
Abstract
Background
Lubiprostone, a bicyclic fatty acid, is used for the treatment of chronic constipation. No published study has addressed the effect of lubiprostone on intestinal ion secretion in vivo.
Aim
The aim of this study was to test the hypothesis that lubiprostone augments duodenal HCO3 − secretion (DBS).
Methods
Rat proximal duodenal loops were perfused with pH 7.0 Krebs, control vehicle (medium-chain triglycerides), or lubiprostone (0.1–10 μM). We measured DBS with flow-through pH and CO2 electrodes, perfusate [Cl−] with a Cl− electrode, and water flux using a non-absorbable ferrocyanide marker. Some rats were pretreated with a potent, selective CFTR antagonist, CFTRinh-172 (1 mg/kg, ip), 1 h before experiments.
Results
Perfusion of lubiprostone concentration dependently increased DBS, whereas net Cl− output and net water output were only increased at 0.1 μM, compared with vehicle. CFTRinh-172 reduced lubiprostone (10 μM)-induced DBS increase, whereas net Cl− output was also unchanged. Nevertheless, CFTRinh-172 reduced basal net water output, which was reversed by lubiprostone. Furthermore, lubiprostone-induced DBS was inhibited by EP4 receptor antagonist, not by an EP1/2 receptor antagonist or by indomethacin pretreatment.
Conclusions
In this first study of the effect of lubiprostone on intestinal ion secretion in vivo, lubiprostone stimulated CFTR-dependent DBS without changing net Cl− secretion. This effect supports the hypothesis that Cl− secreted by CFTR is recycled across the apical membrane by anion exchangers. Recovery of water output during CFTR inhibition suggests that lubiprostone may improve the intestinal phenotype in CF patients. Furthermore, increased DBS suggests that lubiprostone may protect the duodenum from acid-induced injury via EP4 receptor activation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Chronic constipation is a common clinical problem. Although most cases do not reach medical attention due to the plethora of over-the-counter remedies available, severe constipation can be intractable to most remedies and, in its severe form, can be debilitating.
Recently, lubiprostone, a bicyclic fatty acid was approved by the FDA for the treatment of chronic constipation in adults. This compound appears to be in a new therapeutic class and works by a mechanism that has been described in only a few publications. In a detailed investigation of the effect of lubiprostone, Cuppoletti et al. tested the hypothesis that lubiprostone increased the activity of the chloride channel ClC-2. To test the hypothesis, they demonstrated that ClC-2 was expressed in the stomach and in the epithelial cells of the small and large intestine by PCR and in situ hybridization, and demonstrated that ClC-2 was apically expressed in transfected intestinal T84 cells. Electrophysiological measurements in T84 and in ClC-2 transfected human epithelial kidney (HEK) cell monolayers and in patch-clamped individual cells revealed that lubiprostone increased transepithelial and transmembrane currents in T84 only in ClC-2 transfected cells [1]. These data suggest that lubiprostone increases Cl− secretion by activation of the intestinal epithelial cell ClC-2 channel.
Even given these compelling data, the role of ClC-2 channels in epithelial ion secretion is by no means established. First described in 1992 [2], ClC-2 channels are expressed in the stomach, intestine, colon, brain, heart, and muscle. They are part of a family of channels and transporters [3], four of which are plasma membrane expressed, with ClC-2 being the only member to which epithelial transport function is attributed. Its enterocyte expression is thought to be mostly basolateral in situ, based on studies which report localization below the junctional protein ZO-1 [4–6].
The discovery that the mutations of the cystic fibrosis transmembrane regulator (CFTR), an epithelial Cl− channel, are responsible for the disease phenotype has increased interest in the function of all epithelial Cl− channels. With the discovery of ClC-2 channels, investigators hypothesized that ClC-2 function might compensate for CFTR loss-of-function, which might explain the lack of overt pulmonary or pancreatic phenotype in CFTR knockout (KO) transgenic mice. Studies of ClC-2 KO transgenic mice revealed that ClC-2 dysfunction actually increased Cl− secretion, and that double transgenic mice had a survival advantage over CFTR mutant mice, data most compatible with ClC-2 serving to recycle Cl− across the basolateral membrane [7]. Others have implicated ClC-4 in intestinal Cl− secretion [8]. Although apical Cl− channels are hypothesized to facilitate gastric HCl secretion in parietal cells, ClC-2 could not be immunolocalized in the stomach by one group [9], although gastric expression was documented by another [1].
