
Abstract
The fairy shrimp Branchinectella media, because of its passive dispersal capacity and scarce and irregularly distributed habitats (temporary saline aquatic systems), is an intriguing organism from a population genomics and conservation perspective. Stochasticity of dispersal events and the irregular distribution of its habitat might lead to low levels of population connectivity and genetic diversity, and consequently, populations with limited persistence through time. Indeed, by using genomic data (SNPs), we found a strong genetic structure among some of the geographically isolated Iberian populations of B. media. Interestingly, we also obtained high estimates of effective population sizes. Lack of suitable habitat between populations (absence of a “stepping stone” network) and strong genetic differentiation suggest limited dispersal success in B. media. However, the high effective population sizes observed ensure persistence of B. media populations against genetic stochasticity (genetic drift). These results indicate that rescue-effect might not be essential for population persistence if they maintain high effective population sizes able to hold adequate levels of genetic diversity. Should high population sizes be reported in other low dispersing Anostraca, one might be optimistic with regard to their conservation status and fate, provided that their natural habitats remain undisturbed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Genetic diversity is a key factor for species persistence, as it allows individuals to face selection and adapt to environmental changes (Ceballos et al. 2017; Frankham 1996). Genetic variability in a population may decrease through time as a result of non-random mating, genetic drift and/or natural selection (Newman and Pilson 1997; but see Reed 2007). The intensity of genetic drift increases in small and isolated populations (mutations usually have a negligible effect in allele frequencies at short timescale). Indeed, loss of genetic diversity in small and isolated populations may be enhanced by subsequent inbreeding (Crnokrak and Roff 1999; Hedrick and Kalinowski 2000; Jangjoo et al. 2016; Star and Spencer 2013; Wright 1931). Species with fragmented distributions, either due to natural processes like long-distance colonisations (Green et al. 2005; Recuerda et al. 2021; Sánchez-Vialas et al. 2021) and geologic/climatic events (García-Marín et al. 1999; Saunders et al. 1991), or due to human induced habitat destruction (Keller and Largiadèr 2003; Templeton et al. 1990), are therefore, exposed to genetic diversity loss to a larger extent because of connectivity hampering. In contrast, genetic drift will be weak in large populations (with numerous reproductive individuals) because they are expected to harbour more genetic diversity and transfer it through generations to a larger extent. In addition, a larger number of breeders will lead to more mutation events (Méndez et al. 2011; Rich et al. 1979). The number of breeding individuals in a population represent the effective population size (Ne) (Wright 1938). The most frequent definition of Ne is the size of an ideal population that would experience the level of genetic drift or inbreeding observed in the real population. Other calculation procedures gave rise to eigenvalue, mutation and coalescence effective sizes (Caballero 2020). Therefore, isolated populations with large contemporary Ne maintain a high level of genetic diversity in spite of limited gene flow (Palstra and Ruzzante 2008), and will retain their evolutionary potential (Franklin and Frankham 1998).
Dispersal and connectivity between populations in species with geographically disjunct habitats may be further limited when they disperse passively, depending thus on external abiotic and biotic agents like meteorological phenomena or animal vectors (Beladjal and Mertens 2009; Coughlan et al. 2017; Green et al. 2005; Vanschoenwinkel et al. 2008). This may lead to species with very strong genetic structure due to geographic isolation (Makino and Tanabe 2009; Mills et al. 2007; Muñoz et al. 2008; Rodríguez-Flores et al. 2019; Xu et al. 2009) and unlikely rescue effect (Vilà et al. 2003). In the absence of migration, the persistence of populations would thus rely directly on a large Ne. The estimation of Ne is particularly important to evaluate the conservation status of isolated populations, being one of the main objectives in conservation genetics/genomics (Benestan et al. 2016; Carreras et al. 2017; Hohenlohe et al. 2021; Plough 2017).
