
Summary
Prodrugs can have the advantage over parent drugs in increased activation and cellular uptake. The multidrug ETC-L-FdUrd and the duplex drug ETC-FdUrd are composed of two different monophosphate-nucleosides, 5-fluoro-2′deoxyuridine (FdUrd) and ethynylcytidine (ETC), coupled via a glycerolipid or phosphodiester, respectively. The aim of the study was to determine cytotoxicity levels and mode of drug cleavage. Moreover, we determined whether a liposomal formulation of ETC-L-FdUrd would improve cytotoxic activity and/or cleavage. Drug effects/cleavage were studied with standard radioactivity assays, HPLC and LC-MS/MS in FM3A/0 mammary cancer cells and their FdUrd resistant variants FM3A/TK−. ETC-FdUrd was active (IC50 of 2.2 and 79 nM) in FM3A/0 and TK− cells, respectively. ETC-L-FdUrd was less active (IC50: 7 nM in FM3A/0 vs 4500 nM in FM3A/TK−). Although the liposomal formulation was less active than ETC-L-FdUrd in FM3A/0 cells (IC50:19.3 nM), resistance due to thymidine kinase (TK) deficiency was greatly reduced. The prodrugs inhibited thymidylate synthase (TS) in FM3A/0 cells (80–90%), but to a lower extent in FM3A/TK− (10–50%). FdUMP was hardly detected in FM3A/TK− cells. Inhibition of the transporters and nucleotidases/phosphatases resulted in a reduction of cytotoxicity of ETC-FdUrd, indicating that this drug was cleaved outside the cells to the monophosphates, which was verified by the presence of FdUrd and ETC in the medium. ETC-L-FdUrd and the liposomal formulation were neither affected by transporter nor nucleotidase/phosphatase inhibition, indicating circumvention of active transporters. In vivo, ETC-FdUrd and ETC-L-FdURd were orally active. ETC nucleotides accumulated in both tumor and liver tissues. These formulations seem to be effective when a lipophilic linker is used combined with a liposomal formulation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Of many chemotherapeutic agents that have been developed in preclinical studies, only a fraction reaches the clinic. One very successful anticancer drug is 5-fluorouracil (5-FU). The introduction of 5-FU into the clinic has significantly improved the treatment response and survival for cancer patients, particularly in colon, head and neck and breast cancer. However, important therapeutic limitations are a short biological half-life and the induction of resistance. Part of this resistance can be circumvented by the use of 5-FU analogs and/or prodrugs, such as 5-fluoro-2′deoxyuridine (FdUrd) [1], 5′-deoxy-5-fluorodeoxyuridine and ftorafur [2], a constituent of the oral 5-FU formulations UFT and S-1 [3]. 5-FU resistance mechanisms include increased thymidylate synthase (TS) expression levels [4], and for FdUrd a lowered thymidine kinase (TK) expression. Unsuccessful delivery of the nucleoside analogs can be mediated by a decreased cellular uptake by nucleoside transporters [5]. Therefore, modifications of nucleoside analogs by introduction of a lipid side-chain could improve cellular uptake, such as for Ara-C [6]. Other (chemical) modifications are the addition of a lipophilic chain to the monophosphorylated nucleoside rather than to the nucleoside analog itself [7, 8]. This is the so-called pronucleotide approach, which includes the SATE and SGTE compounds. These types of pronucleotides have demonstrated that it is possible to successfully deliver nucleotides into the cells in vitro [9]. Thereby, both uptake and the first activation step, mostly the rate-limiting, can be bypassed. Another strategy to increase the activity of one drug is to apply it in combination with other drugs [10]. Combination therapies are currently the standard for the treatment of various types of cancer, examples are the FOLFOX and FOLFIRI regimens as treatment against colorectal cancer [11–13].
To combine the advantages of these three strategies: (1) circumventing drug uptake by transporters, (2) bypassing the first activation step and (3) combining two drugs to improve their activity, combination-drugs have been developed. These drugs are composed of two different single active drugs, such as 5-FU and chloroethylnitrosourea (CNU) [14]. However, such a drug can also be composed of two different active nucleoside analogs, such as FdUrd and the novel 3′-C-ethynylcytidine (ETC). FdUrd is the 5-FU deoxynucleoside that can be directly monophosphorylated by TK to FdUMP. 5-FU acts by inhibition of TS by forming a stable ternary complex with 5,10CH2-THF mediated by 5-FdUMP. Furthermore, FdUTP can be incorporated into the DNA [1]. ETC is a new anticancer ribonucleoside that can be incorporated into the RNA after activation by specific enzymes (e.g. uridine cytidine kinase, UCK), thereby inducing cell death [7, 8]. ETC has shown preclinical activity in lung, colon and breast cancer and is now under investigation in phase I clinical trials [7, 15–17].
