
Abstract
Background
The aim of this study is to identify the genetic defects in a Chinese family with fundus albipunctatus.
Methods
Complete ophthalmic examinations, including slit-lamp biomicroscopy, dilated indirect ophthalmoscopy, fundus photography, autofluorescence, swept source optical coherence tomography (SS-OCT) and full-field electroretinography (ffERG) were performed. Genomic DNA was extracted from blood samples and whole genome sequencing was performed. Variants were validated with Sanger sequencing.
Results
Six members in this Chinese family, including three affected individuals and three controls, were recruited in this study. The ophthalmic examination of three recruited patients was consistent with fundus albipunctatus. Three variants, a novel frameshift deletion c.39delA [p.(Val14CysfsX47] and a haplotype of two rare missense variants, c.683G > A [p.(Arg228Gln)] along with c.710A > G [p.(Tyr237Cys], within the retinal dehydrogenase 5 (RDH5) gene were found to segregate with fundus albipunctatus in this family in an autosomal recessive matter.
Conclusion
We identified novel compound heterozygous variants in RDH5 responsible for fundus albipunctatus in a large Chinese family. The results of our study further broaden the genetic defects of RDH5 associated with fundus albipunctatus.
Similar content being viewed by others

Background
Fundus albipunctatus (FA; Online Mendelian Inheritance in Man identifier, OMIM #136880), a kind of autosomal recessive form disease, is mainly characterized by nonprogressive night blindness [1]. A lot of small white or pale-yellow spots are scattered in the retina and the macula may or may not be involved [1, 2]. With the increase of age, the shape and number of spots in the retina will change, or even disappear completely [2, 3]. FA was discriminated from a similar disease called retinitis punctata albescens (RPA) [4, 5], and pigmentary degeneration, narrow vasculature and visual field loss are the main characteristics of RPA different from FA. After the standard 30-min dark adaptation, standard full-field electroretinograms (ERGs) show severe reduction in rod responses, while after prolonged dark adaptation for nearly 3 h, the rod responses almost can recover to normal or near-normal levels [3, 6, 7].
The retinol dehydrogenase 5 (RDH5, OMIM 601617) gene, located on chromosome 12q13-q14 and encoding 11-cis-retinol dehydrogenase [8,9,10], is found in abundance in the smooth endoplasmic reticulum of the retinal pigment epithelium (RPE) [11]. This enzyme is a 32-kDa membrane-bound enzyme with 318 amino acids [12, 13]. Variants in RDH5 are associated with fundus albipunctatus and the first identification of clinically significant changes of the RDH5 sequence has been reported in 1999 [14].
In this study, we described the clinical features and molecular genetic results of a large Chinese Han family affected with FA. Novel variants in the RDH5 gene are presented and we expand the spectrum of related genetic defects associated with FA.
Methods
Subject recruitment and clinical examination
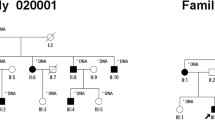
Six members (II:2, II:5, II:8, II:11, III:1, III:8) of the family (Fig. 1) were recruited in Shanghai General Hospital, Shanghai, China. This study was conducted in accordance with the Declaration of Helsinki and was approved by the ethics committee of Shanghai General Hospital. Informed consent was obtained from each member. Non-consanguineous marriages were found in the family. Three of the recruited six members were diagnosed with FA. A full medical history for longitudinal evaluation of the phenotype was obtained for recruited patients. Comprehensive clinical and ophthalmic examination included best corrected visual acuity, intraocular pressure measurement, slit lamp examination, dilated indirect ophthalmoscopy, fundus photography, autofluorescence, swept source optical coherence tomography (SS-OCT) and full-field electroretinography (ffERG), as well as the examination of physical malformations and neurological deficits.

Pedigree of the family with Fundus Albipunctatus. Solid symbols indicate affected individuals, and open symbols indicate unaffected individuals. Arrow indicates the proband of this family
DNA preparation
Genomic DNAs were extracted from peripheral blood using the TruSeq DNA LT Sample Prep kit (Illumina, San Diego, CA) according to the manufacturer’s protocol. DNA samples were stored at − 20 °C until used, and DNA integrity was evaluated by 1% agarose gel electrophoresis [15, 16].