Fairy shrimps (Branchiopoda: Anostraca) are invertebrates that disperse passively across geographically fragmented populations (García-de-Lomas et al. 2015). They are small continental aquatic crustaceans that inhabit temporary ecosystems. In Anostraca, connectivity among populations depends on both habitat availability (Sainz-Escudero et al. 2022) and stochasticity of dispersal events (Kappas et al. 2017; Rodríguez-Flores et al. 2017), often mediated or directed by vertebrates (Rogers 2014a; Sainz-Escudero et al. 2022). Species whose habitats are more continuously distributed have better chances to maintain a connected metapopulation system using a stepping-stone network, keeping higher levels of genetic exchange (Bellin et al. 2020; García-de-Lomas et al. 2017; Hanski and Simberloff 1997). An increase of connecting success might occur when anostracan eggs pass through the digestive tract of dispersing vertebrates (Rogers 2014a). In contrast, less common and distant habitats may result in high inter-population genetic differentiation and strong phylogeographic structure due to the limited connectivity and gene flow, and, consequently, in threatened species (Ketmaier et al. 2008; Lukić et al. 2019; Rodríguez-Flores et al. 2019; but see Kappas et al. 2017). One species likely subjected to these conditions is Branchinectella media (Schmankewitsch 1873), which inhabits temporary saline lakes, ponds and puddles present in steppes scattered along the Palaearctic region (Alonso 2010; Sainz-Escudero et al. 2019) (Fig. 1). In the Iberian Peninsula, this kind of habitat is patchily distributed in separated complexes along the eastern and central areas of Spain (Alcorlo et al. 1997; Alcorlo and Baltanás 1999; García et al. 1997; Pons et al. 2018; Rodríguez-Flores et al. 2016; Sala et al. 2005). These saline lakes are protected under the EU Habitats Directive (Council Directive 92/43/EEC of 21 May 1992 on the conservation of natural habitats and of wild fauna and flora).

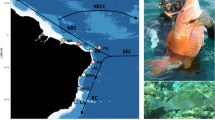
Branchinectella media general habitus and typical habitat of the species. A Adult male from Salada de Piñol (Zaragoza, Spain) photographed ex situ. Note the large size of male eyes, a characteristic trait of Branchinectella. B Adult female from the same locality. C Two young females swimming in shallow pools of Salada de Piñol (Zaragoza, Spain) photographed in situ. D Laguna de Alcahozo (Ciudad Real, Spain) in June; resistance eggs are present in most of the salt surface of the lake. E Laguna de Tirez (Toledo, Spain) in May, when adult fairy shrimps may be still present. F The same perspective of Laguna de Tirez (Toledo, Spain) in June, covered by a salt crust. Photographs by MG-P
Since fairy shrimp conservation, and B. media in particular, depends on knowledge of population demography (i.e., which lakes present long term viable Ne), in this work we used single nucleotide polymorphisms (SNPs) generated by MobiSeq (Rey-Iglesia et al. 2019) to genotype 103 individuals of B. media sampled at five Iberian saline ponds/lakes, and develop a population and conservation genomics study. Our specific aims were (i) to investigate the geographic distribution and levels of genetic diversity in the Iberian populations of B. media and evaluate the existence of isolation, (ii) to estimate contemporary Ne of the resulting genetic units and (iii) to assess the viability and relevance of Ne for B. media populations persistence and the effectiveness of this evolutionary strategy in Anostraca species with limited dispersal capacities.
