
Summary
MEK inhibitors have immunomodulatory activity and potential for synergistic activity when combined with PD-1 inhibitors. We evaluated selumetinib (inhibitor of MEK1/2) plus pembrolizumab (anti‒PD-1 antibody) in patients with advanced/metastatic solid tumors. In this phase 1b study, adults with previously treated advanced/metastatic solid tumors received pembrolizumab 200 mg intravenously every 3 weeks plus selumetinib on days 1‒14 per 3-week cycle (2 weeks on/1 week off); selumetinib dosing began at 50 mg orally twice daily with escalation in 25 mg increments for ≤ 35 cycles. Primary endpoints were dose-limiting toxicities (DLTs), adverse events (AEs), and treatment discontinuations due to AEs. Thirty-two patients were enrolled. Dose escalation was completed up to selumetinib 125 mg twice daily. The target DLT rate of 30% was not reached at any dose level. In the selumetinib 100 mg group, 2/11 patients (18.2%) experienced DLTs (n = 1 grade 3 diarrhea, n = 1 grade 3 fatigue). In the selumetinib 125 mg group, 3/14 (21.4%) experienced DLTs (n = 1 grade 2 retinal detachment, n = 1 grade 3 retinopathy, n = 1 grade 3 stomatitis). Dose-related changes in pharmacokinetic exposures were observed for selumetinib and N-desmethyl selumetinib up to 100 mg (saturation at 125 mg). Two patients achieved partial responses (1 each with selumetinib 75 mg and 125 mg) for an objective response rate of 6%. The study was stopped early because of insufficient efficacy. Although the target DLT rate was not reached at any dose level and no new safety signals were identified, selumetinib plus pembrolizumab had limited antitumor activity in this population. Trial registration: ClinicalTrials.gov, NCT03833427.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The mitogen-activated protein kinase (MAPK) pathway plays a key role in cell cycle regulation, including mechanisms of organ development and tissue homeostasis via mitogen-activated extracellular signal-regulated kinase 1/2 (MEK1/2), receptor tyrosine kinases, RAS and RAF family members, and extracellular signal-regulated kinase 1/2 [1]. Somatic mutations at multiple points within this pathway can lead to cancer [2]. MEK is an attractive therapeutic target as one of the main downstream effectors in the MAPK pathway. Selumetinib is an orally available selective inhibitor of MEK1/2 that is approved in the United States for the treatment of pediatric patients aged 2 years or older with neurofibromatosis type 1 with symptomatic inoperable plexiform neurofibromas [3]. Approval of selumetinib was based on phase 1 and 2 studies that demonstrated efficacy and that selumetinib had no excessive, cumulative, or irreversible toxic effects in this population [4, 5].
Combination therapies involving inhibitors of programmed cell death protein 1 (PD-1) or programmed cell death ligand 1 (PD-L1) are standard of care treatments across multiple advanced solid tumor types [6]. Preclinical evidence suggested that MEK inhibitors have immunomodulatory activity and the potential for improved activity when combined with PD-(L)1 inhibitors [7]. Some preliminary evidence of antitumor activity was suggested with the MEK inhibitor cobimetinib plus the PD-L1 inhibitor atezolizumab in patients with heavily pretreated metastatic colorectal cancer, non‒small-cell lung cancer, or melanoma [8]. In patients with previously untreated, BRAF-mutated advanced melanoma, early reports indicated that the addition of the PD-L1 inhibitor atezolizumab to cobimetinib and vemurafenib in the phase 3 IMspire150 study [9] or the PD-1 inhibitor pembrolizumab to trametinib and dabrafenib in the phase 1/2 KEYNOTE-022 study [10, 11] improved outcomes compared with MEK/BRAF kinase inhibition alone. Triplet therapy with an immune checkpoint, MEK, and BRAF kinase inhibitor significantly improved progression-free survival (PFS) compared with doublet therapy in the IMspire150 study and numerically improved PFS, duration of response, and overall survival (OS) in the KEYNOTE-022 study. Subsequent analyses from the IMspire150 and KEYNOTE-022 studies reported negative results [12, 13].