Whole-genome sequencing
Whole-genome sequencing (WGS) was performed in three patients (II:2, II:5, II:8) and three unaffected family members (II:11, III:1, III:8). The libraries were constructed with the TruSeq Nano DNA LT Sample Prepararion Kit (Illumina, San Diego, CA, USA). Briefly, the genomic DNA was sheared into fragments with length ~ 350 bp using S220 Focused-ultrasonicators (Covaris, USA) [17]. Adapters were ligated onto the 3′ end of the sheared fragments. After polymerase chain reaction (PCR) amplification and purification, the final libraries were sequenced on the Illumina sequencing platform HiSeq X Ten platform (Illumina Inc., San Diego, CA, USA) and 150 bp paired-end reads were generated [18]. The raw data Q30 was above 96.3%, the average sequencing depth was at least 30× and the percentage of the genome region with coverage above 10X was 98.8%.
Bioinformatic analysis
The raw reads were subjected to a quality check and then filtered by fastp (https://github.com/OpenGene/fastp). Reads were aligned to the human genome (hg38) using SpeedSeq [15]. Single nucleotide variants (SNV) and insertions/deletions (Indels) calling was performed using the Genome Analysis Toolkit v4. 1[19]. Structural variants (SVs) and copy number variants (CNVs) were analyzed in SpeedSeq [15]. Annotations of SNVs, InDels, SVs and CNVs were performed with ANNOVAR [20]. Variant filtering was performed as illustrated in Supplementary 1.
Sanger sequencing
In order to verify the variants in RDH5 gene after WGS analysis, primers were designed using Primer3 software (version 4.0, http://bioinfo.ut.ee/primer3-0.4.0/). PCR primer pairs and amplification conditions are available upon request. PCR products were checked by 1% agarose gel electrophoresis and purified with SAP-Exon I kit (USB, USA) [21]. Purified PCR products were directly sequenced in both forward and reverse directions using an ABI 3730xl genetic analyzer (Applied Biosystems, Foster City, CA, USA) per manufacturer’s instructions [16, 21]. DNA sequences were analyzed using Chromas (version 2.22) and DNAMAN (version 7) software [16, 21].
Results
Clinical findings
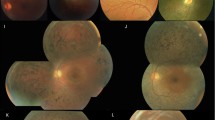
The pedigree of this family is shown in Fig. 1 and suggests an autosomal recessive inheritance. The ophthalmic examination of three recruited patients within this family was consistent with fundus albipunctatus, while other three additional family members were recruited which were unaffected. As illustrated in Table 1, the three affected patients (II:2, II:5, II:8) presented night blindness in both eyes since their early childhood. They received an ophthalmic examination and showed similar clinical symptoms. Representative photos of fundus photography, autofluorescence and SS-OCT of the two patients (II:5, II:8) are shown in Fig. 2. Some non-ocular symptoms, such as intellectual disability, kidney disease, neurological deficits were not found in the patients. Three members, II:11, III:1 and III:8, have no night-blindness phenotype or other major eye diseases.

Representative ophthalmic examination results of the II:8 and II:5 patients with Fundus Albipunctatus. (A1-A4) Fundus photography. (B1-B4) Fundus autofluorescence. (C1-C4) Optical coherence tomography
All patients show a moderate to severe loss of the rod-specific ERG b-wave after a standard period of dark adaptation. Table 2 shows the partial data of ffERG of the three affected patients included in this Chinese Han family.
Variant analysis and verification
Whole genome sequence data of the affected family members was compared with that of other three unaffected family members. Three variants, a novel frameshift deletion NM_002905.5:c.39delA; p.(Val14CysfsX47) and a haplotype of two missense variants [c.683G > A; p.(Arg228Gln) and c.710A > G; p.(Tyr237Cys)], were found in a compound heterozygous state within the RDH5 gene in the three patients. The frameshift variant, p.(Val14CysfsX47) is absent from the Genome Aggregation Database (gnomAD) database and predicted to lead to an early frameshift in protein translation and likely targeted by nonsense medicated decay. The two missense variants [p.(Arg228Gln) and p.(Tyr237Cys)] are mutations linked to one chromosome according to Fig. 3. And they are both located at NAD(P)-binding domain and conserved between species (GERP: 4.88 for both variants), have a Combined Annotation Dependent Depletion (CADD) score of 16.5 and 29.4 respectively, are predicted damaging by Mutation Taster and are present in low frequency in gnomAD, with a minor allele frequency of 0.0005 and 0.00007 in the South East Asian population respectively. These three variants were subsequently confirmed via Sanger sequencing (Fig. 4). The variants were not found either in any of the unaffected members and in the 300 unrelated controls from the same ethnic background. Finally, this three variant were Classifying with likely Pathogenic of the 2015 American College of Medical Genetics and Genomics (ACMG) guidelines by Intervar [22]. More specific variant annotation details can be seen in Supplementary 2 and Supplementary 3.