Materials and methods
Sampling and study area
We sampled five populations of B. media located in saline steppes of La Mancha region (central Spain) and Monegros (northeastern Spain) (Fig. 2). Sampling was designed to evaluate the role of geographic distance limiting gene exchange across populations. In 2015, 16 individuals were collected from a small saline natural rain-puddle (~ 100 m length, ~ 30 m width) next to Longar Salt Lake (“Longar”, hereafter) (39°42′26.78″N-3°18′30.74″W, Lillo, Toledo, Castilla La Mancha; Rodríguez-Flores et al. 2016; Sainz-Escudero et al. 2019). In the same year, half a kilometre away (742 m), 14 individuals were collected from Altillo Chica Salt Lake (“Altillo”, hereafter) (39°42′12.22″N-3°18′05.96″W, Lillo, Toledo, Castilla La Mancha; Alonso 1985, 1998; Boronat-Chirivella 2003; Rodríguez-Flores et al. 2016). These two populations are separated by 18 km from Tirez Lake (“Tirez”, hereafter) (39°32′37.58″N-3°20′59.37″W, Villacañas, Toledo, Castilla La Mancha; Rodríguez-Flores et al. 2016), where 30 individuals were collected in 2015. Around 50 km apart, 30 individuals from Alcahozo Salt Lake (“Alcahozo”, hereafter) (39°23′33.84″N-2°52′30.74″W, Pedro Muñoz, Ciudad Real, Castilla La Mancha; Boronat-Chirivella 2003; Margalef 1948; Pons et al. 2018) were collected in 2019. Finally, 30 individuals were collected in 2016 from Piñol lake (“Piñol”, hereafter) (41°24′31.9″N-0°15′10.5″W, Sástago, Zaragoza, Aragón; Alcorlo 2004; Alcorlo and Baltanás 1999; Alcorlo et al. 1997; Boronat-Chirivella 2003; Brehm and Margalef 1948; Martino 1988; Sala et al. 2005), located around 320 km away from the other sites. Specimens were captured with an aquarium net of 15 cm mouth width and 1 mm mesh pore size, then they were georeferenced and stored at −20ºC at the Museo Nacional de Ciencias Naturales (MNCN, CSIC; Madrid, Spain). The map (Fig. 2) was constructed in ArcMap 10.3 (ESRI, Inc.). The relief layer is a web map belonging to Cartographic Service of Universidad Autónoma de Madrid (SCUAM). Saline steppes layer is a web map of MITECO Spanish Government Cartographic Service.

Reported distribution of B. media in the Iberian Peninsula from Sainz-Escudero et al. (2019). White dots represent unsampled populations while coloured ones represent populations analysed in the present work
Illumina library preparation and sequencing
High quality genomic DNA was extracted from thoracopods of the 120 individuals of B. media using a Zymo Research Quick-DNA Microprep Plus Kit (Zymo Research Corporation, Portugal) following the manufacturer’s protocol. DNA quantity was assessed by using a Qubit Fluorometer (Thermo Fisher Scientific®) and 4 individuals (1 from Altillo and 3 from Alcahozo) did not achieve the minimum DNA amount required for sequencing. All laboratory processing from the low coverage whole genome sequencing of one individual and candidate region selection of a transposable element (TE) to libraries sequencing was carried out by All Genetics & Biology SL (www.allgenetics.eu) following the reduced representation library protocol of MobiSeq (Rey-Iglesia et al. 2019). Once the TE was chosen, the conserved region of the TE was used to design the primer MobiBranchi_578R (5′GAAGCTCATTCTCGTAGGC3′), which was selected for the MobiSeq experiment. Libraries and amplification of selected genomic regions were performed. The resulting libraries were quantified and sequenced in a fraction of a NovaSeq PE150 flow cell.
Raw data processing, SNP genotyping and filtering
Bioinformatic processing from raw data curation to SNP calling was also carried out by AllGenetics & Biology SL. Illumina paired-end raw data (between 1,358,794 and 5,123,706 paired-end reads per individual/library) consisted of forward (R1) and reverse (R2) reads. Quality of raw fastq files was checked in FastQC (v.0.11.5) (Andrews 2010) and summarized in MultiQC (Ewels et al. 2016). The clumplify.sh script from the BBmap package 38.90 (Bushnell B. — https://sourceforge.net/projects/bbmap/) was used to remove exact forward and reverse duplicated reads. Trimmomatic 0.39 (Bolger et al. 2014) was employed to remove the adapters (ILLUMINACLIP option) and low-quality regions (AVGQUAL:26). Cutadapt 2.9 (Martin 2011) was used to remove reads that did not contain the primer sequences (allowing up to 1 mismatches). De novo assembly of loci (reference catalogue construction), read mapping and SNP calling was performed through dDocent pipeline (Puritz et al. 2014). Previous difficulties in the primer selection, and the high levels of obtained PCR duplicates indicated high levels of nucleotide polymorphisms. This led to the use of dDocent script, which takes full advantage of both paired-end reads and performs a quality trimming instead of the quality filtering carried out by STACKS (Catchen et al. 2011, 2013).