Based on the promising preclinical findings and preliminary results from the IMspire150 and KEYNOTE-022 clinical studies described above, the current phase 1b MK-5618-001 study (ClinicalTrials.gov, NCT03833427) was conducted to evaluate the safety and tolerability of selumetinib plus pembrolizumab in patients with advanced or metastatic solid tumors and to establish a recommended phase 2 dose of selumetinib when used in combination with pembrolizumab. The study also evaluated the pharmacokinetics of selumetinib and the antitumor activity of this combination.
Methods
This study was done in accordance with local and/or national regulations and the ethical principles in the Declaration of Helsinki. An institutional review board or independent ethics committee approved the study protocol before the study was initiated at each site. Patients provided written informed consent before entering the study.
Patients
Eligible patients were ≥ 18 years of age and had histologically or cytologically confirmed advanced or metastatic solid tumors with progression on, or intolerance to, all other treatments known to confer benefit. Other inclusion criteria included measurable disease as determined by Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1, an Eastern Cooperative Oncology Group performance status of 0 or 1, and adequate organ function. Patients were ineligible if they had received chemotherapy, definitive radiation, or biological cancer therapy ≤ 4 weeks (≤ 2 weeks for palliative radiation) prior to the first dose of study treatment or if they were still experiencing grade ≥ 2 adverse events from a cancer therapy administered > 4 weeks earlier. Other exclusion criteria included clinically active central nervous system metastases, carcinomatous meningitis, active infection requiring therapy, history of noninfectious pneumonitis requiring steroids or current pneumonitis, active autoimmune disease requiring systemic treatment in the last 2 years, diagnosis of immunodeficiency or receipt of immunosuppressive therapy ≤ 7 days before treatment allocation, and grade > 1 peripheral neuropathy or paresthesia at baseline.
Study design
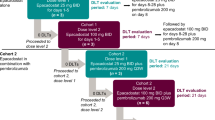
This was a phase 1b, open-label, multicenter, multiple-dose, dose-escalation study. Patients received pembrolizumab 200 mg intravenously every 3 weeks in combination with oral selumetinib. The recommended phase 2 dose for selumetinib monotherapy is 75 mg orally twice daily administered on a continuous basis [14]. At this dose, enhanced MEK inhibition is proposed to be associated with greater response, as well as an escalation in dose-dependent adverse events. To mitigate dose-dependent adverse events associated with MEK inhibition, this study chose an intermittent dosing schedule for selumetinib (2 weeks on/1 week off) and began treatment at a dose below the recommended phase 2 dose. The starting dose of selumetinib was 50 mg twice daily on days 1 to 14 of each 3-week treatment cycle, which was escalated in 25 mg increments up to a prespecified maximum of 300 mg twice daily on days 1 to 14 of each cycle. Treatment was continued until documented radiographic disease progression, as assessed by the investigator per RECIST version 1.1 and then confirmed by modified RECIST for immune-based therapeutics (iRECIST) [15], unacceptable toxicity, intercurrent illness, investigator decision, or completion of 35 treatment cycles (~2 years). Patients with unconfirmed disease progression may have continued treatment at the discretion of the investigator until progression was confirmed using iRECIST; if repeat imaging did not confirm progressive disease per iRECIST, as assessed by the investigator, and the patient continued to be clinically stable, study treatment could have continued.
At least 3 patients were enrolled at each dose level; the modified toxicity probability interval (mTPI) design [16], with a target dose-limiting toxicity (DLT) rate of ~30%, was used to identify a potential maximum tolerated dose. Dose escalation and de-escalation decisions were based on the mTPI design and were dependent on the number of patients enrolled and number of DLTs observed at the particular dose level (Online Resource 1). Dose finding was considered complete after 14 patients were enrolled at any of the tested dose levels and the decision was made to stay at that dose level.
Assessments
Patients were monitored for DLTs (see Online Resource 2 for definition) during the first 21 days of treatment. Adverse events were monitored throughout the study and for 30 days after cessation of study treatment (90 days for serious adverse events) and were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0. A full ophthalmic examination was performed on day 1 of cycle 2 and every 8 weeks thereafter through treatment discontinuation, as selumetinib monotherapy is known to cause ocular toxicity, including blurred vision, photophobia, cataracts, and ocular hypertension [3, 17, 18].
