
Abstract
Background
Oguchi disease is a rare autosomal recessive form of congenital stationary night blindness caused by disease-causing variants in the rhodopsin kinase gene (GRK1) or the arrestin gene (SAG). Our study aims to describe the clinical features and identify the genetic defects for three Chinese patients with Oguchi disease.
Methods
We conducted detailed ophthalmologic examinations for three patients from three unrelated non-consanguineous Chinese families. Targeted next-generation sequencing (targeted NGS) and copy number variations (CNVs) analysis were applied to screen pathogenic variants. Sanger sequencing validation, quantitative real-time PCR (qPCR), and segregation analysis were further performed for confirmation. Subsequently, a combined genetic and structural biology approach was used to infer the likely functional consequences of novel variants.
Results
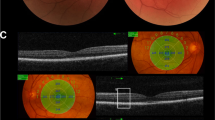
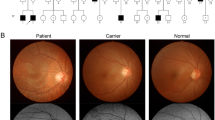
All three patients presented with typical clinical features of Oguchi disease, including night blindness, characteristic fundus appearance (Mizuo-Nakamura phenomenon), attenuated rod responses, and negative ERG waveforms. Their visual acuity and visual field were normal. Genetic analysis revealed two pathogenic variants in SAG and four pathogenic variants in GRK1. Patient 1 was identified to harbor compound heterozygous SAG variants c.874C > T (p.R292*) and exon2 deletion. Compound heterozygous GRK1 variants c.55C > T (p.R19*) and c.1412delC (p.P471Lfs*52) were found in patient 2. In patient 3, compound heterozygous GRK1 variants c.946C > A (p.R316S) and c.1388 T > C (p. L463P) were detected.
Conclusions
We reported the first two Chinese Oguchi patients with novel GRK1 pathogenic variants (P471Lfs*52, R316S, L463P) and one Oguchi case with SAG, indicating both GRK1 and SAG are important causative genes in Chinese Oguchi patients.




Similar content being viewed by others

Availability of data and materials
This work was conducted at Peking Union Medical College Hospital (PUMCH). The case’s data are available at PUMCH.
References
Fuchs S, Nakazawa M, Maw M, Tamai M, Oguchi Y, Gal A (1995) A homozygous 1–base pair deletion in the arrestin gene is a frequent cause of Oguchi disease in Japanese. Nat Genet 10(3):360–362
Yamamoto S, Sippel KC, Berson EL, Dryja TP (1997) Defects in the rhodopsin kinase gene in the Oguchi form of stationary night blindness. Nat Genet 15(2):175–178
Kühn H, Wilden U (1987) Deactivation of photoactivated rhodopsin by rhodopsin-kinase and arrestin. J Recept Res 7(1–4):283–298. https://doi.org/10.3109/10799898709054990
Hoare SRJ, Tewson PH, Sachdev S, Connor M, Hughes TE, Quinn AM (2021) Quantifying the kinetics of signaling and arrestin recruitment by nervous system G-protein coupled receptors. Front Cell Neurosci 15:814547. https://doi.org/10.3389/fncel.2021.814547
Moore CAC, Milano SK, Benovic JL (2007) Regulation of receptor trafficking by GRKs and arrestins. Annu Rev Physiol 69:451–482
Gurevich VV, Gurevich EV (2019) GPCR signaling regulation: the role of GRKs and arrestins. Front Pharmacol 10:125. https://doi.org/10.3389/fphar.2019.00125
Palczewski K, Rispoli G, Detwiler PB (1992) The influence of arrestin (48K protein) and rhodopsin kinase on visual transduction. Neuron 8(1):117–126. https://doi.org/10.1016/0896-6273(92)90113-r
Oguchi C (1907) Ueber eine Abart von Hemeralopie. Nippon Ganka Gakkai Zasshi (Acta Soc Ophthalmol Jpn) 11:123–134