The decomposed VCF (variant calling format) file obtained was filtered using VCFtools v4.1 (Danecek et al. 2011). Indels (insertions and deletions) were removed and bi-allelic SNPs were kept, as well as loci with a missing data proportion equal or less than 15% and with a minimum allelic frequency (MAF) of 0.02. There were 13 individuals with more than 75% of missing data for all the loci, so they were discarded keeping a total of 103 individuals (15 from Longar, 12 from Altillo, 23 from Alcahozo, 23 from Tirez and 30 from Piñol). In order to detect loci under balancing or purifying selection (outlier loci) and remove them from the neutral dataset, we used the Bayesian-based software BayeScan v2.1 (Foll 2012; Foll and Gaggiotti 2008). We ran the program with the default 5000 iterations (sample size), with a thinning interval of 200, 20 pilot runs of 10,000 iterations each, and a burn-in of 100,000. Prior odds for the model were set at 100. After the convergence checking, outlier loci were selected through a false discovery rate of 0.1, obtaining 13,689 neutral SNPs and 98 outliers. In order to remove library and sequencing artefacts or allelic dropout (Waples 2015), we removed loci departing from Hardy–Weinberg Equilibrium (HWE) following the “out all” approach, which avoids the removal of informative loci for population structure through the choice of a locus as departed from HWE when it did in all populations (Pearman et al. 2021). We used the function gl.hwe.pop of the R package “HardyWeinberg” 1.7.5 (Graffelman 2015), and 151 loci were detected at disequilibrium in each of the five populations. Finally, our dataset consisting of 13,538 loci grouped in 1771 contigs, was thinned to keep one locus per contig (O’Leary et al. 2018) to avoid possible linked loci due to proximity, resulting in 1771 SNPs as final dataset. This dataset was used to describe the relatedness degree among individuals, population genetic diversity parameters, population genetic structure and Ne.
Genetic diversity and relatedness
The usual descriptors of genetic diversity parameters were estimated for each population (Table 1): allelic richness (AR), observed and expected heterozygosities (Ho and He) and inbreeding coefficient (FIS). These parameters were estimated with the function basic.stats of the R package “hierfstat” 0.5–11 (Goudet 2005). We also reported the population average of the inbreeding coefficient estimates (FL) calculated for each individual using the two likelihood methods implemented in COANCESTRY 1.0.1.10 (Wang 2011). Both likelihood methods produced the same results.
Relatedness coefficients and 95% confidence intervals were calculated per pair of individuals using both the dyadic (Milligan 2003) and triadic (Wang 2007) likelihood methods, assuming inbreeding and default parameters as implemented in COANCESTRY. Pairwise relatedness results were represented as heatmaps through the corrplot function of the R package “corrplot” 0.92 (Wei et al. 2021). Both methods produced similar results, so only triadic results are shown.
Population genetic structure
Population structure was inferred through three different methods. We used PGDSPIDER (Lischer and Excoffier 2012) to convert the VCF file to the different formats required by each approach.
We used the Bayesian clustering method carried out by the software STRUCTURE 2.3.4 (Pritchard et al. 2000). We ran a preliminary analysis to infer the lambda value with K = 1, and once lambda was fixed, the analysis was configuring assuming admixture and correlated allele frequencies (Falush et al. 2003) between populations. The analysis was run five times per K value, each one including a burnin of 50,000 iterations and 100,000 iterations after the burnin. The first analysis included all populations and K values ranged from 1 to 5. Separately, we ran a specific analysis of La Mancha populations to improve the structure resolution in this region and K values ranged from 1 to 4. The STRUCTURE plots were generated using CLUMPAK (Kopelman et al. 2015) and the number of clusters was explored by the rate of change in the log probability of data [Pr(X|K]) between successive K values (∆K) (Evanno et al. 2005).