Plasma samples for analyzing the pharmacokinetics of selumetinib were collected on day 1 of cycle 1 (predose; 1, 2, 4, and 6 h postdose; and 8–12 h postdose [before evening dosing]). Additional predose samples were collected on days 1 and 14 of cycle 2 and on days 1 and 14 of cycle 5.
Tumor imaging was performed at screening, every 9 weeks for 12 months, every 12 weeks during months 12 to 24, and at treatment discontinuation. For patients who discontinued study treatment without disease progression, an effort was made to continue tumor imaging (same schedule as during treatment) until the start of new anticancer treatment, disease progression, pregnancy, death, withdrawal of consent, or the end of the study.
Biomarkers were assessed at baseline and included PD-L1 combined positive score (CPS), T-cell‒inflamed gene expression profile (Tcellinf GEP), tumor mutational burden (TMB), microsatellite instability (MSI), and KRAS/BRAF mutations [19,20,21].
Endpoints
Primary endpoints were the occurrence of DLTs, adverse events, and study treatment discontinuations due to adverse events. Pharmacokinetic parameters of selumetinib were assessed as secondary endpoints. An exploratory endpoint was the objective response rate (ORR) per RECIST version 1.1 as assessed by the investigator, defined as the proportion of patients with a confirmed complete or partial response. Biomarkers were an additional exploratory endpoint.
Statistical analysis
The sample size was planned at 50 to 84 patients and depended on the observed DLT rate. Analyses of adverse events were based on the safety population, which included all patients who received ≥ 1 dose of study treatment. The DLT population included all patients in the safety population who completed cycle 1 without a DLT or who experienced a DLT in cycle 1. Estimates of DLT rates across dose levels in each treatment arm were analyzed using isotonic regression with the pooled adjacent violators algorithm [16]. Pharmacokinetic analyses were based on all patients who complied with the protocol sufficiently to ensure that their data would likely reflect treatment effects. Efficacy analyses were based on all patients with a baseline scan who had measurable disease by investigator assessment and who received ≥ 1 dose of study treatment. No formal hypothesis testing was planned.
Results
Patients
The study was conducted between March 18, 2019, and June 28, 2022. The study was terminated early because of insufficient efficacy; dose escalation of selumetinib was completed up to 125 mg. A total of 32 patients were enrolled (n = 4 in 50 mg group, n = 3 in 75 mg group, n = 11 in 100 mg group, n = 14 in 125 mg group) at 6 study sites in the United States and Canada. Patient disposition is summarized in Online Resource 3. At the database cutoff date of July 21, 2022, the median (range) duration of follow-up (defined as time from first dose to database cutoff date) was 31.2 (22.6–39.6) months overall, 39.2 (38.7‒39.6) months in the selumetinib 50 mg group, 37.3 (36.8‒37.6) months in the selumetinib 75 mg group, 33.0 (30.8‒35.7) months in the selumetinib 100 mg group, and 26.3 (22.6‒30.0) months in the selumetinib 125 mg group. Across these dose levels, patients received a median (range) of 2 (1‒7) cycles, 2 (2‒12) cycles, 4 (1‒11) cycles, and 3 (1‒35) cycles of selumetinib, respectively. Pembrolizumab was administered for a median (range) of 2 (1‒7) doses, 9 (2‒12) doses, 3 (1‒11) doses, and 3.5 (1‒35) doses, respectively.
Patient demographics and baseline disease characteristics were generally similar across the dose levels (Table 1). Overall, 22 patients (69%) were female, and median age was 56 years. Twenty-one patients (66%) had an Eastern Cooperative Oncology Group performance status of 1, and 18 patients (56%) had received ≥ 3 previous lines of therapy. The most common cancer types were breast cancer, colorectal cancer, and ovarian cancer (n = 4 each).
Safety
The target DLT rate of 30% was not reached at any dose level. DLTs were reported for 0 patients in the selumetinib 50 mg and 75 mg groups. In the selumetinib 100 mg group, 2 of 11 patients (18.2% [80% CI, 7.0%‒34.9%]) experienced DLTs (n = 1 grade 3 diarrhea, n = 1 grade 3 fatigue). In the selumetinib 125 mg group, 3 of 14 patients (21.4% [80% CI, 10.2%‒36.7%]) experienced DLTs (n = 1 grade 2 retinal detachment, n = 1 grade 3 retinopathy, n = 1 grade 3 stomatitis).