The comparison of high throughput sequencing between one affected (II:2) and one unaffected (II:11) member by Integrative Genomics Viewer (IGV)

Partial Sanger traces of the RDH5 gene. The arrow indicates the variant position. (A) A novel frameshift deletion c.39delA;p.(Val14CysfsX47); (B and C) Missense variants c.683G > A;p.(Arg228Gln) and c.710A > G; p.(Tyr237Cys) which are present on the same haplotype
Discussion
Fundus albipunctatus (FA) is a kind of flecked retinal syndrome, which also includes benign familial fleck retina, fleck retina of Kandori, Oguchi disease, retinitis punctate albescens (RPA), and vitamin A deficiency. As a rare autosomal recessive disorder, FA is characterized by impaired dark adaptation and the widely-distributed yellow or white dot lesions in the retina [1, 2]. Patients with fundus albipunctatus have suffered from night blindness since childhood. The main clinical manifestation of them is delayed dark adaptation, which means that they are difficult to adapt to conditions from bright light to dark, usually taking 2–3 h to adapt. The flecks, between the outer limiting membrane (OLM) and the outer aspect of RPE, are widely distributed at the outer edge of the retina [6]. Affected individuals typically have normal visual acuity with severely abnormal rod ERGs, but cone ERG abnormalities, macular dysfunction, and disease progression also have been reported [1, 23,24,25,26,27,28,29,30]. These patients in our study were diagnosed as fundus albipunctatus because reduced rod b waves could be found after a short time of dark-adaptation, and then the rod b waves improved after a prolonged dark-adaptational period. They also had white punctata which closely resembled those of fundus albipunctatus.
In this family and other reported cases, FA is caused by variants of RDH5 gene. However, other two genes, retinaldehyde binding protein 1 (RLBP1) gene [31, 32] and RPE-specific protein (RPE65) gene [33], can also lead to FA. Mutations in RLBP1 gene are also associated with RPA [34,35,36]. Furthermore, 11-cis retinol and 11-cis retinaldehyde as its ligands, RLBP1 is expressed in the RPE cells and Müller cells [37]. As the isomerase of the visual cycle and important role in 11-cis retinal production [38], RPE65 mutations also have been associated in FA-like change [39]. Therefore, molecular evaluation of RDH5 gene plays an important role in distinguishing FA and RPA.
This study described the clinical characteristics and phenotypic variation of a Chinese Han family with newly identified compound heterozygous RDH5 variants. The RDH5, encoding the 11-cis retinol dehydrogenase, is predominantly expressed in the smooth endoplasmic reticulum of the RPE [12]. RPE cells participate in the uptake and metabolism of retinoids in the retinoid cycle and play significant roles in maintaining normal visual function [11, 40]. The 11-cis retinol dehydrogenase can oxidize 11-cis-retinol to 11-cis-retinal [41], which is then transported to adjacent photoreceptors as chromophore in rhodopsin, and in the cones under dark-adapted condition [42]. RDH5 variants resulted in a significant reduction of the stability and the activities of this enzyme [43, 44]. Table 3 summarizes some mutations in the RDH5 gene that have been reported in families with FA.
According to some long-term follow-up reports, usually there is no progression in rod response in patients with this kind of night blindness, but some patients, especially the elderly, cone dystrophy is progressive [26, 28, 29]. It has been estimated that more than 30% of FA patients would be affected by cone dysfunction [6, 29, 49]. Lidén et al. suggested that cone dystrophy may be the result of RPE function impairment caused by RDH5 gene mutation, or it may be the direct result of a reduced supply of 11-cis retinal to cones [44]. The ability to complete recovery of retinal function after prolonged dark-adaptation provided a new idea that RDH5-related disease may be one of suitable candidates for gene therapy.