As an alternative to the Bayesian clustering method, we used the multivariate analyses of Principal Components (PCA) (Hotelling 1933) and Discriminant Analysis of Principal Components (DAPC) (Jombart et al. 2010) in order to summarize the overall variability between individuals and to visualize more clearly the relationships between different clusters, respectively. PCA was performed with the function glPCA implemented in the R package “adegenet” (Jombart 2008). For DAPC analysis (retaining 50 principal components and 1 discriminant function) we used the find.clusters and dapc functions in the R package “adegenet” to identify the number of clusters or genetic groups (K’s) under a Bayesian Criterion Inference (BIC) and to describe the relation between and within these clusters (Jombart et al. 2010), respectively. The first DAPC analysis included all populations. An additional analysis included only La Mancha populations to improve the genetic structure resolution of this group of lakes.
Differentiation between populations was estimated by pairwise FST (Weir and Cockerham 1984). Point estimates and their associated p-values were calculated using stamppFst function in the R package “StAMPP” 1.6.3 (Pembleton et al. 2013).
Effective population size estimation
Contemporary Ne was estimated for each genetic unit using a bias-corrected version of the linkage-disequilibrium (LD) single-sample method by (Waples 2006; Waples and Do 2008) implemented in NeESTIMATOR 2.01 (Do et al. 2014). Although missing data in our final dataset is not high (less than 15% per site), NeEstimator v2 accounts adequately for the lack of missing data (Peel et al. 2013). Due to the close proximity between Longar and Altillo (0.7 km apart) and the total absence of genetic differentiation among them, they were considered as a single population to avoid the effect of low population sizes in estimations and obtain the “metapopulation” Ne (Waples and Do 2010). We used PGDSPIDER 2.1.1.5 (Lischer and Excoffier 2012) to convert the VCF file into the “genepop” file required for the software. The analysis was set to allow for random mating due to the polygyny mating behaviour of anostracans (Belk 1991) and to calculate confidence intervals (lower and upper bounds).
Due to the absence of a reference genome, we could not ensure the absence of physically linked loci in our dataset (LD not caused by genetic drift but by physical proximity within chromosomes), which would downwardly bias Ne estimates (Waples et al. 2016). Downwardly biased Ne (^Ne) was estimated with two different datasets. We used the unpruned dataset of 13,538 SNPs (no LD filter applied), and also the thinned dataset (1 SNP per contig) in which possible physically linked loci are minimized to a larger extent to reduce bias. To estimate Ne (unbiased), we took into account the number of chromosomes as suggested by Waples et al. (2016): ^Ne/Ne = 0.098 + 0.219 * ln (n° chromosomes). Number of chromosomes was set to 24 due to B. media belonging to Chirocephalidae (Kořínková and Godyn 2011). Parametric and non-parametric (jackknife—Waples and Do 2008) methods were used to calculate confidence intervals.
Results
Genetic diversity and relatedness
Allelic richness was similar in all populations (ranging from 1.363 to 1.383) (Table 1). Expected (0.135–0.139) and observed (0.044–0.067) heterozygosities were lower in Longar, Altillo and Alcahozo and higher in Tirez and Piñol. Hardy–Weinberg deviations were obtained for all the populations, being expected heterozygosity higher than the observed one (Table 1). The inbreeding coefficients were fairly high, showing Piñol the lowest values (FIS = 0.379, FL = 0.441; Table 1).
Values of genetic relatedness between pairs of individuals within and between populations were obtained by the two likelihood methods implemented in COANCESTRY were similar. Global genetic relatedness values between pairs of individuals ranged from 0 to 0.25 (Fig. 3). Relationships in the whole sample included parent-offspring, full siblings, half siblings, avuncular, grandparent-grandchild, double first cousins, first cousins, in addition to unrelated individuals. However, no twins were present in the sample (relatedness value equal or higher than 0.5) (Wang 2011). Close relations were very frequent within Piñol with relatedness values between 0.125 and 0.25, meaning that most of pairs of this population showed half sibs, avuncular, grandparent-grandchild and double-first cousin relations (relatedness = 0.125–0.25), and only two pairs of full siblings (relatedness ≥ 0.25) (Fig. 3). These close relationships also appeared within La Mancha populations. About half of the pairs of individuals within each population show relatedness values between 0.06 and 0.25, meaning the existence of half sibs, avuncular, grandparent-grandchild and double-first cousins while the other half are unrelated. Pairs of individuals between Longar, Altillo, Tirez and Alcahozo shared moderate kinship, with the strongest relatedness among Longar, Altillo and Alcahozo. Most pairs of individuals from La Mancha and Piñol populations are unrelated, although a few pairs showed closer kinship with each other.