Treatment-related adverse events are shown by dose level and grade in Table 2. Overall, 30 of 32 patients (94%) in the safety population experienced treatment-related adverse events. The most common events were dermatitis acneiform (47%), fatigue (34%), diarrhea (31%), and maculopapular rash (22%). Twelve patients (38%) experienced grade 3 or 4 treatment-related adverse events; those occurring in > 1 patient included increased alanine aminotransferase (n = 1 in 100 mg group, n = 3 in 125 mg group), increased aspartate aminotransferase (n = 2 in 125 mg group), diarrhea (n = 1 in 50 mg group, n = 1 in 100 mg group), and retinopathy (n = 1 in 100 mg group, n = 1 in 125 mg group). Eight patients (25%) discontinued study treatment (selumetinib, pembrolizumab, or both) due to treatment-related adverse events; the only event that led to discontinuation of study treatment in > 1 patient was pneumonitis (n = 2 in 125 mg group [both selumetinib and pembrolizumab were discontinued]). Four patients had selumetinib dose reductions due to treatment-related AEs; there were no pembrolizumab dose reductions due to AEs. No treatment-related adverse events led to death.
Pharmacokinetics
On day 1 of cycle 1, plasma exposures of selumetinib and its metabolite N-desmethyl selumetinib (area under the concentration‒time curve and maximum concentration) increased as dose increased up to 100 mg, with saturation observed at 125 mg (Table 3). On day 1 of cycle 1, plasma concentrations of selumetinib and N-desmethyl selumetinib peaked between 1 and 2 h postdose at each dose level and steadily declined thereafter through 8 h postdose (Fig. 1A, B). The observed t1/2 ranged from 2.82 h to 3.82 h for selumetinib and 3.07 h to 3.94 h for N-desmethyl selumetinib (Table 3).

Arithmetic mean ± standard deviation plasma concentration‒time profiles for A selumetinib and B N-desmethyl selumetinib on day 1 of cycle 1. Database cutoff date: July 21, 2022
Efficacy
Best overall response is shown in Table 4. No complete responses were observed. Partial responses were observed in 1 of 3 patients (33%) in the selumetinib 75 mg group and in 1 of 14 patients (7%) in the selumetinib 125 mg group; no partial responses were seen in the selumetinib 50 mg and 100 mg groups. The durations of response for these 2 patients were 6.0 months (colorectal cancer) and 25.1 months (anaplastic thyroid carcinoma), respectively. The patient with a duration of response of 25.1 months discontinued selumetinib after the first cycle and completed 35 cycles of pembrolizumab. Progressive disease was observed in 2 of 4 patients (50%) in the selumetinib 50 mg group, 1 of 3 patients (33%) in the selumetinib 75 mg group, 4 of 11 patients (36%) in the selumetinib 100 mg group, and 6 of 14 patients (43%) in the selumetinib 125 mg group. A reduction of ≥ 30% in target lesion size was observed in 1 patient receiving selumetinib 75 mg (colorectal cancer), 1 receiving selumetinib 100 mg (esophageal cancer), and 3 receiving selumetinib 125 mg (anaplastic thyroid carcinoma, pancreatic adenocarcinoma, and cervical cancer) (Online Resource 4).
Biomarkers
The prevalence of biomarkers at baseline is shown in Online Resource 5. Thirteen patients (41%) had tumors with PD-L1 CPS ≥ 10, 16 patients (50%) had Tcellinf GEP low tumors, 24 patients (75%) had non–TMB-high tumors (< 10 mut/Mb), 25 patients (78%) had microsatellite stable (MSS) tumors, and no patient had known microsatellite instability-high/mismatch repair deficient tumors. Five patients (16%) had KRAS mutations; none had BRAF mutations. Biomarkers in the patient with a PR in the selumetinib 75 mg group included PD-L1 CPS 90, Tcellinf GEP non-low, TMB-high (≥ 10 mut/Mb), MSS, and KRAS/BRAF wild type (Online Resource 6). The patient with a PR in the selumetinib 125 mg group did not have any biomarker data available.