Conclusion
Variants in the RDH5 gene cause autosomal recessive fundus albipunctatus, a rare form of night blindness that is characterized by a delay in the regeneration of cone and rod photopigments. The present study expands our knowledge of RDH5-related retinal dysfunction. We identified three variants in RDH5, a novel frameshift deletion Val14CysfsX47, and a haplotype of rare missense variants (Arg228Gln + Tyr237Cys), are responsible for fundus albipunctatus patients in this Chinese family. These results of our study further broaden the genetic defects of RDH5 associated with fundus albipunctatus.
Availability of data and materials
The datasets generated during the current study are available in the National Genomics Data Center (NGDC) repository, the accession number is HRA000883 and the persistent web link is https://ngdc.cncb.ac.cn/gsa-human/s/OXeY408H.
Abbreviations
- FA:
-
Fundus albipunctatus
- RDH5:
-
Retinol dehydrogenase 5
- RPE:
-
Retinal pigment epithelium
- RPA:
-
Retinitis punctata albescens
- BCVA:
-
Best corrected visual acuity
- OCT:
-
Optical coherence tomography
- WGS:
-
Whole genome sequencing
- PCR:
-
Polymerase chain reaction
- OCT:
-
Optical coherence tomography
- ffERG:
-
Full-field electroretinography
References
Nakamura M, Lin J, Miyake Y. Young monozygotic twin sisters with fundus albipunctatus and cone dystrophy. Arch Ophthalmol. 2004;122(8):1203–7.
Sekiya K, Nakazawa M, Ohguro H, Usui T, Tanimoto N, Abe H. Long-term fundus changes due to Fundus albipunctatus associated with mutations in the RDH5 gene. Arch Ophthalmol. 2003;121(7):1057–9.
Dryja TP. Molecular genetics of Oguchi disease, fundus albipunctatus, and other forms of stationary night blindness: LVII Edward Jackson Memorial Lecture. Am J Ophthalmol. 2000;130(5):547–63.
Healy JJ. RETINITIS PUNCTATA ALBESCENS. Br J Ophthalmol. 1921;5(1):18–9.
Ramdani T, Sekhsoukh R. Retinitis punctata albescens. Pan Afr Med J. 2016;25:39.
Sergouniotis PI, Sohn EH, Li Z, McBain VA, Wright GA, Moore AT, et al. Phenotypic variability in RDH5 retinopathy (Fundus Albipunctatus). Ophthalmology. 2011;118(8):1661–70.
Carr RE. Congenital stationary nightblindness. Trans Am Ophthalmol Soc. 1974;72:448–87.
Driessen CA, Winkens HJ, Hoffmann K, Kuhlmann LD, Janssen BP, Van Vugt AH, et al. Disruption of the 11-cis-retinol dehydrogenase gene leads to accumulation of cis-retinols and cis-retinyl esters. Mol Cell Biol. 2000;20(12):4275–87.
Farjo KM, Moiseyev G, Takahashi Y, Crouch RK, Ma JX. The 11-cis-retinol dehydrogenase activity of RDH10 and its interaction with visual cycle proteins. Invest Ophthalmol Vis Sci. 2009;50(11):5089–97.
Jang GF, Van Hooser JP, Kuksa V, McBee JK, He YG, Janssen JJ, et al. Characterization of a dehydrogenase activity responsible for oxidation of 11-cis-retinol in the retinal pigment epithelium of mice with a disrupted RDH5 gene. A model for the human hereditary disease fundus albipunctatus. J Biol Chem. 2001;276(35):32456–65.
Simon A, Romert A, Gustafson AL, McCaffery JM, Eriksson U. Intracellular localization and membrane topology of 11-cis retinol dehydrogenase in the retinal pigment epithelium suggest a compartmentalized synthesis of 11-cis retinaldehyde. J Cell Sci. 1999;112(Pt 4):549–58.
Simon A, Lagercrantz J, Bajalica-Lagercrantz S, Eriksson U. Primary structure of human 11-cis retinol dehydrogenase and organization and chromosomal localization of the corresponding gene. Genomics. 1996;36(3):424–30.