Huang L, Li W, Tang W, Zhu X, Ou-yang P, Lu G (2012) A Chinese family with Oguchi’s disease due to compound heterozygosity including a novel deletion in the arrestin gene. Mol Vis 18:528
Liu X, Gao L, Wang G, Long Y, Ren J, Fujinami K et al (2020) Oguchi disease caused by a homozygous novel SAG splicing alteration associated with the multiple evanescent white dot syndrome: a 15-month follow-up. Doc Ophthalmol 141(3):217–226
Deng Z, Fan F, Tang D, Wu Y, Shu Y, Wu K (2022) A compound heterozygous mutation in the S-Antigen Visual Arrestin SAG gene in a Chinese patient with Oguchi type one: a case report. BMC Ophthalmol 22(1):99. https://doi.org/10.1186/s12886-022-02307-z
Robson AG, Frishman LJ, Grigg J, Hamilton R, Jeffrey BG, Kondo M et al (2022) ISCEV Standard for full-field clinical electroretinography (2022 update). Doc Ophthalmol 144(3):165–177. https://doi.org/10.1007/s10633-022-09872-0
Lodowski DT, Tesmer VM, Benovic JL, Tesmer JJ (2006) The structure of G protein-coupled receptor kinase (GRK)-6 defines a second lineage of GRKs. J Biol Chem 281(24):16785–16793
Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M (2019) CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 47(D1):D886–D894
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P et al (2010) A method and server for predicting damaging missense mutations. Nat Methods 7(4):248–249
Siepel A, Pollard KS, Haussler D (2006) New methods for detecting lineage-specific selection. Springer, Berlin. 190–205
Sim N-L, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC (2012) SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res 40(W1):W452–W457
Schwarz JM, Cooper DN, Schuelke M, Seelow D (2014) MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 11(4):361–362
Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S (2010) Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput Biol 6(12):e1001025
Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K et al (2005) Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15(8):1034–1050
Garber M, Guttman M, Clamp M, Zody MC, Friedman N, Xie X (2009) Identifying novel constrained elements by exploiting biased substitution patterns. Bioinformatics 25(12):i54–i62
Aryan H, Bahadori A, Farhud DD, Yeganeh MZ, Pourkalhor H (2020) A Homozygote mutation in S-Antigen Visual Arrestin SAG gene in an iranian patient with Oguchi type One: a case report. Iran J Public Health 49(5):995
Ballios BG, Weisbrod D, Kohly R, Muni RH, Wright T, Yan P (2020) Wide-field true-colour imaging and clinical characterization of a novel GRK1 mutation in Oguchi disease. Doc Ophthalmol 141(2):181–185. https://doi.org/10.1007/s10633-020-09759-y
Mirshahi A, Hassanpoor N, Khojasteh H, Baradaran MR, Faghihi H, Lashay A (2021) Oguchi disease associated with keratoconus. J Ophthalmic Vis Res 16(1):137–139. https://doi.org/10.18502/jovr.v16i1.8262
Ilhan C, Citirik M, Teke MY, Dulger SC (2020) Clinical findings in four siblings with genetically proven Oguchi disease. J Curr Ophthalmol 32(4):390–394. https://doi.org/10.4103/joco.Joco_155_20
Poulter JA, Gravett MSC, Taylor RL, Fujinami K, De Zaeytijd J, Bellingham J et al (2021) New variants and in silico analyses in GRK1 associated Oguchi disease. Hum Mutat 42(2):164–176. https://doi.org/10.1002/humu.24140
Arnold K, Bordoli L, Kopp J, Schwede T (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22(2):195–201