The multidrug tested in the present study is composed of the mononucleotide of ETC, coupled with a glycerophospholipid bridge to FdUrd (ETC-L-FdUrd). The duplex drug is composed of the mononucleoside of ETC linked with a phosphodiester to FdUrd (ETC-FdUrd) (Fig. 1). When these drugs are enzymatically cleaved behind the phosphate group, the mononucleotides (FdUMP or ETCMP) will be released. Therefore, the specific activating enzymes TK and UCK will be redundant and resistance due to deficiency in these key enzymes can be overcome. The lipid group of the multidrug may have the advantage to facilitate cell permeation through passive diffusion or possibly via a vesicle mediated uptake, bypassing the nucleoside transporter. When the drug is inside the cell, specific enzymes can further activate it. For another multidrug, AraC-L-FdUrd, a partial reversal of resistance to one drug was found [18]. However, we previously concluded that the efficacy of the two drugs was too different in order to exert a cytotoxic effect of both drugs [19]. Another strategy to increase drug uptake is by a liposomal formulation, which has already shown promising results with cytarabine and doxorubicin [20, 21]. To evaluate whether a liposomal formulation could increase drug activity, we tested the multidrug ETC-L-FdUrd in a liposomal formulation [22].

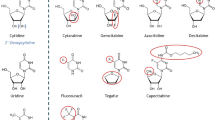
Structural formulas of the duplex drug ETC-FdUrd (3’-C-Ethynylcytidylyl-(5’➔5’)-5-fluoro-2’-deoxyuridine) and the multidrug ETC-L-FdUrd (3’-C-Ethynylcytidylyl-(5’➔1)-2-O-octadecyl-glycerylyl-(3➔5’)-5-fluoro2’-deoxyuridine)
The aim of this study was to compare the multi- and duplex drug and the liposomal formulation of the multidrug on cytotoxicity both in vitro and in vivo and to analyze whether the drugs are cleaved in- or outside the cell and which cleavage products are obtained.
Materials & methods
Chemicals
The investigated duplex drug ETC-FdUrd (3′-C-Ethynylcytidylyl-(5′➔5′)-5-fluoro-2′-deoxyuridine) and the multidrug, ETC-L-FdUrd (3′-C-Ethynylcytidylyl-(5′➔1)-2-O-octadecyl-glycerylyl-(3➔5′)-5-fluoro-2′-deoxyuridine) were synthesized by Prof. Dr. H. Schott [23]. FdUrd (5-fluoro-2′-deoxyuridine) and alkaline phosphatase were purchased from Sigma-Aldrich Chemicals (Zwijndrecht, The Netherlands). [5-3H]-2′-deoxycytidine (3H-dCyd) and [6-3H]-5-fluoro-2′-deoxyuridine-monophosphate (3H-FdUMP), were obtained from Moravek Biochemicals Inc. (Brea, CA, USA). All other chemicals used were of standard quality and commercially available.
Cell culture
The murine breast cancer suspension cell lines FM3A/0 and FM3A/TK− were a gift from Prof. Dr. J. Balzarini at the Rega Institute (Leuven, Belgium) and Dr Seno (Japan) [24]. The original FM3A/TK− cells are deficient in cytosolic TK1 and can not activate FdUrd. FM3A cells were cultured in RPMI, supplemented with 10% heat inactivated FCS and 20 mM HEPES. The 5-FU resistant colon carcinoma cell line HT29 was cultured in DMEM, supplemented with 10% heat inactivated FCS and 20 mM HEPES. All cells were maintained in a humidified atmosphere at 37 °C and 5% CO2.
Drug cytotoxicity assays
Drug cytotoxicity in FM3A/0 and FM3A/TK− cells was determined using the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide) assay for suspension cell lines as described previously [25]. In brief, 10 000 cells/well were seeded, after which drugs were added at increasing concentrations. After 72 h of continuous drug exposure, 10 µl MTT (5 mg/ml) was added to each well for 3 h at 37 °C. Thereafter, formazan crystals were dissolved in 100 µL of 0.04 N HCl-isopropyl alcohol and the optical density (OD) was measured at 495–540 nm. Drug cytotoxicity in HT-29 cells was determined using the sulphorhodamine B (SRB)-assay as described previously [25]. Cells (7000/well) were seeded, after which the drugs were added at increasing concentrations. After 72 h of continuous drug exposure, cells were fixed in trichloroacetic acid (TCA) and stained with SRB. After diluting the SRB-dye in 150 µl 10 mM Tris-solution, the OD was measured at 495–540 nm. IC50 values were defined as the concentrations that correspond to a reduction of cell growth by 50 % when compared to values of untreated control cells and are given in means ± SEM.
Drug cleavage
To determine intra- and extracellular drug cleavage, cells were pre-treated with the equilibrative nucleoside transporter inhibitor (ENT) dipyridamole (1 µM), the concentration that was previously shown to prevent nucleoside entry [26] To determine whether the drug is cleaved (outside the cell) into a nucleotide and subsequently degraded to a nucleoside, cells were also treated with a nucleotidase (2.5 mM α,ß-methylene-ADP) and a phosphatase inhibitor (15 mM 2-glycerol-phosphate), at concentrations previously shown to prevent nucleotidase or phosphatase mediated breakdown of (fluorinated) pyrimidine analogues [27, 28]. The protection factor was determined by dividing the growth of cells treated with inhibitor(s) by the growth of cells treated without inhibitor(s).