Wada Y, Abe T, Fuse N, Tamai M. A frequent 1085delC/insGAAG mutation in the RDH5 gene in Japanese patients with fundus albipunctatus. Invest Ophthalmol Vis Sci. 2000;41(7):1894–7.
Yamamoto H, Simon A, Eriksson U, Harris E, Berson EL, Dryja TP. Mutations in the gene encoding 11-cis retinol dehydrogenase cause delayed dark adaptation and fundus albipunctatus. Nat Genet. 1999;22(2):188–91.
Chiang C, Layer RM, Faust GG, Lindberg MR, Rose DB, Garrison EP, et al. SpeedSeq: ultra-fast personal genome analysis and interpretation. Nat Methods. 2015;12(10):966–8.
Qian T, Chen C, Li C, Gong Q, Liu K, Wang G, et al. Xu X: A novel 4.25 kb heterozygous deletion in PAX6 in a Chinese Han family with congenital aniridia combined with cataract and nystagmus. BMC Ophthalmol. 2021;21(1):353.
Díaz-de Usera A, Lorenzo-Salazar JM, Rubio-Rodríguez LA, Muñoz-Barrera A, Guillen-Guio B, Marcelino-Rodríguez I, et al. Evaluation of Whole-Exome Enrichment Solutions: Lessons from the High-End of the Short-Read Sequencing Scale. J Clin Med. 2020;9(11).
Nair SS, Luu PL, Qu W, Maddugoda M, Huschtscha L, Reddel R, et al. Guidelines for whole genome bisulphite sequencing of intact and FFPET DNA on the Illumina HiSeq X Ten. Epigenetics & chromatin. 2018;11(1):24.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164.
Cen Z, Jiang Z, Chen Y, Zheng X, Xie F, Yang X, et al. Intronic pentanucleotide TTTCA repeat insertion in the SAMD12 gene causes familial cortical myoclonic tremor with epilepsy type 1. Brain J Neurol. 2018;141(8):2280–8.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Yamamoto H, Yakushijin K, Kusuhara S, Escaño MF, Nagai A, Negi A. A novel RDH5 gene mutation in a patient with fundus albipunctatus presenting with macular atrophy and fading white dots. Am J Ophthalmol. 2003;136(3):572–4.
Nakamura M, Miyake Y. Macular dystrophy in a 9-year-old boy with fundus albipunctatus. Am J Ophthalmol. 2002;133(2):278–80.
Nakamura M, Hotta Y, Tanikawa A, Terasaki H, Miyake Y. A high association with cone dystrophy in Fundus albipunctatus caused by mutations of the RDH5 gene. Invest Ophthalmol Vis Sci. 2000;41(12):3925–32.
Wada Y, Abe T, Sato H, Tamai M. A novel Gly35Ser mutation in the RDH5 gene in a Japanese family with fundus albipunctatus associated with cone dystrophy. Arch Ophthalmol. 2001;119(7):1059–63.
Hotta K, Nakamura M, Kondo M, Ito S, Terasaki H, Miyake Y, et al. Macular dystrophy in a Japanese family with fundus albipunctatus. Am J Ophthalmol. 2003;135(6):917–9.
Nakamura M, Skalet J, Miyake Y. RDH5 gene mutations and electroretinogram in fundus albipunctatus with or without macular dystrophy: RDH5 mutations and ERG in fundus albipunctatus. Doc Ophthalmol Adv Ophthalmol. 2003;107(1):3–11.
Niwa Y, Kondo M, Ueno S, Nakamura M, Terasaki H, Miyake Y. Cone and rod dysfunction in fundus albipunctatus with RDH5 mutation: an electrophysiological study. Invest Ophthalmol Vis Sci. 2005;46(4):1480–5.
Wang C, Nakanishi N, Ohishi K, Hikoya A, Koide K, Sato M, et al. Novel RDH5 mutation in family with mother having fundus albipunctatus and three children with retinitis pigmentosa. Ophthalmic Genet. 2008;29(1):29–32.
Katsanis N, Shroyer NF, Lewis RA, Cavender JC, Al-Rajhi AA, Jabak M, et al. Fundus albipunctatus and retinitis punctata albescens in a pedigree with an R150Q mutation in RLBP1. Clin Genet. 2001;59(6):424–9.