In one of the few published clinical studies addressing the intestinal effects of lubiprostone, Camilleri et al. [10] found that lubiprostone inhibited gastric emptying, but accelerated small and large intestinal transit, which is associated with postprandial fullness. Subsequently, investigators have reported a beneficial effect of lubiprostone in subjects with chronic constipation and functional bowel disease on the basis of clinical trials [11, 12].
To date, one full-length published study has addressed the effects of lubiprostone on gut fluid secretion. Fei et al. reported that lubiprostone increased I sc in Ussing-chambered guinea pig distal ileum and colon [13]. Although the response was unaffected by the neurotoxin tetrodotoxin, or by EP receptor antagonists, consistent with a non-neurally and non-prostaglandin mediated mechanism, serosal application also produced a robust response, calling into question the role of ClC-2 if it was indeed apically expressed. Furthermore, the response was also inhibited by mucosal application of anion channel blockers such as glibenclamide and DIDS.
Given the aforementioned Ussing chamber studies, functional confirmation in situ in vivo would help further understand the effect of the compound on intestinal fluid secretion. We thus propose to examine the effect of lubiprostone on fluid and electrolyte secretion in the duodenum, a locus of enterocyte anion secretion, with a focus on HCO3 − secretion, which protects the mucosa from injury due to luminal acid [14]. Furthermore, given the possible confounding effect of CFTR-mediated secretion, we intend to examine the effect of acute CFTR inhibition on duodenal fluid and electrolyte secretion with a systemic, pharmacologic highly selective CFTR inhibitor.
Methods
Chemicals and Animals
Lubiprostone (10 mg/ml in 0.04% medium-chain triglycerides (MCT) diluent Miglyol 812 N) and vehicle (MCT diluent alone) was provided from Takeda Pharmaceuticals North America, Inc. (Deerfield, IL). CFTRinh-172 was synthesized by Dr. Samedy Ouk in the Department of Chemistry, UCLA [15]. AH6809, AH23848, indomethacin, sodium ferrocyanide ([Fe(CN)6]4−), HEPES and other chemicals were obtained from Sigma Chemical (St. Louis, MO, USA). Krebs solution contained (in mM) 136 NaCl, 2.6 KCl, 1.8 CaCl2, and 10 HEPES at pH 7.0. All studies were performed with approval of the Veterans Affairs Institutional Animal Care and Use Committee (VA IACUC). Male Sprague-Dawley rats weighing 200–250 g (Harlan, San Diego, CA, USA) were fasted overnight, but had free access to water.
Measurement of Duodenal HCO3 − Secretion
Duodenal loops were prepared and perfused as previously described [16, 17]. Under isoflurane anaesthesia (2.0%), the proximal duodenal loop (perfused length 2 cm) was perfused with pH 7.0 Krebs buffer by using a peristaltic pump (Fisher Scientific, Pittsburgh, PA, USA) at 1 ml/min. The perfusate was bubbled with 100% O2, and stirred and warmed at 37°C with a heating stirrer (Barnstead Int., Dubuque, IA, USA). To eliminate the buffer action of perfusing chemicals, which would over- or under-estimate the titration volume using pH-stat, two sets of flow-through pH and CO2 electrodes (Lazar Lab, Los Angeles, CA) were connected in the perfusion loop where pH and CO2 concentration ([CO2]) were simultaneously and continuously measured. Since the input (perfusate) [CO2] is approximately zero, the effluent [CO2] and pH were used to calculate the total CO2 output equivalent to the secreted HCO3 − as previously described [16, 17]. HCO3 − secretion was expressed as total CO2 output (μmol/min/cm). The effluent was collected every 5 min in a sterilized tube on ice for water output measurement as described below. After stabilization with continuous perfusion of pH 7.0 Krebs for about 30 min, the time was set as t = 0. The duodenal loop was perfused with pH 7.0 Krebs from t = 0 min until t = 10 min (basal period). The perfusate was then changed to pH 7.0 Krebs buffer containing lubiprostone (0.1–10 μM) or vehicle from t = 10 min until t = 35 min (challenge period), with or without antagonists.