Discussion
This phase 1b study investigated the safety and tolerability of the combination of selumetinib and pembrolizumab in patients with advanced or metastatic solid tumors after intolerance to or disease progression during all available approved therapies. Dose escalation of selumetinib was completed up to 125 mg, at which point the study was terminated by the sponsor. The target DLT rate of 30% was not reached at any dose level, suggesting that the maximum tolerable dose level for selumetinib exceeds 125 mg. Pharmacokinetic results showed that plasma levels of selumetinib and N-desmethyl selumetinib increased in a dose-related manner up to 100 mg, and that saturation was met at 125 mg. No new safety concerns were observed, and the safety profile of the combination of selumetinib plus pembrolizumab was consistent with the reported safety profiles of the individual agents. Selumetinib plus pembrolizumab demonstrated limited antitumor activity, with partial responses observed in only 6% of patients. One of the 2 patients with a partial response had a tumor with TMB ≥ 10 mut/Mb, which is a known predictive biomarker for response to pembrolizumab. Biomarker data were available for 1 of the 2 PRs and were consistent with what would be expected for response to pembrolizumab monotherapy. Efficacy findings were not sufficient to pursue further development of this combination.
Early reports of efficacy across multiple trials indicated that the combination of an anti-PD-(L)1 antibody with one or more inhibitors along the MAPK-MEK-BRAF signaling axis may be a promising therapeutic approach in select advanced solid tumors [8,9,10,11]. Following initiation of the MK-5618-001 study, clinical trials that evaluated MEK/BRAF kinase inhibitors combined with PD-(L)1 inhibitors for BRAF-mutant advanced melanoma published more mature, negative results. The second interim analysis of the IMspire150 study showed no significant improvement in OS with triplet therapy (atezolizumab, cobimetinib, and vemurafenib) compared with MEK/BRAF kinase inhibition alone [12] and, in the phase 3 COMBI-i study, the addition of the PD-1 inhibitor spartalizumab to trametinib plus dabrafenib did not significantly improve PFS and led to increased toxicity compared with trametinib plus dabrafenib [22]. In a phase 1 study, DLTs of dermatitis (n = 5), QTc prolongation (n = 1), and arthralgias (n = 1) were reported among 5 patients treated with the triplet combination of pembrolizumab, cobimetinib, and vemurafenib, and the safety findings led to early closure of the study [23].
Similar findings have been reported for BRAF-wild type or BRAF-unselected melanoma and other advanced solid tumors. In the phase 3 IMspire170 study, patients with BRAF wild-type advanced melanoma had no PFS benefit with cobimetinib plus atezolizumab relative to pembrolizumab alone (HR, 1.15 [95% CI, 0.88‒1.50]), and rates of adverse events and treatment discontinuations or dose modifications due to adverse events were higher with combination therapy than monotherapy [24]. In the KEYNOTE-022 study, limited antitumor activity (ORR, 17%) was demonstrated with trametinib plus pembrolizumab in patients with BRAF wild-type advanced melanoma or advanced solid tumors regardless of BRAF mutation; safety was manageable, although there was no control arm [13]. Lastly, the phase 3 IMblaze370 study evaluated cobimetinib plus atezolizumab versus monotherapy with atezolizumab or the multikinase inhibitor regorafenib in patients with predominantly microsatellite-stable, unresectable locally advanced or metastatic colorectal cancer [25]. Overall survival was similar among the 3 treatment groups and rates of serious adverse events and treatment discontinuations due to adverse events were more common with combination therapy than monotherapy [25].
Interpretation of the current study results is limited by the small sample size, particularly given that cohort expansion was not initiated. Since the pharmacokinetic data showed saturation at the 125 mg dose, it was concluded that further escalation of the selumetinib dose was unlikely to improve efficacy. Due to early study termination and small sample size, no further biomarker analysis is planned.
In conclusion, selumetinib plus pembrolizumab had manageable safety but did not demonstrate sufficient antitumor activity in patients with advanced or metastatic solid tumors. Accumulating evidence suggests that despite a mechanistic rationale for combining MAPK inhibition with immune checkpoint inhibition, such regimens have modest efficacy and limited utility for patients with advanced or metastatic solid tumors.