TS in situ activity assay
Inhibition of thymidylate synthase (TS) in intact FM3A/0 and FM3A/TK− cells was determined by measuring the conversion of [3H]-dCyd to 3H2O, which is catalyzed by TS as described previously [28, 29]. Briefly, 1 × 106 cells were seeded and incubated with the different drugs for 22 h. Subsequently, [3H]-dCyd (final concentration: 1 µM, specific activity 4.9 Ci/mmol) was added to each sample for 2 h at 37 °C. Blanks consisted of culture medium only and as controls untreated cells were used. The reaction was stopped by adding TCA and unconverted [3H]-dCyd was removed by activated charcoal. After centrifugation, the supernatant was transferred to a liquid scintillation vial and counted.
FdUMP assay
The level of FdUMP accumulation was determined with an isotope-dilution assay, based on the binding of [6-3H]-FdUMP to L. casei TS [30, 31]. Cells (2x106) were incubated for 24 h with 0.05 µM of the different drugs. After the incubation period, samples were deproteinized by TCA and neutralized. Cell extract (75 µl) was added to 10 µl assay buffer (200 mM Tris, 100 mM sodium fluoride, 15 mM CMP, 20 mM β-mercaptoethanol, pH 7.4), 10 µl 6.4 mM folate (CH2-THF), 10 µl 0.01 µCi [6-3H]-FdUMP (specific activity of 53 mCi/mmol) and 10 µl 25 nM TS and subsequently incubated at 30 °C for 2 h. The assay was stopped by adding ice-cold 3% charcoal to each sample. Samples were centrifuged for 15 min at 12000 rpm and counted in Optima Gold liquid scintillation fluid in a liquid scintillation counter (Packard Instrument B.V., Chemical Operations, Groningen, The Netherlands).
HPLC analysis of nucleosides
To determine the total amount of extracellular nucleosides (FdUrd and ETC), cells were exposed to 100 µM of the drugs for 24 h. Since FM3A/TK− cells may accumulate less active nucleosides, this higher concentration was chosen in order to allow reliable measurement. After the drug exposure, a sample was taken from the medium above the cells (after centrifugation), which was extracted as described for the FdUMP assay. HPLC-UV detection was performed as described previously with the nucleoside assay [32].
LC-MS/MS detection of ETC-nucleotides
To determine the total amount of intracellular mono- di- and triphosphates of the ETC-nucleosides, cells were exposed to 100 µM of the drugs for 24 h, after which they were extracted as described for the FdUMP assay. Thereafter, neutralized extracts were treated with alkaline phosphatase and subsequently precipitated with ice cold isopropyl alcohol. After centrifugation (21 000 g at 4 °C, 10 min), supernatants were transferred to a new vial and evaporated via freeze drying. The sample was reconstituted in ethyl acetate and back-extracted with 50 µl water. After centrifugation (21 000 g at 4 °C), the aqueous layer was transferred to a 96 well plate for LC-MS/MS analysis (TSI-API3000 mass spectrometer) essentially as described earlier for gemcitabine [33]. For ETC the molecular ion [M+H]+ 268.2 was observed, fragmentation of this ion resulted in the ion of 112.2 amu corresponding to the nucleoside base, this is a typical fragmentation pattern for nucleosides. The resulting transition of 268.2/112.2 was used for a multi-reaction mode (MRM) detection method. Chromatography consisted of a gradient system using 100% aqueous formic acid (0.1%) as eluent A and 40% aqueous formic acid (0.1%) / 60% methanol for eluent B. The gradient consisted of an initial 2 min hold at 96% A: 4% B followed by an increase to 100% B over 10 min (Phenomenex 100 × 2.0 mm ODS3, 3 µm particle size column maintained at 30 °C). Using the transition parameters of 268.2/112.2 chromatography demonstrate retention times of 0.6 min.
In vivo effect
In vivo testing of the compounds was performed using 5-FU resistant HT-29 colon cancer cells, which were transplanted subcutaneously into NMRI:nu/nu female mice [34]. This model was chosen because it is more representative for studying the anti-tumor effect related to humans. Treatment of mice started at palpable tumor size and was performed in a q4dx3 schedule. Mice were sacrificed when tumors reached a size of more than 1 cm3. The tumor volumes of treated tumors (T) were compared to those of controls (C), which were PBS-treated. White blood cell counts (WBC) were determined in peripheral blood, 4 days after initiation of therapy with a Coulter Counter.
Results
Drug sensitivity
All compounds were active in FM3A/0 cells (Table 1). The duplex, ETC-FdUrd, was active in FM3A/0, but 36-fold less active in the TK deficient cells FM3A/TK−. This indicates that TK may be important for the activity of ETC-FdUrd, thus this drug is possibly cleaved to the FdUrd nucleoside. The multidrug, ETC-L-FdUrd, was active in FM3A/0, but not in FM3A/TK− cells. In this cell line, cell growth by ETC-L-FdUrd was inhibited at a comparable level as FdUrd itself. When cells were exposed to the liposomal formulation of ETC-L-FdUrd, FM3A/TK− cells became 60-fold more sensitive, compared to ETC-L-FdUrd. However, FM3A/0 cells were less sensitive, compared to ETC-L-FdUrd with a 3-fold increase in IC50. In FM3A/TK− cells, the liposomal ETC-L-FdUrd was only 4-fold less active, indicating that TK was less important for the activity of this drug, either by circumvention of active transport or cleavage to ETC. Cells were exposed to the drugs for 72 h, which requires relatively low levels of the drugs, which remain below the K m for ENT mediated uptake, which is usually > 100 µM [35, 36]. At higher concentrations of these drugs, passive diffusion will become more important. Therefore we limited these experiments to a long exposure.