Measurement of Duodenal Cl− Secretion
Cl− concentration ([Cl−]) in the duodenal effluent was measured using a flow-through Cl−-selective electrode (Lazar Lab), connected in series with the flow-through effluent unit of CO2 electrode. A standard curve for Cl− solution (2.8, 28, 280 mM) was used to calculate the effluent [Cl−]. Since perfusate [Cl−] is stable, the perfusate (input) [Cl−] was measured separately using the same electrode. Net Cl− secretion was calculated by subtracting the perfusate [Cl−] from the effluent [Cl−] with adjustment for water output, and expressed as net Cl− output (μmol/min/cm).
Measurement of Duodenal Water Flux
Water flux from the duodenal loop was measured with a non-absorbable, minimally bioreactive marker [18], [Fe(CN)6]4− (10 mM), contained in the perfusate. [Fe(CN)6]4− concentration was measured by a spectrophotometer at 320 nm absorbance. Water flux was expressed as net water output (μl/min/cm). Cl− output was adjusted according to the calculated water output.
Effect of Lubiprostone With or Without CFTR Inhibition on HCO3 −, Cl−, and Water Output
In order to examine the effect of lubiprostone on duodenal HCO3 − secretion, Cl− secretion and water flux, we first perfused lubiprostone (0.1, 1, or 10 μM) or vehicle in pH 7.0 Krebs buffer containing [Fe(CN)6]4− (10 mM).
Some animals were pre-treated with the potent selective CFTR inhibitor CFTRinh-172 (1 mg/kg, ip) 1 h prior to the experiments. Pre-treatment with CFTRinh-172 at this concentration eliminates acid-induced HCO3 − secretion in rat duodenum [15].
Role of the Prostaglandin Pathway in Lubiprostone-Induced HCO3 − Secretion
We investigated the contribution of the prostaglandin (PG) pathway involving cyclooxygenase (COX) and E-type PG receptors (EP) towards lubiprostone-induced HCO3 − secretion. Lubiprostone (10 μM) was co-perfused with an EP1/EP2 receptor antagonist AH6809 (0.1 mM) or a potent EP4 receptor antagonist AH23848 (0.1 mM). Some animals were pre-treated with the non-selective COX inhibitor indomethacin (5 mg/kg, ip) 1 h prior to the experiments as previously described [19].
Statistics
All data are expressed as means ± SEM. Data were derived from six rats in each group. Comparisons between groups were made by one-way ANOVA followed by Fischer’s least significant difference test. P values of 0.05 were taken as significant.
Results
Effect of Lubiprostone on Duodenal Ion Secretion
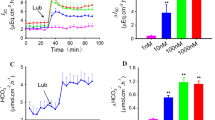
Lubiprostone (0.1−10 μM) concentration-dependently increased duodenal HCO3 − secretion in comparison with the vehicle group (Fig. 1a). In contrast, net Cl− output was increased only by a low concentration (0.1 μM) of lubiprostone (Fig. 1b), accompanied by increased net water output (Fig. 1c). This result demonstrated that a low concentration (0.1 μM) of lubiprostone affects Cl− secretion, whereas a high concentration (10 μM) stimulates HCO3 − secretion. Lubiprostone may thus either reciprocally activate HCO3 − and Cl− secretion, or augmented HCO3 − secretion may mask stimulated Cl− secretion.