Data availability
Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA (MSD) is committed to providing qualified scientific researchers access to anonymized data and clinical study reports from the company’s clinical trials for the purpose of conducting legitimate scientific research. MSD is also obligated to protect the rights and privacy of trial participants and, as such, has a procedure in place for evaluating and fulfilling requests for sharing company clinical trial data with qualified external scientific researchers. The MSD data sharing website (available at: http://engagezone.msd.com/ds_documentation.php) outlines the process and requirements for submitting a data request. Applications will be promptly assessed for completeness and policy compliance. Feasible requests will be reviewed by a committee of MSD subject matter experts to assess the scientific validity of the request and the qualifications of the requestors. In line with data privacy legislation, submitters of approved requests must enter into a standard data-sharing agreement with MSD before data access is granted. Data will be made available for request after product approval in the US and EU or after product development is discontinued. There are circumstances that may prevent MSD from sharing requested data, including country or region-specific regulations. If the request is declined, it will be communicated to the investigator. Access to genetic or exploratory biomarker data requires a detailed, hypothesis-driven statistical analysis plan that is collaboratively developed by the requestor and MSD subject matter experts; after approval of the statistical analysis plan and execution of a data-sharing agreement, MSD will either perform the proposed analyses and share the results with the requestor or will construct biomarker covariates and add them to a file with clinical data that is uploaded to an analysis portal so that the requestor can perform the proposed analyses.
References
Wagle MC, Kirouac D, Klijn C, Liu B, Mahajan S, Junttila M, Moffat J, Merchant M, Huw L et al (2018) A transcriptional MAPK Pathway Activity Score (MPAS) is a clinically relevant biomarker in multiple cancer types. NPJ Precis Oncol 2:7. https://doi.org/10.1038/s41698-018-0051-4
Heppner DE, Eck MJ (2021) A structural perspective on targeting the RTK/Ras/MAP kinase pathway in cancer. Protein Sci 30:1535–1553. https://doi.org/10.1002/pro.4125
Koselugo (Selumetinib) (2021) Full prescribing information. AstraZeneca Pharmaceuticals LP, Wilmington, DE
Dombi E, Baldwin A, Marcus LJ, Fisher MJ, Weiss B, Kim A, Whitcomb P, Martin S, Aschbacher-Smith LE et al (2016) Activity of selumetinib in neurofibromatosis type 1-related plexiform neurofibromas. N Engl J Med 375:2550–2560. https://doi.org/10.1056/NEJMoa1605943
Gross AM, Wolters PL, Dombi E, Baldwin A, Whitcomb P, Fisher MJ, Weiss B, Kim A, Bornhorst M et al (2020) Selumetinib in children with inoperable plexiform neurofibromas. N Engl J Med 382:1430–1442. https://doi.org/10.1056/NEJMoa1912735
Yi M, Zheng X, Niu M, Zhu S, Ge H, Wu K (2022) Combination strategies with PD-1/PD-L1 blockade: current advances and future directions. Mol Cancer 21:28. https://doi.org/10.1186/s12943-021-01489-2
Dennison L, Mohan AA, Yarchoan M (2021) Tumor and systemic immunomodulatory effects of MEK inhibition. Curr Oncol Rep 23:23. https://doi.org/10.1007/s11912-020-01008-4
Hellmann MD, Kim TW, Lee CB, Goh BC, Miller WH Jr, Oh DY, Jamal R, Chee CE, Chow LQM et al (2019) Phase Ib study of atezolizumab combined with cobimetinib in patients with solid tumors. Ann Oncol 30:1134–1142. https://doi.org/10.1093/annonc/mdz113
Gutzmer R, Stroyakovskiy D, Gogas H, Robert C, Lewis K, Protsenko S, Pereira RP, Eigentler T, Rutkowski P et al (2020) Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAF(V600) mutation-positive melanoma (IMspire150): primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 395:1835–1844. https://doi.org/10.1016/S0140-6736(20)30934-X