Transport into the cells
To determine whether the nucleoside transporter is circumvented by the multi- and duplex drugs, the inhibitor dipyridamole was used (Table 2). When dipyridamole decreases the drug cytotoxicity, these drugs are likely to be dependent on the equilibrative nucleoside transporter to enter the cells. As expected, in both cell lines the cytotoxicity of both ETC and FdUrd decreased after addition of dipyridamole. Since dipyridamole inhibited the sensitivity to the parent drugs, ENT seems to be the most important nucleoside transporter and the concentrative nucleoside transporter (CNT) is probably not. Dipyridamole did not protect against FdUrd cytotoxicity in FM3A/TK− cells, which can be explained by the inactivity of FdUrd in this cell line and the high concentration of FdUrd used in the assay, at which the drug may enter the cells via passive diffusion. The protection factor by dipyridamole against ETC-L-FdUrd was very low, in contrast to that of ETC-FdUrd of which the cytotoxicity was highly decreased. The activity of the liposomal formulation was not influenced at all by dipyridamole.
Extracellular cleavage of the drugs
Addition of nucleotidase and phosphatase inhibitors to the cell cultures resulted in a 5.5–8.3 fold decreased cytotoxicity of ETC-FdUrd (Table 2), indicating that ETC-FdUrd was cleaved outside the cells. By contrast, the cytotoxicity of ETC-L-FdUrd was hardly affected by nucleotidases or phosphatases. Also, the cytotoxicity of the liposomal formulation was hardly affected. Surprisingly, inhibition of nucleotide breakdown also reduced the sensitivity to ETC.
To demonstrate extracellular cleavage, we measured the concentration of the prodrugs, ETC and FdUrd by HPLC after 24 h incubation (Fig. 2). More than 60% of the parent ETC-FdUrd drug was still present in the medium. The concentrations of ETC and FdUrd were about 20 µM, which was sufficient to exert cytotoxic effects (Fig. 2). Extracellular ETC-L-FdUrd could not be detected with the applied HPLC method, but ETC was found in the medium at about 5 µM and FdUrd at concentrations below 1 µM. Similar low concentrations of ETC were detected after incubation with the liposomal formulation, whereas extracellular FdUrd was hardly detected. This is in line with the findings that the liposomal formulation was not affected by the inhibitors.

Cleavage of ETC-L-FdUrd, ETC-FdUrd and the liposome of ETC-L-FdUrd to the single nucleosides outside the cells after 24 h incubation with 100 µM with the prodrugs. Uncleaved forms of ETC-L-FdUrd and ETC-L-FdUrd liposome were not detectable by the applied HPLC assay and are therefore not included in the graph. Values represent means of 3–5 independent experiments ± SEM
FdUMP accumulation
After exposure to FdUrd, FdUMP accumulated in FM3A/0 cells, and as expected hardly in FM3A/TK− cells (Table 3). Exposure to ETC-FdUrd led to some accumulation of FdUMP in FM3A/0 cells, but hardly in TK− cells. ETC-L-FdUrd treatment resulted in a lower level of FdUMP accumulation in FM3A/0 cells than FdUrd alone, but accumulation was somewhat higher in FM3A/TK− cells. After exposure to the ETC-L-FdUrd liposomes, FdUMP accumulated at comparable levels in both FM3A/0 and TK− cells. These levels were comparable to the FdUMP concentration after treatment with ETC-L-FdUrd in FM3A/0 cells, indicating that this drug is at least partially cleaved to FdUMP.
TS in situ activity
Since the direct effect of FdUMP accumulation is TS inhibition, intracellular TS activity was measured after treatment with the different drugs. As shown in Fig. 3, TS activity was inhibited almost completely in FM3A/0 cells after treatment with FdUrd. TS activity of FM3A/0 cells was also inhibited effectively after treatment with both the multi- and duplex drugs. The extent of TS inhibition was FdUrd > ETC-FdUrd > ETC-L-FdUrd > liposomal ETC-L-FdUrd, respectively. In FM3A/TK− cells, a lower extent of TS inhibition was found. The degree of TS inhibition in these cells was FdUrd > liposome ETC-L-FdUrd > ETC-FdUrd > ETC-L-FdUrd.