Blum M, Chang H-Y, Chuguransky S, Grego T, Kandasaamy S, Mitchell A et al (2021) The InterPro protein families and domains database: 20 years on. Nucleic Acids Res 49(D1):D344–D354
Venselaar H, Tebeek TAH, Kuipers RKP, Hekkelman ML, Vriend G (2010) Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinform 11:548. https://doi.org/10.1186/1471-2105-11-548
Gasteiger E, Hoogland C, Gattiker A, Duvaud Se, Wilkins MR, Appel RD, et al. (2005) Protein identification and analysis tools on the ExPASy server. In: Walker JM, editor. The Proteomics Protocols Handbook. Totowa, NJ: Humana Press. p. 571–607
Cheng J, Randall A, Baldi P (2006) Prediction of protein stability changes for single‐site mutations using support vector machines. Proteins Struct, Funct, Bioinform 62(4):1125–1132
Capriotti E, Fariselli P, Casadio R (2005) I-Mutant2.0 predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res 33 W306-W10
Singh P, Wang B, Maeda T, Palczewski K, Tesmer JJ (2008) Structures of rhodopsin kinase in different ligand states reveal key elements involved in G protein-coupled receptor kinase activation. J Biol Chem 283(20):14053–14062
Morgan AA, Rubenstein E (2013) Proline: the distribution, frequency, positioning, and common functional roles of proline and polyproline sequences in the human proteome. PLoS ONE 8(1):e53785. https://doi.org/10.1371/journal.pone.0053785
Godara P, Cooper RF, Sergouniotis PI, Diederichs MA, Streb MR, Genead MA et al (2012) Assessing retinal structure in complete congenital stationary night blindness and Oguchi Disease. Am J Ophthalmol 154(6):987-1001.e1. https://doi.org/10.1016/j.ajo.2012.06.003
Raghuram A, Hansen RM, Moskowitz A, Fulton AB (2013) Photoreceptor and postreceptor responses in congenital stationary night blindness. Invest Ophthalmol Vis Sci 54(7):4648–4658. https://doi.org/10.1167/iovs.13-12111
Nishiguchi KM, Oguchi Y, Nakazawa T (2020) Progression from classical Oguchi disease to retinitis pigmentosa after 50 years. Ophthalmology 127(1):51. https://doi.org/10.1016/j.ophtha.2019.09.015
Nishiguchi KM, Ikeda Y, Fujita K, Kunikata H, Akiho M, Hashimoto K et al (2019) Phenotypic features of Oguchi disease and retinitis pigmentosa in patients with S-Antigen mutations: a long-term follow-up study. Ophthalmology 126(11):1557–1566. https://doi.org/10.1016/j.ophtha.2019.05.027
Hayashi T, Tsuzuranuki S, Kozaki K, Urashima M, Tsuneoka H (2011) Macular dysfunction in Oguchi disease with the frequent mutation 1147delA in the <i>SAG</i> Gene. Ophthalmic Res 46(4):175–180. https://doi.org/10.1159/000325024
Hayashi T, Gekka T, Takeuchi T, Goto-Omoto S, Kitahara K (2007) A novel homozygous GRK1 mutation (P391H) in 2 siblings with Oguchi Disease with markedly reduced cone responses. Ophthalmology 114(1):134–41.e1. https://doi.org/10.1016/j.ophtha.2006.05.069
Morris TA, Fong SL (1993) Characterization of the gene encoding human cone transducin alpha-subunit (GNAT2). Genomics 17(2):442–448. https://doi.org/10.1006/geno.1993.1345
Piriev NI, Viczian AS, Ye J, Kerner B, Korenberg JR, Farber DB (1995) Gene structure and amino acid sequence of the human cone photoreceptor cGMP-phosphodiesterase alpha’ subunit (PDEA2) and its chromosomal localization to 10q24. Genomics 28(3):429–435
Feshchenko EA, Andreeva SG, Suslova VA, Smirnova EV, Zagranichny VE, Lipkin VM (1996) Human cone-specific cGMP phosphodiesterase alpha’ subunit: complete cDNA sequence and gene arrangement. FEBS Lett 381(1–2):149–152