Naz S, Ali S, Riazuddin SA, Farooq T, Butt NH, Zafar AU, et al. Mutations in RLBP1 associated with fundus albipunctatus in consanguineous Pakistani families. Br J Ophthalmol. 2011;95(7):1019–24.
Schatz P, Preising M, Lorenz B, Sander B, Larsen M, Rosenberg T. Fundus albipunctatus associated with compound heterozygous mutations in RPE65. Ophthalmology. 2011;118(5):888–94.
Torres-Costa S, Ferreira CS, Grangeia A, Santos-Silva R, Brandão E, Estrela-Silva S, et al. A novel homozygous frameshift variant in the cellular retinaldehyde-binding protein 1 (RLBP1) gene causes retinitis punctata albescens. Eur J Ophthalmol. 2021;31(3):Np74–np80.
Burstedt MS, Sandgren O, Holmgren G, Forsman-Semb K. Bothnia dystrophy caused by mutations in the cellular retinaldehyde-binding protein gene (RLBP1) on chromosome 15q26. Invest Ophthalmol Vis Sci. 1999;40(5):995–1000.
Morimura H, Berson EL, Dryja TP. Recessive mutations in the RLBP1 gene encoding cellular retinaldehyde-binding protein in a form of retinitis punctata albescens. Invest Ophthalmol Vis Sci. 1999;40(5):1000–4.
Sparkes RS, Heinzmann C, Goldflam S, Kojis T, Saari JC, Mohandas T, et al. Assignment of the gene (RLBP1) for cellular retinaldehyde-binding protein (CRALBP) to human chromosome 15q26 and mouse chromosome 7. Genomics. 1992;12(1):58–62.
Moiseyev G, Chen Y, Takahashi Y, Wu BX, Ma JX. RPE65 is the isomerohydrolase in the retinoid visual cycle. Proc Natl Acad Sci U S A. 2005;102(35):12413–8.
Li S, Xiao X, Yi Z, Sun W, Wang P, Zhang Q. RPE65 mutation frequency and phenotypic variation according to exome sequencing in a tertiary centre for genetic eye diseases in China. Acta ophthalmologica. 2020;98(2):e181–90.
Lhor M, Salesse C. Retinol dehydrogenases: membrane-bound enzymes for the visual function. Biochem Cell Biol. 2014;92(6):510–23.
Hirose E, Inoue Y, Morimura H, Okamoto N, Fukuda M, Yamamoto S, et al. Mutations in the 11-cis retinol dehydrogenase gene in Japanese patients with Fundus albipunctatus. Invest Ophthalmol Vis Sci. 2000;41(12):3933–5.
Liu X, Liu L, Li H, Xu F, Jiang R, Sui R. RDH5 retinopathy (fundus albipunctatus) with preserved rod function. Retina (Philadelphia, Pa). 2015;35(3):582–9.
Cideciyan AV, Haeseleer F, Fariss RN, Aleman TS, Jang GF, Verlinde C, et al. Rod and cone visual cycle consequences of a null mutation in the 11-cis-retinol dehydrogenase gene in man. Vis Neurosci. 2000;17(5):667–78.
Lidén M, Romert A, Tryggvason K, Persson B, Eriksson U. Biochemical defects in 11-cis-retinol dehydrogenase mutants associated with fundus albipunctatus. J Biol Chem. 2001;276(52):49251–7.
Yang G, Liu Z, Xie S, Li C, Lv L, Zhang M, et al. Genetic and phenotypic characteristics of four Chinese families with fundus albipunctatus. Sci Rep. 2017;7:46285.
Kuehlewein L, Nasser F, Gloeckle N, Kohl S, Zrenner E. FUNDUS ALBIPUNCTATUS ASSOCIATED WITH CONE DYSFUNCTION. Retin Cases Brief Rep. 2017;11(Suppl 1):S73–s76.
Wang NK, Chuang LH, Lai CC, Chou CL, Chu HY, Yeung L, et al. Multimodal fundus imaging in fundus albipunctatus with RDH5 mutation: a newly identified compound heterozygous mutation and review of the literature. Doc Ophthalmol Adv Ophthalmol. 2012;125(1):51–62.