Effect of lubiprostone on ion secretion in rat duodenum. a Duodenal HCO3 − secretion was measured as total CO2 output using pH and CO2 electrodes. b Cl− secretion was measured with a Cl−-selective electrode and expressed as net Cl− output. c Water secretion was measured using [Fe(CN)6]4− as a non-absorbable marker and expressed as net water output. Perfusion of lubiprostone (0.1–10 μM) concentration dependently increased total CO2 output (a), whereas only low concentration of lubiprostone (0.1 μM) increased net Cl− and water output (b, c). Data are expressed as mean ± SEM (n = 6). * P < 0.05 versus vehicle group
Effect of CFTR Inhibition on Lubiprostone-Induced Ion Secretion
We pretreated rats with CFTRinh-172 in order to examine the function of CFTR in HCO3 − secretion in response to perfusion of high concentration lubiprostone (10 μM). CFTR inhibition reduced lubiprostone-induced HCO3 − secretion (Fig. 2a), suggesting that lubiprostone-induced HCO3 − secretion is partially CFTR-dependent. CFTR inhibition had no effect on net Cl− output (Fig. 2b). Interestingly, CFTR inhibition decreased net water output during the basal period, with lubiprostone perfusion increasing net water output to the same level as measured in the vehicle group (Fig. 2c). This result suggests that lubiprostone reverses the effect of CFTR inhibition on water output.

Effect of CFTR inhibition on lubiprostone-induced HCO3 − secretion in rat duodenum. a CFTR was inhibited by CFTRinh-172 (1 mg/kg, ip) 1 h prior to the experiment. Lubiprostone (10 μM)-induced HCO3 − secretion was reduced by CFTR inhibition. b CFTR inhibition had no effect on Cl− secretion. c CFTR inhibition decreased water secretion and lubiprostone reversed the effect of CFTR inhibition. Data are expressed as mean ± SEM (n = 6). * P < 0.05 versus vehicle group, † P < 0.05 versus lubiprostone group
In contrast, CFTR inhibition partially suppressed low concentration lubiprostone (0.1 μM)-induced Cl− output (Fig. 3b), but had no effect on HCO3 − secretion (Fig. 3a). Again, CFTR inhibition decreased net water output during the basal period, and inhibited low concentration lubiprostone-induced water output (Fig. 3c), suggesting that lubiprostone-induced Cl− secretion is partially CFTR-dependent.

Effect of CFTR inhibition on lubiprostone-induced Cl− secretion in rat duodenum. CFTR inhibition tended to decrease lubiprostone (0.1 μM)-induced Cl− secretion (b), but had no effect on HCO3 − secretion (a). Lubiprostone reversed CFTR inhibition-induced decrease of water output (c). Data are expressed as mean ± SEM (n = 6). * P < 0.05 versus vehicle group, † P < 0.05 versus lubiprostone group
Effect of EP Receptor Antagonist or COX Inhibition on Lubiprostone-Induced HCO3 − Secretion
We next examined whether lubiprostone has PGE2 agonist-like effects. Lubiprostone (10 μM)-induced HCO3 − secretion was abolished by the co-perfusion of the potent EP4 receptor antagonist AH23848 (0.1 mM), whereas an EP1/EP2 receptor antagonist AH6809 (0.1 mM) had no effect (Fig. 4a). Furthermore, indomethacin pretreatment (5 mg/kg, ip) had no effect on lubiprostone (10 μM)-induced HCO3 − secretion (Fig. 4b). These results suggest that lubiprostone may directly stimulate EP4 receptors, increasing HCO3 − secretion, rather than activating COX.

Effect of prostaglandin receptor antagonist and cyclooxygenase inhibitor on lubiprostone-induced HCO3 − secretion in rat duodenum. a AH6809 (0.1 mM) had no effect on lubiprostone (10 μM)-induced HCO3 − secretion, whereas AH23848 (0.1 mM) abolished the effect of lubiprostone. Data are expressed as mean ± SEM (n = 6). * P < 0.05 versus vehicle group, † P < 0.05 versus lubiprostone group. b Indomethacin (Indo, 5 mg/kg, sc) was given 1 h prior to the experiment. Indomethacin pretreatment had no effect on lubiprostone (10 μM)-induced HCO3 − secretion. Data are expressed as mean ± SEM (n = 6). * P < 0.05 versus vehicle group
Discussion
We demonstrated that luminally-perfused lubiprostone increased ion secretion with a concentration-related manner; a high concentration (10 μM) increased HCO3 − secretion, whereas a low concentration (0.1 μM) increased Cl− and water secretion. Lubiprostone-induced HCO3 − secretion was partially CFTR-dependent, whereas Cl− secretion was not apparently CFTR-dependent, presumably involving non-CFTR mediated Cl− secretion. High concentration lubiprostone may directly stimulate EP4 receptors, increasing duodenal HCO3 − secretion. CFTR inhibition decreased net water output, reversed by a high or low concentration of lubiprostone, consistent with CFTR-independent Cl− secretion. This is the first study demonstrating that lubiprostone stimulates duodenal ion secretion in vivo. Furthermore, lubiprostone may act as a dual activator of CFTR-independent Cl− secretion and as a PG receptor agonist.
