi.org/10.1007/s12600-015-0484-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12600-015-0484-z