Heatmap of pairwise relatedness between specimens of B. media as revealed by the triadic likelihood method implemented in COANCESTRY. Individuals appear in the same order in the horizontal and diagonal axes. Relatedness coefficient ranged from 0 to 0.25 (see colour scale)
Genetic structure
STRUCTURE revealed strong genetic differentiation between Piñol and La Mancha complex (Fig. 4). When all populations were included in the analysis, the optimal number of clusters was K = 2. A hierarchical reanalysis removing Piñol from the dataset revealed substantial substructure within La Mancha, i.e., between Tirez and the remaining sites, resulting in an optimal number of clusters of K = 2.

Genetic structure inferred by STRUCTURE for 103 individuals of B. media based on 1771 SNPs (thinned dataset). Each vertical bar represents and individual coloured according to that individual membership assigned to each cluster. Substructure within La Mancha region (hierarchical analysis) is shown at the bottom of the figure. Asterisks (*) indicate the “optimal K” following Evanno’s method
Most of the genetic variability was described in the first two principal components of the PCA analysis. Population scores from the first and second principal components of the PCA were plotted on two axes (PC1 and PC2), which cumulatively explained 8.67% of the total genetic variation (PC1 = 7.20%, PC2 = 1.47%) (Fig. 5A). The first component separated two clusters represented by La Mancha complex and Piñol population (Fig. 5A). The second component showed a slightly separated cluster represented by Tirez population (Fig. 5A). Intra-population genetic variability was high in all populations (Fig. 5A). DAPC analysis identified the lowest BIC value at K = 2 (463.092) in the dataset including all populations and at K = 1 (327.129) in that including only La Mancha populations, so only the result of the first dataset is shown (Fig. 5B). DAPC grouped individuals into two well differentiated genetic clusters corresponding to La Mancha complex and Piñol population (Fig. 5B).

Genetic structure represented by multivariate analyses of A PCA and B DAPC for all B. media populations. Principal Component Analysis represent the distribution of the sample genetic variation in the two first axes. Discriminant Analysis of Principal Components identifies and describes clusters of genetically related individuals. Optimal number of clusters is identified by the lowest Bayesian Information Criterion (BIC). PCA eigenvalues represent the number of principal components retained in the data transformation step.
The highest significant FST values were obtained between Piñol and La Mancha complex (0.120–138). Within La Mancha the only differentiated population is Tirez, as the p-values for pairwise comparisons between Longar, Altillo and Alcahozo were ≥ 0.18 (Table 2).
Effective population size
Both the thinned and unpruned datasets yielded downwardly biased estimations of contemporary Ne (^Ne) due to an excess of LD (specially the unpruned one). Biased Ne obtained with the thinned dataset ranged from 490.3 (Longar + Altillo) to 4405.7 (Alcahozo) (Table 3). The lower bound of parametric and non-parametric confidence intervals showed the same trend, but the higher bounds were infinite for Longar + Altillo, Tirez and Alcahozo. The unpruned dataset produced finite intervals for the four sites when calculated by parametric method, however, higher bounds were infinite with jackknife method. Results indicated a significantly lower ^Ne for Longar + Altillo (280.1), while the other three populations had approximately twice this size. We applied the correction suggested by Waples et al. (2016) obtaining a bias of 0.794. Corrected Ne became larger in all populations after bias adjustment. In the two cases, Longar + Altillo has the lowest Ne value, approximately half the size of the remaining populations.