TS in situ activity after 24 h exposure to 0.5 µM of FdUrd, ETC-FdUrd, ETC-L-FdUrd or the ETC-L-FdUrd liposome. Values represent means of three independent experiments ± SEM
In vivo sensitivity and accumulation of (phospho)nucleosides
In order to determine whether the in vitro activity would translate to an antitumor effect in vivo, we tested the drugs in the human HT29 xenograft model. HT29 was one of the cell lines most resistant to 5-FU in vitro [37], although this resistance was not due to an increased TS expression. HT29 cells have a normal ENT expression [38], but this is most likely not important for 5-FU since nucleoside transporters do not play a role in base transport. A major difference found in this cell line compared to other colon cancer cell lines is its high expression of dUTPase, which can prevent incorporation of FdUTP and dUTP into DNA (unpublished results).
The in vitro IC50 concentrations in these cells of the compounds are listed in Table 4. HT29 cells were 5-FU resistant, but not cross-resistant to FdUrd and they were more sensitive to ETC than to FdUrd. Both ETC-FdUrd and ETC-L-FdUrd had similar activity as the parent compounds. As summarized in Table 4, both i.v. and oral administration of the multidrug were effective in the HT29-xenograft, in contrast to the parental compounds, which were orally inactive. Both ETC-FdUrd and ETC-L-FdUrd were active after i.v. and oral treatment. Treatment with either ETC-L-FdUrd or ETC-FdUrd did not result in any haematological side effects. The antitumor effect was not dose dependent (data not shown). As a control, mice were treated with 5-FU, showing that HT-29 was resistant to this agent.
After 2 h treatment with ETC-FdUrd, the ETC-nucleoside and especially the ETC-nucleotides accumulated to a high extent in the tumor tissue (Fig. 4). ETC derived from ETC-FdUrd accumulated in the liver tissue, although the phosphate-forms were present at lower levels than in the tumor tissue. After 4 h, ETC or ETC-nucleotides were hardly present in either the tumor or the liver tissue (data not shown). After 2 h treatment with the lipophilic ETC-L-FdUrd, both ETC-nucleoside and the active nucleotide were detectable at comparable levels in the tumor tissue. Both ETC and ETC-nucleotides were present in the liver tissue, but to a lower extent than after exposure to ETC-FdUrd. After 4 h, an increase in ETC-nucleotides was detectable in the liver tissue, however at lower levels than detected with ETC-FdUrd (data not shown). In addition, the difference in the presence of ETC and ETC-nucleotides between ETC-FdUrd and ETC-L-FdUrd did not result in a difference in anti-tumor response. This may be related to a more rapid and direct action by ETC-FdUrd and a slower action by the liphophilic ETC-L-FdUrd due to a slow release or cleavage mechanism.

ETC nucleoside and nucleotide accumulation in human HT-29-xenograft (tumor) and liver tissues, 2 h after administration of the ETC-FdUrd and ETC-L-FdUrd. ETC and ETC nucleotides were measured with LC-MS/MS detection. Values represent means of three independent experiments ± SEM
Discussion
In the present study, we show that combining several advantages in one drug can be successful. The prodrugs were designed with the primary aim to circumvent decreased activation catalyzed by phosphorylation, while the ETC-L-FdURD might also circumvent transport to some extent. In vitro, ETC-FdUrd had the highest cytotoxic activity, compared to ETC-L-FdUrd, however it did not circumvent the nucleoside transporters, and it can be cleaved extracellularly. The long lipophilic chain of ETC-L-FdUrd can cause circumvention of the transporters, however without leading to an increased cellular uptake. When administered as a liposome, ETC-L-FdUrd was protected from extracellular cleavage, underlining the advantage of the use of liposomes as a delivery system.
To circumvent drug resistance, many types of new drugs are under development, including several prodrugs. Some examples of such prodrugs include a lipophilic derivative of Ara-C (NOAC) and a heteronucleoside phosphate dimer (NOAC-AraC), which have previously shown to exert promising anticancer cytotoxicity [39] CP-4055 (Elacyt; Clavis Pharma), an Ara-C elaidic acid derivative was reported to be active in in vitro and in vivo models in which Ara-C itself was inactive [40]. Elacyt can bypass the nucleoside transporter, but requires dCK for activation [41]. Moreover, CP-4055 had a different metabolism and intracellular accumulation than its parent compound, which underlines the potency of lipophilic prodrugs [8]. A different type of prodrug is FdUMP[10], a 10-mer of FdUMP, which has shown a higher activity than 5-FU and FdUrd and also has a higher activity in cells that overexpress TS [42, 43]. Other pronucleotide strategies have shown promising in vitro results, however many have been unsuccessful in vivo [8]. These different prodrugs are either synthesized using the nucleoside analogs or the mononucleotide of the parent drug. The latter can then bypass the first phosphorylation step. In order to combine the (pharmacologic and therapeutic) advantages of two drugs, new multi- and duplex drugs have been synthesized. The multidrug Ara-C-L-FdUrd has previously shown strong antitumor effects in vitro and could also overcome Ara-C and FdUrd resistance [18]. However, Ara-C-L-FdUrd is cleaved outside the cells to a high extent, limiting its cytotoxic activity [19]. Extracellular cleavage of ETC-L-FdUrd was found in the present study, indicating that the type of lipophilic chain does not determine the fate of the drug.