Shimizu-Matsumoto A, Itoh K, Inazawa J, Nishida K, Matsumoto Y, Kinoshita S et al (1996) Isolation and chromosomal localization of the human cone cGMP phosphodiesterase gamma cDNA (PDE6H). Genomics 32(1):121–124
Wissinger B, Müller F, Weyand I, Schuffenhauer S, Thanos S, Kaupp UB et al (1997) Cloning, chromosomal localization and functional expression of the gene encoding the alpha-subunit of the cGMP-gated channel in human cone photoreceptors. Eur J Neurosci 9(12):2512–2521
Kohl S, Baumann B, Broghammer M, Jägle H, Sieving P, Kellner U et al (2000) Mutations in the CNGB3 gene encoding the beta-subunit of the cone photoreceptor cGMP-gated channel are responsible for achromatopsia (ACHM3) linked to chromosome 8q21. Hum Mol Genet 9(14):2107–2116
Sundin OH, Yang JM, Li Y, Zhu D, Hurd JN, Mitchell TN et al (2000) Genetic basis of total colourblindness among the Pingelapese islanders. Nat Genet 25(3):289–293
Colombo L, Abeshi A, Maltese PE, Frecer V, Miertuš J, Cerra D et al (2019) Oguchi type I caused by a homozygous missense variation in the SAG gene. Eur J Med Genet 62(9):103548. https://doi.org/10.1016/j.ejmg.2018.09.015
Fujinami K, Tsunoda K, Nakamura M, Oguchi Y, Miyake Y (2011) Oguchi disease with unusual findings associated with a heterozygous mutation in the SAG gene. Arch Ophthalmol 129(10):1375–1376. https://doi.org/10.1001/archophthalmol.2011.300
Saga M, Mashima Y, Kudoh J, Oguchi Y, Shimizu N (2004) Gene analysis and evaluation of the single founder effect in Japanese patients with Oguchi disease. Jpn J Ophthalmol 48(4):350–352. https://doi.org/10.1007/s10384-004-0070-2
Sonoyama H, Shinoda K, Ishigami C, Tada Y, Ideta H, Ideta R et al (2011) Oguchi disease masked by retinitis pigmentosa. Doc Ophthalmol 123(2):127–133. https://doi.org/10.1007/s10633-011-9286-x
Maw M, Kumaramanickavel G, Kar B, John S, Bridges R, Denton M (1998) Two Indian siblings with Oguchi disease are homozygous for an arrestin mutation encoding premature termination. Hum Mutat 11(S1):S317–S319. https://doi.org/10.1002/humu.1380110199
Waheed NK, Qavi AH, Malik SN, Maria M, Riaz M, Cremers FP et al (2012) A nonsense mutation in S-antigen Glu306* causes Oguchi disease. Mol Vis 18:1253
Nakamura M, Yamamoto S, Okada M, Ito S, Tano Y, Miyake Y (2004) Novel mutations in the arrestin gene and associated clinical features in Japanese patients with Oguchi’s disease. Ophthalmology 111(7):1410–1414
Mucciolo DP, Sodi A, Murro V, Passerini I, Palchetti S, Pelo E et al (2018) A novel GRK1 mutation in an Italian patient with Oguchi disease. Ophthalmic Genet 39(1):137–138
Teke MY, Citirik M, Kabacam S, Demircan S, Alikasifoglu M (2016) A novel missense mutation of the GRK1 gene in Oguchi disease. Mol Med Rep 14(4):3129–3133
Azam M, Collin RW, Khan MI, Shah STA, Qureshi N, Ajmal M et al (2009) A novel mutation in GRK1 causes Oguchi disease in a consanguineous Pakistani family. Mol Vis 15:1788
Zhang Q, Zulfiqar F, Riazuddin SA, Xiao X, Yasmeen A, Rogan PK et al (2005) A variant form of Oguchi disease mapped to 13q34 associated with partial deletion of GRK1 gene. Mol Vis 11:977–985
Oishi A, Akimoto M, Kawagoe N, Mandai M, Takahashi M, Yoshimura N (2007) Novel mutations in the GRK1 gene in Japanese patients With Oguchi disease. Am J Ophthalmol 144(3):475–477
Skorczyk-Werner A, Kocięcki J, Wawrocka A, Wicher K, Krawczyńiski MR (2015) The first case of Oguchi disease, type 2 in a Polish patient with confirmed GRK1 gene mutation. Klin Oczna 117(1):27–30