The mechanism by which lubiprostone stimulates intestinal Cl− secretion selectively via ClC-2 is controversial. Lubiprostone activates ClC-2 via a CFTR-independent mechanism in T84 cells [1]. In Ussing-chambered ileum and colon, lubiprostone increases Cl− secretion by a CFTR-independent mechanism [13]. Furthermore, the localization of ClC-2 in the intestinal epithelial cells is also controversial. Transfected T84 and MDCK cells express ClC-2 on the apical membrane [1], whereas immunohistochemistry on intact intestinal sections reveals the expression of ClC-2 on the enterocyte basolateral membrane [6, 20] or in the region of the intercellular tight junctions [4]. Since the duodenum secretes Cl− and HCO3 − via a CFTR-dependent mechanism, whereas ClC-2 is localized on the basolateral membrane of duodenocytes [6], we assessed the involvement of CFTR in duodenal lubiprostone-induced ion secretion. Our results demonstrated that at high concentration, lubiprostone stimulated HCO3 − secretion via a CFTR-dependent mechanism, whereas a low concentration increased Cl− secretion via a CFTR-independent mechanism.
Furthermore, the site of lubiprostone action is still controversial. Since lubiprostone is derived from PGE1 [21], it may activate EP receptors. Indeed, EP receptor antagonists diminished lubiprostone-induced contraction of rat gastric and colonic smooth muscle with an apparent pKB of 6.7–7.6 [22]. In contrast, the lubiprostone-induced I sc increase in guinea pig colon was not affected by EP receptor antagonists [13]. Our results show that high concentration lubiprostone-induced HCO3 − secretion is mediated via the EP4 receptor, but not via EP1/EP2 or endogenous PGs by COX activation. Since EP4 activation increases intracellular cAMP [23], and elevated cAMP activates CFTR [24], CFTR-dependent HCO3 − secretion by lubiprostone is consistent with EP4 receptor activation by lubiprostone.
Taken together, our results support the hypothesis that high concentration lubiprostone directly stimulates EP4 receptors, activating CFTR and increasing HCO3 − secretion in rat duodenum. Existing controversies regarding the CFTR dependence of lubiprostone action might reflect species, intestinal segment, and concentration differences. Our results also support the hypothesis that low concentration lubiprostone activates Cl− secretion via non-CFTR channels. Unfortunately, the lack of selective ClC-2 inhibitors impedes further characterization of the mechanism by which low concentration lubiprostone augments Cl− secretion. Very recent data generated from mucosal biopsies obtained from normal mice and humans and those homozygous for CFTR loss-of-function mutations, and CFTR null mice strongly implicated the CFTR in lubiprostone-stimulated anion secretion. Furthermore, this secretion was significantly reduced by EP4 receptor antagonists, as is consistent with our data [25].
Increased duodenal HCO3 − secretion induced by a high concentration of lubiprostone (10 μM) may protect the mucosa from acid-induced injury. Although the exact luminal concentration of lubiprostone in humans is not available, the recommended dose (24 μg; ~60 nmol) may produce duodenal luminal concentrations to the dose we tested (10 nmol/min). Since duodenal luminal concentrations are no doubt higher than in the distal gut, the 24 μg dose may protect the gastroduodenal mucosa. In the distal gut, the lower lubiprostone concentrations may increase Cl− secretion.