Nucleoside analogs require phosphorylation to their active forms, for FdUrd and ETC to FdUMP and ETCMP, respectively. Our data indicate that both ETC-L-FdUrd and ETC-FdUrd still require TK in order to be active, although ETC-L-FdUrd to a lesser extent. FdUrd may therefore be the most prominently released compound of this formulation.
Drug delivery systems, such as liposomes, lead to improved chemical stability of drugs, enhanced accumulation in tumors and decreased unwanted toxicity [44–46]. To enhance cellular drug uptake, the multidrug ETC-L-FdUrd was administered in a liposomal formulation. In vitro cytotoxicity levels in FM3A/TK− cells indicated that this formulation, when compared to ETC-L-FdUrd alone, is probably cleaved inside the cells behind the phosphate group, thereby circumventing TK. The decreased activity in the wild type FM3A/0 cells may be explained by a decreased permeability of the lipophilic drug because of accumulation in the lipophilic cell membrane [47]. Both i.v. and oral administration of ETC-L-FdUrd and ETC-FdUrd were effective, in contrast to the parental analogues which were inactive when administered orally. This underlines the advantage that these drugs have over the parental nucleoside analogs. ETC derived from ETC-L-FdUrd accumulated to a high extent in the liver, which has been described in previous studies using lipid drugs [8]. Optimized liposomal formulations could result in a decreased uptake/accumulation in the liver, such as polyethylene glycol (PEG) coated liposomes. PEG liposomes have shown an increased accumulation at tumor sites and have been evaluated in more than 20 clinical trials [8]. These liposomes may increase the activity of the multidrug used in this study. Moreover, these PEGylated liposomes were reported in various studies to be able to cross the blood brain barrier and to have an increased delivery at brain-tumor sites [8, 47]. Combining the attractive features of these drugs with lipophilic multidrugs may result in a higher efficacy and potency to these drugs.
In conclusion, ETC-FdUrd was active but could not completely circumvent resistance mechanisms, while ETC-L-FdUrd could only bypass the nucleoside transporter. The liposomal formulation of ETC-L-FdUrd showed the best potency and could circumvent important resistance mechanisms.
References
van Laar JA, Rustum YM, Ackland SP, van Groeningen CJ, Peters GJ (1998) Comparison of 5-fluoro-2′-deoxyuridine with 5-fluorouracil and their role in the treatment of colorectal cancer. Eur J Cancer 34:296–306
Köhne CH, Peters GJ (2000) UFT: mechanism of drug action. Oncology (Williston Park) 14:13–18
Folprecht G, Köhne CH (2005) Drug Insight: Metastatic colorectal cancer-oral fluoropyrimidines and new perspectives in the adjuvant setting. Nat Clin Pract Oncol 2:578–587
Peters GJ, Backus HH, Freemantle S, van Triest B, Codacci-Pisanelli G, van der Wilt CL et al (2002) Induction of thymidylate synthase as a 5-fluorouracil resistance mechanism. Biochim Biophys Acta 1587:194–205
Pastor-Anglada M, Cano-Soldado P, Molina-Arcas M, Lostao MP, Larráyoz I, Martínez-Picado J (2005) Cell entry and export of nucleoside analogues. Virus Res 107:151–164
Bergman AM, Kuiper CM, Voorn DA, Comijn EM, Myhren F, Sandvold ML et al (2004) Antiproliferative activity and mechanism of action of fatty acid derivatives of arabinofuranosylcytosine in leukemia and solid tumor cell lines. Biochem Pharmacol 67:503–511
Galmarini CM, Popowycz F, Joseph B (2008) Cytotoxic nucleoside analogues: different strategies to improve their clinical efficacy. Curr Med Chem 15:1072–1082
Adema AD, Bijnsdorp IV, Sandvold ML, Verheul HM, Peters GJ (2009) Innovations and opportunities to improve conventional (deoxy)nucleoside and fluoropyrimidine analogs in cancer. Cur Med Chem 16:4632–4643
Peyrottes S, Egron D, Lefebvre I, Gosselin G, Imbach JL, Périgaud C (2004) SATE pronucleotide approaches: an overview. Mini Rev Med Chem 4:395–408
Peters GJ, van der Wilt CL, van Moorsel CJ, Kroep JR, Bergman AM, Ackland SP (2000) Basis for effective combination cancer chemotherapy with antimetabolites. Pharmacol Ther 87:227–253
de Gramont A, Figer A, Seymour M, Homerin M, Hmissi A, Cassidy J et al (2000) Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J Clin Oncol 18:2938–2947