Kuroda M, Hirami Y, Nishida A, Jin Z-B, Ishigami C, Takahashi M et al (2011) A case of Oguchi disease with disappearance of golden tapetal-like fundus reflex after vitreous resection. Nippon Ganka Gakkai Zasshi 115(10):916–923
Tawfik CA, Elbagoury NM, Khater NI, Essawi ML (2022) Mutation analysis reveals novel and known mutations in SAG gene in first two Egyptian families with Oguchi disease. BMC Ophthalmol 22(1):217. https://doi.org/10.1186/s12886-022-02444-5
Pilotto E, Trevisson E, Nacci EB, Longhin E, Guidolin F, Midena E (2021) Two novel compound heterozygous mutations in an Italian patient with Oguchi disease: A genetic and multimodal retinal imaging study. Eur J Ophthalmol 32 11206721211027422 doi: https://doi.org/10.1177/11206721211027422
Li L, Chen Y, Jiao X, Jin C, Jiang D, Tanwar M et al (2017) Homozygosity mapping and genetic analysis of autosomal recessive retinal dystrophies in 144 Consanguineous Pakistani Families. Invest Ophthalmol Vis Sci 58(4):2218–2238. https://doi.org/10.1167/iovs.17-21424
Sergouniotis PI, Davidson AE, Sehmi K, Webster AR, Robson AG, Moore AT (2011) Mizuo-Nakamura phenomenon in Oguchi disease due to a homozygous nonsense mutation in the SAG gene. Eye (Lond) 25(8):1098–1101. https://doi.org/10.1038/eye.2011.88
Arencibia JM, Pastor-Flores D, Bauer AF, Schulze JO, Biondi RM (2013) AGC protein kinases: from structural mechanism of regulation to allosteric drug development for the treatment of human diseases. Biochim Et Biophys Acta (BBA)-Proteins and Proteomics 1834(7):1302–21
Hirsch JA, Schubert C, Gurevich VV, Sigler PB (1999) The 2.8 A crystal structure of visual arrestin: a model for arrestins regulation. Cell. 97(2):257–70
Acknowledgements
We thank all the patients and their family members for participating in the study.
Funding
This work was supported by CAMS Innovations Fund for Medical Sciences (CIFMS 2021-I2M-1–003) and the National Natural Science Foundation of China 81873687.
Author information
Authors and Affiliations
Contributions
XW wrote the main manuscript. RS designed and supported the study, analyzed data and revised the paper. XW, RS, HL, SW and TZ examined the patients. XW, SW and TZ performed the genetic analysis. All authors reviewed the results and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Ethics approval
This study was complied with the Guidance on Sample Collection of Human Genetic Diseases by the Ministry of Public Health of China and the tenets of the Declaration of Helsinki. Institutional Review Board of PUMCH (No. JS-2059) approved the study.
Consent to publication
Informed consent was obtained from each patient’s guardians in the study.
Statement of human rights
All procedures performed in the study were in accordance with the Guidance on Sample Collection of Human Genetic Diseases by the Ministry of Public Health of China and the tenets of the Declaration of Helsinki. The study was approved by the Institutional Review Board of PUMCH (No. JS-2059).
Statement on the welfare of animals
This study did not involve the use of animal subjects.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wei, X., Li, H., Wu, S. et al. Genetic analysis and clinical features of three Chinese patients with Oguchi disease. Doc Ophthalmol 146, 17–32 (2023). https://doi.org/10.1007/s10633-022-09910-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10633-022-09910-x