Ascierto PA, Ferrucci PF, Fisher R, Del Vecchio M, Atkinson V, Schmidt H, Schachter J, Queirolo P, Long GV et al (2019) Dabrafenib, trametinib and pembrolizumab or placebo in BRAF-mutant melanoma. Nat Med 25:941–946. https://doi.org/10.1038/s41591-019-0448-9
Ferrucci PF, Di Giacomo AM, Del Vecchio M, Atkinson V, Schmidt H, Schachter J, Queirolo P, Long GV, Stephens R et al (2020) KEYNOTE-022 part 3: a randomized, double-blind, phase 2 study of pembrolizumab, dabrafenib, and trametinib in BRAF-mutant melanoma. J Immunother Cancer. https://doi.org/10.1136/jitc-2020-001806
Ascierto PA, Stroyakovskiy D, Gogas H, Robert C, Lewis K, Protsenko S, Pereira RP, Eigentler T, Rutkowski P et al (2023) Overall survival with first-line atezolizumab in combination with vemurafenib and cobimetinib in BRAF(V600) mutation-positive advanced melanoma (IMspire150): second interim analysis of a multicentre, randomised, phase 3 study. Lancet Oncol 24:33–44. https://doi.org/10.1016/S1470-2045(22)00687-8
Maio M, Carlino MS, Joshua AM, McWhirter E, Ribas A, Ascierto PA, Miller WH Jr, Butler MO, Ferrucci PF et al (2022) KEYNOTE-022: pembrolizumab with trametinib in patients with BRAF wild-type melanoma or advanced solid tumours irrespective of BRAF mutation. Eur J Cancer 160:1–11. https://doi.org/10.1016/j.ejca.2021.09.024
Banerji U, Camidge DR, Verheul HM, Agarwal R, Sarker D, Kaye SB, Desar IM, Timmer-Bonte JN, Eckhardt SG et al (2010) The first-in-human study of the hydrogen sulfate (Hyd-sulfate) capsule of the MEK1/2 inhibitor AZD6244 (ARRY-142886): a phase I open-label multicenter trial in patients with advanced cancer. Clin Cancer Res 16:1613–1623. https://doi.org/10.1158/1078-0432.ccr-09-2483
Seymour L, Bogaerts J, Perrone A, Ford R, Schwartz LH, Mandrekar S, Lin NU, Litière S, Dancey J et al (2017) iRECIST: guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol 18:e143–e152. https://doi.org/10.1016/S1470-2045(17)30074-8
Ji Y, Li Y, Nebiyou Bekele B (2007) Dose-finding in phase I clinical trials based on toxicity probability intervals. Clin Trials 4:235–244. https://doi.org/10.1177/1740774507079442
Mendez-Martinez S, Calvo P, Ruiz-Moreno O, Pardinas Baron N, Lecinena Bueno J, Gil Ruiz MDR, Pablo L (2019) Ocular adverse events associated with MEK inhibitors. Retina 39:1435–1450. https://doi.org/10.1097/IAE.0000000000002451
Carvajal RD, Sosman JA, Quevedo JF, Milhem MM, Joshua AM, Kudchadkar RR, Linette GP, Gajewski TF, Lutzky J et al (2014) Effect of selumetinib vs chemotherapy on progression-free survival in uveal melanoma: a randomized clinical trial. JAMA 311:2397–2405. https://doi.org/10.1001/jama.2014.6096
Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, Albright A, Cheng JD, Kang SP et al (2017) IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest 127:2930–2940. https://doi.org/10.1172/JCI91190
Emancipator K (2020) Keytruda and PD-L1: a real-world example of co-development of a drug with a predictive biomarker. AAPS J 23:5. https://doi.org/10.1208/s12248-020-00525-1
Wei B, Kang J, Kibukawa M, Arreaza G, Maguire M, Chen L, Qiu P, Lang L, Aurora-Garg D et al (2022) Evaluation of the TruSight Oncology 500 assay for routine clinical testing of tumor mutational burden and clinical utility for predicting response to pembrolizumab. J Mol Diagn 24:600–608. https://doi.org/10.1016/j.jmoldx.2022.01.008
Dummer R, Long GV, Robert C, Tawbi HA, Flaherty KT, Ascierto PA, Nathan PD, Rutkowski P, Leonov O et al (2022) Randomized phase III trial evaluating spartalizumab plus dabrafenib and trametinib for BRAF V600-mutant unresectable or metastatic melanoma. J Clin Oncol 40:1428–1438. https://doi.org/10.1200/JCO.21.01601