Köhne CH, van Cutsem E, Wils J, Bokemeyer C, El-Serafi M, Lutz MP et al (2005) Phase III study of weekly high-dose infusional fluorouracil plus folinic acid with or without irinotecan in patients with metastatic colorectal cancer: European organisation for research and treatment of cancer gastrointestinal group study 40986. J Clin Oncol 23:4856–4865
Douillard JY, Cunningham D, Roth AD, Navarro M, James RD, Karasek P et al (2000) Irinotecan combined with Fluorouracil compared with Fluorouracil alone as First-line treatment for metastatic colorectal cancer: a multicentre randomized trial. Lancet 355:1041–1047
McElhinney RS, McCormick JE, Bibby MC, Double JA, Atassi G, Dumont P et al (1989) Nucleoside analogues: 7. Effect on colon, breast and lung tumours in mice of 5-fluorouracil/nitrosourea molecular combinations incorporating alkoxy and oxidized sulphur functions. Anticancer Drug Des 3:255–269
Naito T, Yokogawa T, Kim HS, Futagami M, Wataya Y, Matsuda A, et al (2002) Anticancer mechanisms of 1-(3-C-ethynyl-beta-D-ribo-pentofuranosyl) cytosine (ECyd, TAS-106). Nucleic Acids Res Suppl (2):241–2
Hattori H, Tanaka M, Fukushima M, Sasaki T, Matsuda A (1996) Nucleosides and nucleotides. 158. 1-(3-C-ethynyl-beta-D-ribo-pentofuranosyl)-cytosine, 1-(3-C-ethynyl-beta-D-ribo-pentofuranosyl)uracil, and their nucleobase analogues as new potential multifunctional antitumor nucleosides with a broad spectrum of activity. J Med Chem 39:5005–5011
Shimamoto Y, Kazuno H, Murakami Y, Azuma A, Koizumi K, Matsuda A et al (2002) Cellular and biochemical mechanisms of the resistance of human cancer cells to a new anticancer ribo-nucleoside, TAS-106. Jpn J Cancer Res 93:445–452
Saiko P, Horvath Z, Illmer C, Madlener S, Bauer W, Hoechtl T et al (2005) Cytotoxic effects of novel amphiphilic dimers consisting of 5-fluorodeoxyuridine and arabinofuranosylcytosine in cross-resistant H9 human lymphoma cells. Leuk Res 29:785–791
Bijnsdorp IV, Schwendener RA, Schott H, Fichtner I, Smid K, Schott S et al (2007) In vivo and in vitro activity and mechanism of action of the multidrug cytarabine-L-glycerylyl-fluorodeoxyuridine. Nucleosides Nucleotides Nucleic Acids 26:1619–1624
Phuphanich S, Maria B, Braeckman R, Chamberlain M (2007) A pharmacokinetic study of intra-CSF administered encapsulated cytarabine (DepoCyt) for the treatment of neoplastic meningitis in patients with leukemia, lymphoma, or solid tumors as part of a phase III study. J Neurooncol 81:201–208
O’Shaughnessy JA (2003) Pegylated liposomal doxorubicin in the treatment of breast cancer. Clin Breast Cancer 4:318–328
Schwendener RA (2007) Liposomes in biology and medicine. Adv Exp Med Biol 620:117–128
Schott H, Schott S, Schwendener RA (2009) Synthesis and in vitro activities of new anticancer duplex drugs linking 2'-deoxy-5-fluorouridine (5-FdU) with 3'-C-ethynylcytidine (ECyd) via a phosphodiester bonding. Bioorg Med Chem 17:6824–6831
Balzarini J, de Clercq E, Ayusawa D, Seno T (1985) Murine mammary FM3A carcinoma cells transformed with the herpes simplex virus type 1 thymidine kinase gene are highly sensitive to the growth-inhibitory properties of (E)-5-(2-bromovinyl)-2'-deoxyuridine and related compounds. FEBS Lett 185:95–100
Keepers YP, Pizao PE, Peters GJ, van Ark-Otte J, Winograd B, Pinedo HM (1991) Comparison of the sulforhodamine B protein and tetrazolium (MTT) assays for in vitro chemosensitivity testing. Eur J Cancer 27:897–900
Wu W, Sigmond J, Peters GJ, Borch RF (2007) Synthesis and biological activity of a gemcitabine phosphoramidate prodrug. J Med Chem 50:3743–3746
Peters GJ, Laurensse E, Leyva A, Lankelma J, Pinedo HM (1986) Sensitivity of human, murine, and rat cells to 5-fluorouracil and 5'-deoxy-5-fluorouridine in relation to drug-metabolizing enzymes. Cancer Res 46:20–28
Rots MG, Pieters R, Kaspers GJL, van Zantwijk CH, Noordhuis P, Mauritz R et al (1999) Differential methotrexate resistance in childhood T- versus Common/PreB-Acute lymphoblastic Leukemia can be measured by an in situ thymidylate synthase inhibition assay, but not by the MTT assay. Blood 93:1067–1074
van Triest B, Pinedo HM, Telleman F, van der Wilt CL, Jansen G, Peters GJ (1997) Cross-resistance to antifolates in multidrug resistant cell lines with P-glycoprotein or multidrug resistance protein expression. Biochem Pharmacol 53:1855–1866
Spears CP, Gustavsson BG, Berne M, Frösing R, Bernstein L, Hayes AA (1988) Mechanisms of innate resistance to thymidylate synthase inhibition after 5-fluorouracil. Cancer Res 48:5894–5900