CFTR inhibition decreased water output, presumably explained by the reciprocal activation of Na+/H+ exchanger-3 (NHE3) on the apical membrane [26] mediating Na+ absorption followed by water absorption. Recovery of water output by low concentration lubiprostone during CFTR inhibition is consistent with CFTR-independent Cl− secretion. ClC-2 channels, due to their basolateral location in the duodenum, may increase duodenal Cl− absorption during HCO3 − secretion as an alternative to CFTR-mediated Cl− recycling across the apical membrane. Furthermore, lubiprostone stimulates CFTR-dependent HCO3 − secretion without changing net Cl− secretion. This effect supports the hypothesis that Cl− secreted by CFTR is recycled across the apical membrane by anion exchangers. Interestingly, the non-specific anion exchange inhibitor DIDS inhibits lubiprostone-induced I sc increase in guinea pig ileum [13], also supporting this hypothesis.
CFTR-dependent and -independent actions of lubiprostone may be beneficial for other organs, which manifest the CF phenotype. Recently, lubiprostone was reported to increase I sc and mucus secretion via CFTR-dependent and -independent pathways in airway submucosal glands, suggesting it might be of benefit outside of the gastrointestinal tract [27].
In conclusion, lubiprostone stimulates duodenal HCO3 − and Cl− secretion via different pathways, dependent on its concentration. Increased duodenal HCO3 − secretion suggests that lubiprostone may protect the duodenum from acid-induced injury. Restoration of impaired water output during CFTR inhibition suggests that lubiprostone may improve the intestinal phenotype in CF patients.
References
Cuppoletti J, Malinowska DH, Tewari KP, et al. SPI-0211 activates T84 cell chloride transport and recombinant human ClC-2 chloride currents. Am J Physiol Cell Physiol. 2004;287:C1173–C1183.
Thiemann A, Grunder S, Pusch M, Jentsch TJ. A chloride channel widely expressed in epithelial and non-epithelial cells. Nature. 1992;356:57–60.
Jentsch TJ, Neagoe I, Scheel O. CLC chloride channels and transporters. Curr Opin Neurobiol. 2005;15:319–325.
Gyömörey K, Yeger H, Ackerley C, Garami E, Bear CE. Expression of the chloride channel ClC-2 in the murine small intestine epithelium. Am J Physiol Cell Physiol. 2000;279:C1787–C1794.
Moeser AJ, Haskell MM, Shifflett DE, et al. ClC-2 chloride secretion mediates prostaglandin-induced recovery of barrier function in ischemia-injured porcine ileum. Gastroenterology. 2004;127:802–815.
Peña-Münzenmayer G, Catalan M, Cornejo I, et al. Basolateral localization of native ClC-2 chloride channels in absorptive intestinal epithelial cells and basolateral sorting encoded by a CBS-2 domain di-leucine motif. J Cell Sci. 2005;118:4243–4252.
Zdebik AA, Cuffe JE, Bertog M, Korbmacher C, Jentsch TJ. Additional disruption of the ClC-2 Cl− channel does not exacerbate the cystic fibrosis phenotype of cystic fibrosis transmembrane conductance regulator mouse models. J Biol Chem. 2004;279:22276–22283.
Mohammad-Panah R, Ackerley C, Rommens J, et al. The chloride channel ClC-4 co-localizes with cystic fibrosis transmembrane conductance regulator and may mediate chloride flux across the apical membrane of intestinal epithelia. J Biol Chem. 2002;277:566–574.
Hori K, Takahashi Y, Horikawa N, et al. Is the ClC-2 chloride channel involved in the Cl− secretory mechanism of gastric parietal cells? FEBS Lett. 2004;575:105–108.
Camilleri M, Bharucha AE, Ueno R, et al. Effect of a selective chloride channel activator, lubiprostone, on gastrointestinal transit, gastric sensory, and motor functions in healthy volunteers. Am J Physiol Gastrointest Liver Physiol. 2006;290:G942–G947.
Drossman DA, Chey WD, Johanson JF, et al. Clinical trial: lubiprostone in patients with constipation-associated irritable bowel syndrome—results of two randomized, placebo-controlled studies. Aliment Pharmacol Ther. 2009;29:329–341.