Discussion
Population genomics and biology of Anostraca
We found that the genetic diversity (allelic richness and heterozygosity) of Iberian B. media populations is generally low with respect to other non-Anostraca branchiopods for mitochondrial, nuclear or microsatellite markers (Cesari et al. 2007; Horn et al. 2014). However, studies of Branchiopoda or other phylogenetically close crustaceans, in which genetic diversity indices are estimated based on SNPs are still scarce (e.g., Muñoz et al. 2016; Schwentner et al. 2018). Accordingly, obtained inbreeding levels are higher (Rogers 2015; Rowe et al. 2007; Velonà et al. 2009), which may be the reason for the resulted deviations from HWE. These comparatively (with Branchiopoda) low levels of genetic variability within anostracan populations may be a consequence of partial hatching during life cycles (Wahlund effect) (Brendonck et al. 2000a, b; Vanschoenwinkel et al. 2010; Wahlund 1928), abortive hatching and/or habitat monopolization by highly successful individuals following genetic drift or founder effects (Rogers 2015). In the case of B. media we cannot attribute low genetic diversity to limited dispersal and reduced gene flow because the lowest estimates do not concur with the most isolated populations (Ortego et al. 2010). Anyway, the values observed so far in Anostraca are not particularly low if compared to those of many other passively dispersed organisms (Montero-Pau et al. 2017; Unal and Bucklin 2010). In fact, genetic diversity estimates in B. media concur with those obtained so far in other Anostraca species (Bohonak 1998; Muñoz et al. 2009). High levels of inbreeding, and thus, low levels of heterozygosity in Anostraca might not be rare (Davies et al. 1997). Generation overlapping (cohabitation with relatives) and partial eggs hatching might lead to a reduction of heterozygosity in populations. Another possibility could be that inbreeding estimates were biased by the existence of loci with non-Mendelian inheritance contained in our dataset. However, some filters such as thinning (linkage disequilibrium) and keeping bi-allelic SNPs (eliminating loci with multiple alleles) decrease their occurrence probability.
Level of genetic differentiation (as measured by FST) between our populations is intermediate if compared to previous analyses on Anostraca (Boileau et al. 1992; Brendonck et al. 2000a, b; Ketmaier et al. 2008, 2012). Nevertheless, the three approaches performed in this study concurred that the strongest genetic differentiation occurs between Piñol (part of the Ebro Basin population complex) and La Mancha population complex. The strong genetic structure and large geographic distance between Piñol and La Mancha point to two independent and historically isolated genetic clusters. We can rule out such a pattern to be an artefact caused by the higher relatedness found in Piñol, as sequence data from the cox1 mitochondrial marker show a 4% of genetic distance (p) between Piñol and La Mancha complex and 0% among La Mancha lakes (unpublished data). These estimates indicate that obtained differentiation between these two subunits has been caused by a significant isolation through time, and not by the high inbreeding of Piñol. Piñol lake is in the Ebro Basin while La Mancha complex is in the Tajo Basin. These two basins, although of ancient origins (Tertiary Period), are currently covered by Quaternary sediments (Benito-Calvo et al. 2009). The time since the origin of the lakes seem to be sufficiently long to separate their populations as genetic units. The distinct climatic and morphogenetic nature of these two areas (northern and central of Iberian Peninsula) could have been supported the differentiation of the units (Rogers 2014b). With respect to La Mancha complex, all the methods agree that populations of Longar, Altillo and Alcahozo conform a single subunit which also correspond to the same geomorphological structure. Conversely, the different genetic composition of Tirez revealed by STRUCTURE (Fig. 4), the partially non-overlapped distribution of individuals of this population displayed in the PCA (Fig. 5A in red) and its intermediate FST values compared to the other La Mancha populations, may indicate this lake as a different genetic subunit. This population is settled in an artificially cut off adjacent section of the lake, which together with its smaller size may influence the frequency of visits by the dispersal agents, likely large birds. Consequently, dispersal events may become rarer (Boronat-Chirivella 2003; Rogers 2014a). This could imply that the population inhabiting the contiguous unsampled natural lake may likely share the genetic structure displayed by the other studied lakes (Longar, Altillo and Alcahozo) but additional analyses are needed to test for this hypothesis. Either way, this artificial population does conform a slightly different genetic unit, and therefore must be considered for conservation management. Therefore, Iberian B. media populations conform a minimum of three genetic subunits corresponding to (1) Longar, Altillo and Alcahozo, (2) Tirez (artificial pond) and (3) Piñol. The existence of a certain level of kinship between Piñol and La Mancha populations could be indicating some incidental dispersal. These results show that geographical distance affects B. media dispersal capacity at large scale (~ 300 km), but not at small or intermediate ones (~ 50 km). Longar, Altillo and Alcahozo are practically identical in genetic composition, which implies that dispersal and consequently, gene flow among these populations are very high. Connectivity among these separated lakes seem to be consequence of passive vectors, birds like flamingos or ducks (Amat et al. 2005; Figuerola et al. 2003; Green et al. 2005).