Skorczyk-Werner A, Pawłowski P, Michalczuk M, Warowicka A, Wawrocka A, Wicher K, et al. Fundus albipunctatus: review of the literature and report of a novel RDH5 gene mutation affecting the invariant tyrosine (p.Tyr175Phe). J Appl Genet. 2015;56(3):317–27.
Pras E, Pras E, Reznik-Wolf H, Sharon D, Raivech S, Barkana Y, et al. Fundus albipunctatus: novel mutations and phenotypic description of Israeli patients. Mol Vis. 2012;18:1712–8.
Iannaccone A, Tedesco SA, Gallaher KT, Yamamoto H, Charles S, Dryja TP. Fundus albipunctatus in a 6-year old girl due to compound heterozygous mutations in the RDH5 gene. Doc Ophthalmol Adv Ophthalmol. 2007;115(2):111–6.
Schatz P, Preising M, Lorenz B, Sander B, Larsen M, Eckstein C, et al. Lack of autofluorescence in fundus albipunctatus associated with mutations in RDH5. Retina (Philadelphia, Pa). 2010;30(10):1704–13.
Rüther K, Janssen BP, Kellner U, Janssen JJ, Bohne M, Reimann J, et al. Clinical and genetic findings in a patient with fundus albipunctatus. Der Ophthalmologe : Zeitschrift der Deutschen Ophthalmologischen Gesellschaft. 2004;101(2):177–85.
Sato M, Oshika T, Kaji Y, Nose H. A novel homozygous Gly107Arg mutation in the RDH5 gene in a Japanese patient with fundus albipunctatus with sectorial retinitis pigmentosa. Ophthalmic Res. 2004;36(1):43–50.
Kuroiwa S, Kikuchi T, Yoshimura N. A novel compound heterozygous mutation in the RDH5 gene in a patient with fundus albipunctatus. Am J Ophthalmol. 2000;130(5):672–5.
Gonzalez-Fernandez F, Kurz D, Bao Y, Newman S, Conway BP, Young JE, et al. 11-cis retinol dehydrogenase mutations as a major cause of the congenital night-blindness disorder known as fundus albipunctatus. Mol Vis. 1999;5:41.
Hajali M, Fishman GA, Dryja TP, Sweeney MO, Lindeman M. Diagnosis in a patient with fundus albipunctatus and atypical fundus changes. Doc Ophthalmol Adv Ophthalmol. 2009;118(3):233–8.
Ajmal M, Khan MI, Neveling K, Khan YM, Ali SH, Ahmed W, et al. Novel mutations in RDH5 cause fundus albipunctatus in two consanguineous Pakistani families. Mol Vis. 2012;18:1558–71.
Driessen CA, Janssen BP, Winkens HJ, Kuhlmann LD, Van Vugt AH, Pinckers AJ, et al. Null mutation in the human 11-cis retinol dehydrogenase gene associated with fundus albipunctatus. Ophthalmology. 2001;108(8):1479–84.
Acknowledgments
We thank the all family members for their participation. All authors attest that they meet the current ICMJE criteria for authorship.
Funding
Supported by the National Natural Science Foundation of China (No.81800878 & No.82101132).
Author information
Authors and Affiliations
Contributions
W.W and I.S had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Conception and design: T.Q and Q. G. Data collection: T.Q, Q.G, H.S, C.L and X.X. Analysis and interpretation: T.Q, Q.G, C.L, G.W, W.W and I.S. Drafting the manuscript: T.Q, Q.G and I.S. Critical revision of the manuscript: T.Q, C.L, I.S and W.W. Supervision: I.S and W.W. The authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was conducted in accordance with the Declaration of Helsinki and was approved by the ethics committee of Shanghai General Hospital. A written informed consent was obtained from each participant.
Consent for publication
A written informed consent was obtained from each participant for the publication of this research and any accompanying images.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Supplementary 1. The process of whole-genome sequencing and variants filtration.
Additional file 2.
RDH5 mutation results.
Additional file 3.
RDH5 Sequences.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Qian, T., Gong, Q., Shen, H. et al. Novel variants in the RDH5 Gene in a Chinese Han family with fundus albipunctatus. BMC Ophthalmol 22, 69 (2022). https://doi.org/10.1186/s12886-022-02301-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12886-022-02301-5