van der Wilt CL, van Laar JA, Smid K, Rustum YM, Peters GJ (1994) Comparison of 5-fluoro-2'-deoxyuridine and 5-fluorouracil in the treatment of murine colon cancer; effects on thymidylate synthase. Adv Exp Med Biol 370:109–114
Laurensse EJ, Pinedo HM, Peters GJ (1988) A sensitive non-radioactive assay for pyrimidine nucleoside phosphorylase using reversed-phase high performance liquid chromatography. Clin Chim Acta 178:71–78
Honeywell R, Laan AC, van Groeningen CJ, Strocchi E, Ruiter R, Giaccone G et al (2007) The determination of gemcitabine and 2'-deoxycytidine in human plasma and tissue by APCI tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 847:142–152
Fichtner I, Slisow W, Gill J, Becker M, Elbe B, Hillebrand T et al (2004) Anticancer drug response and expression of molecular markers in early-passage xenotransplanted colon carcinomas. Eur J Cancer 40:298–307
Pastor-Anglada M, Molina-Arcas M, Casado FJ, Bellosillo B, Colomer D, Gil J (2004) Nucleoside transporters in chronic lymphocytic leukaemia. Leukemia 18:385–393
Huber-Ruano I, Pastor-Anglada M (2009) Transport of nucleoside analogs across the plasma membrane: a clue to understanding drug-induced cytotoxicity. Curr Drug Metab 10:347–358
van Triest B, Pinedo HM, van Hensbergen Y, Smid K, Telleman F, Schoenmakers PS et al (1999) Thymidylate synthase level as the main predictive parameter for sensitivity to 5-fluorouracil, but not for folate-based thymidylate synthase inhibitors, in 13 nonselected colon cancer cell lines. Clin Cancer Res 5:643–654
Serova M, Galmarini CM, Ghoul A, Benhadji K, Green SR, Chiao J et al (2007) Antiproliferative effects of sapacitabine (CYC682), a novel 2'-deoxycytidine-derivative, in human cancer cells. Br J Cancer 97:628–636
Horber DH, Cattaneo-Pangrazzi RM, von Ballmoos P, Schott H, Ludwig PS, Eriksson S et al (2000) Cytotoxicity, cell-cycle perturbations and apoptosis in human tumor cells by lipophilic N4-alkyl-1-beta-D-arabinofuranosylcytosine derivatives and the new heteronucleoside phosphate dimer arabinocytidylyl-(5'- > 5')-N4-octadecyl-1-beta-D-arabinofuranosylcytosine. J Cancer Res Clin Oncol 126:311–319
Breistøl K, Balzarini J, Sandvold ML, Myhren F, Martinsen M, De Clercq E et al (1999) Antitumor activity of P-4055 (elaidic acid-cytarabine) compared to cytarabine in metastatic and s.c. human tumor xenograft models. Cancer Res 59:2944–2949
Bergman AM, Kuiper CM, Voorn DA, Comijn EM, Myhren F, Sandvold ML et al (2004) antiproliferative activity and mechanism of action of fatty acid derivatives of arabinofuranosylcytosine in leukemia and solid tumor cell lines. Biochem Pharmacol 67:503–511
Bijnsdorp IV, Comijn EM, Padron JM, Gmeiner WH, Peters GJ (2007) Mechanisms of action of FdUMP[10]: metabolite activation and thymidylate synthase inhibition. Oncol Rep 18:287–291
Gmeiner WH, Trump E, Wei C (2004) Enhanced DNA-directed effects of FdUMP[10] compared to 5FU. Nucleosides Nucleotides Nucleic Acids 23:401–410
Gabizon A, Catane R, Uziely B, Kaufman B, Safra T, Cohen R et al (1994) Prolonged circulation time and enhanced accumulation in malignant exudates of doxorubicin encapsulated in polyethylene-glycol coated liposomes. Cancer Res 54:987–992
Newman MS, Colbern GT, Working PK, Engbers C, Amantea MA (1999) Comparative pharmacokinetics, tissue distribution, and therapeutic effectiveness of cisplatin encapsulated in long-circulating, pegylated liposomes (SPI-077) in tumor-bearing mice. Cancer Chemother Pharmacol 43:1–7
Minko T, Pakunlu RI, Wang Y, Khandare JJ, Saad M (2006) New generation of liposomal drugs for cancer. Anticancer Agents Med Chem 6:537–552
Béduneau A, Saulnier P, Benoit JP (2007) Active targeting of brain tumors using nanocarriers; Biomaterials 28:4947–4967
Peters GJ, van Triest B, Backus HH, Kuiper CM, van der Wilt CL, Pinedo HM (2000) Molecular downstream events and induction of thymidylate synthase in mutant and wild-type p53 colon cancer cell lines after treatment with 5-fluorouracil and the thymidylate synthase inhibitor raltitrexed. Eur J Cancer 36:916–924
Acknowledgement
This study was supported by a minigrant from the Drug Development Committee of the EORTC-PAMM group.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Bijnsdorp, I.V., Schwendener, R.A., Schott, H. et al. Cellular pharmacology of multi- and duplex drugsconsisting of ethynylcytidine and 5-fluoro-2′-deoxyuridine. Invest New Drugs 29, 248–257 (2011). https://doi.org/10.1007/s10637-009-9353-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-009-9353-2