Johanson JF, Drossman DA, Panas R, Wahle A, Ueno R. Clinical trial: phase 2 study of lubiprostone for irritable bowel syndrome with constipation. Aliment Pharmacol Ther. 2008;27:685–696.
Fei G, Wang YZ, Liu S, et al. Stimulation of mucosal secretion by lubiprostone (SPI-0211) in guinea pig small intestine and colon. Am J Physiol Gastrointest Liver Physiol. 2009;296:G823–G832.
Allen A, Flemström G. Gastroduodenal mucus bicarbonate barrier: protection against acid and pepsin. Am J Physiol Cell Physiol. 2005;288:C1–19.
Akiba Y, Jung M, Ouk S, Kaunitz JD. A novel small molecule CFTR inhibitor attenuates HCO3 − secretion and duodenal ulcer formation in rats. Am J Physiol Gastrointest Liver Physiol. 2005;289:G753–G759.
Mizumori M, Meyerowitz J, Takeuchi T, et al. Epithelial carbonic anhydrases facilitate PCO2 and pH regulation in rat duodenal mucosa. J Physiol. 2006;573:827–842.
Akiba Y, Mizumori M, Guth PH, Engel E, Kaunitz JD. Duodenal brush border intestinal alkaline phosphatase activity affects bicarbonate secretion in rats. Am J Physiol Gastrointest Liver Physiol. 2007;293:G1223–G1233.
Sadowski DC, Meddings JB. Luminal nutrients alter tight-junction permeability in the rat jejunum: an in vivo perfusion model. Can J Physiol Pharmacol. 1993;71:835–839.
Akiba Y, Furukawa O, Guth PH, et al. Sensory pathways and cyclooxygenase regulate mucus gel thickness in rat duodenum. Am J Physiol Gastrointest Liver Physiol. 2001;280:G470–G474.
Lipecka J, Bali M, Thomas A, et al. Distribution of ClC-2 chloride channel in rat and human epithelial tissues. Am J Physiol Cell Physiol. 2002;282:C805–C816.
Ginzburg R, Ambizas EM. Clinical pharmacology of lubiprostone, a chloride channel activator in defecation disorders. Expert Opin Drug Metab Toxicol. 2008;4:1091–1097.
Bassil AK, Borman RA, Jarvie EM, et al. Activation of prostaglandin EP receptors by lubiprostone in rat and human stomach and colon. Br J Pharmacol. 2008;154:126–135.
Regan JW. EP2 and EP4 prostanoid receptor signaling. Life Sci. 2003;74:143–153.
Li C, Naren AP. Macromolecular complexes of cystic fibrosis transmembrane conductance regulator and its interacting partners. Pharmacol Ther. 2005;108:208–223.
Bijvelds MJ, Bot AG, Escher JC, De Jonge HR. Activation of intestinal Cl− secretion by lubiprostone requires the cystic fibrosis transmembrane conductance regulator. Gastroenterology. 2009 (in press).
Mizumori M, Choi Y, Guth PH, et al. CFTR inhibition augments NHE3 activity during luminal high CO2 exposure in rat duodenal mucosa. Am J Physiol Gastrointest Liver Physiol. 2008;294:G1318–G1327.
Joo NS, Wine JJ, Cuthbert AW. Lubiprostone stimulates secretion from tracheal submucosal glands of sheep, pigs, and humans. Am J Physiol Lung Cell Mol Physiol. 2009;296:L811–L824.
Acknowledgments
We thank Jenifer Kugler for her assistance with manuscript preparation. This work was supported by a research grant from Takeda North America No. 07-014L, Department of Veterans Affairs Merit Review Award, NIH-NIDDK R01 DK54221 (J. Kaunitz), and the animal core of NIH-NIDDK P30 DK0413 (J. E. Rozengurt).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Mizumori, M., Akiba, Y. & Kaunitz, J.D. Lubiprostone Stimulates Duodenal Bicarbonate Secretion in Rats. Dig Dis Sci 54, 2063–2069 (2009). https://doi.org/10.1007/s10620-009-0907-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-009-0907-0