Shaikh SS, Zang Y, Hanmer J, Wang H, Lin Y, Davar D, Zarour HM, Kirkwood JM, Najjar YG (2022) Phase I trial of pembrolizumab plus vemurafenib and cobimetinib in patients with metastatic melanoma. Front Oncol 12:1022496. https://doi.org/10.3389/fonc.2022.1022496
Gogas H, Dreno B, Larkin J, Demidov L, Stroyakovskiy D, Eroglu Z, Francesco Ferrucci P, Pigozzo J, Rutkowski P et al (2021) Cobimetinib plus atezolizumab in BRAF(V600) wild-type melanoma: primary results from the randomized phase III IMspire170 study. Ann Oncol 32:384–394. https://doi.org/10.1016/j.annonc.2020.12.004
Eng C, Kim TW, Bendell J, Argilés G, Tebbutt NC, Di Bartolomeo M, Falcone A, Fakih M, Kozloff M et al (2019) Atezolizumab with or without cobimetinib versus regorafenib in previously treated metastatic colorectal cancer (IMblaze370): a multicentre, open-label, phase 3, randomised, controlled trial. Lancet Oncol 20:849–861. https://doi.org/10.1016/s1470-2045(19)30027-0
Acknowledgements
We thank the patients and their families and caregivers for participating in this study. We also thank Lillian L. Siu, MD, FRCPC, along with all investigators and site personnel. Medical writing assistance was provided by Autumn Kelly, MA, and Michael S. McNamara, MS, of ICON plc (Blue Bell, PA, USA). This assistance was funded by Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA.
Funding
Funding for this research was provided by Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA.
Author information
Authors and Affiliations
Contributions
Heng Zhou contributed towards the conception, design, or planning of the study. Lin-Chi Chen contributed towards the drafting of the manuscript. Maxime Chénard-Poirier, Martin E. Gutierrez, Drew Rasco, Yan Xing, Manish R. Sharma provided study materials/patients. Maxime Chénard-Poirier, Aaron R. Hansen, Martin E. Gutierrez, Drew Rasco, Yan Xing, Lin-Chi Chen, Heng Zhou, Andrea L. Webber, Tomoko Freshwater, and Manish R. Sharma contributed to the acquisition, analysis, or interpretation of the data, reviewed the manuscript for important intellectual content, and agreed to submit the manuscript for publication.
Corresponding author
Ethics declarations
Ethics approval
All procedures performed in this study were in accordance with local and/or national regulations and with the Declaration of Helsinki. The study protocol was approved by the institutional review board or independent ethics committee at each site.
Consent
Written informed consent was obtained from all individual patients included in the study.
Competing interests
Maxime Chénard-Poirier: Advisory board: Incyte, Merck & Co., Inc., Rahway, NJ, USA, Pfizer, and Bristol Myers Squibb. Aaron R. Hansen: Honoraria from Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA (MSD), AstraZeneca, MedImmune, Pfizer, GSK, Novartis, and Bristol Myers Squibb; Consulting or advisory role for Genentech, MSD, GSK, Bristol Myers Squibb, Novartis, Boston Biomedical, and Boehringer Ingelheim; Research funding to institution from Karyopharm Therapeutics, MSD, Bristol Myers Squibb, Boehringer Ingelheim, GSK, Novartis, Genentech, and Janssen. Martin E. Gutierrez: Advisory Board: Sanofi, Incyte, Boehringer Ingelheim. Consultant: Guardant. Stock/Shares: COTA/OMI. Drew Rasco: Research funding from Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA to support study conduct. Yan Xing: None disclosed. Lin-Chi Chen, Heng Zhou, Andrea L. Webber, Tomoko Freshwater: Employees of Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA, and stockholders in Merck & Co., Inc., Rahway, NJ, USA. Manish R. Sharma: Stockholder in Merck & Co., Inc., Rahway, NJ, USA.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chénard-Poirier, M., Hansen, A.R., Gutierrez, M.E. et al. A phase 1 trial of the MEK inhibitor selumetinib in combination with pembrolizumab for advanced or metastatic solid tumors. Invest New Drugs 42, 241–251 (2024). https://doi.org/10.1007/s10637-024-01428-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-024-01428-0