On the other hand, the apparent inconsistent pattern of high relatedness and low inbreeding estimates in Piñol (Fig. 3), may have several explanations. The irregular filling of this lake might account for this seemingly contradictory pattern. The marginal areas of Piñol do not fill every year, so some eggs in the dry area will not hatch (diapause). When the filling is complete and the diapause eggs hatch, there will be co-occurrence of different generations (Rogers 2014a, b). Therefore, it may be more likely to have sampled related individuals at Piñol after complete filling increased the degree of relatedness. In addition, inconsistent values of relatedness and inbreeding (FIS, FL) are feasible to be found due to the different genetic data used for their estimation (allele frequencies for inbreeding—Wright 1951- and proportions of modes of identity by descent for relatedness -Wang 2011). Observed differences in inbreeding between Piñol and La Mancha populations (these last with a high inbreeding level) might be a consequence of the high concentration of additional salt lakes across surrounding area of Piñol (e.g., at least eleven known populations in a 25 km radius, Alcorlo and Baltanás 1999). These unstudied populations might be a constant source of new immigrants to Piñol contributing to decrease FIS values.
Contemporary Ne values in B. media are high compared with estimates in other crustacean lineages, such as freshwater (Ada 2021) and marine decapods (Anagnostou and Schubart 2017; Cabezas et al. 2012; Heras et al. 2019), amphipods (Gergs et al. 2019) and isopods (Guzik et al. 2012). The highest Ne estimates were obtained with the thinned dataset, which seems to be reasonable because of the excess of removed LD (Waples and Do 2010). Conversely, the non-LD pruned dataset, which contains the whole LD, physical linkage and that originated by genetic drift, produces lower Ne estimates. In both cases, the excess of LD is balanced with Waples et al. (2016) correction, which increases Ne estimates. Highest Ne concur with the largest sized lakes (Tirez, Alcahozo and Piñol). The populations of Longar and Altillo are located in small to medium-size ponds near a large lake, so a lower Ne is expected in relation to the possible smaller population size (Nunney and Elam 1994). However, these values are still high, which suggests that habitat size does not considerably influence the Ne of Anostraca populations. Large population sizes have been previously registered in small habitats like rock pools or those from wheel tracks (Brendonck et al. 2000a, b; Timms 2006; Vanschoenwinkel et al. 2013). Comparing with standardized Ne thresholds, a minimum value of 50–70 is needed to minimize inbreeding depression, and 500 to maintain an adequate evolutionary potential (Franklin 1980; Caballero et al. 2017). Accordingly, the large observed Ne is buffering the effect of relatedness and genetic drift on genetic diversity levels (Giesel 1971; Keller and Waller 2002). In addition, high Ne estimates suggest a balanced sex ratio and reproductive success within all populations (Caballero 1994; Frankham 1995; Nunney and Elam 1994). These results proved invaluable for a first assessment of the conservation status of B. media, as well as to infer that this species likely maintains large Ne to compensate limited gene flow and subsequent inbreeding effects and maintain adequate levels of genetic diversity. However, we must highlight that the estimation of Ne in anostracans following LD method infringes the assumption of non-overlapping generations that can be underestimating the results (Waples et al. 2014), which is confirmed with the high level of relatedness obtained in all the study sites. This means that our estimates of contemporary Ne should be taken with caution. Future studies should confirm our estimates by using the temporal approach, proved to be more robust to scenarios with overlapping generations (reviewed by Wang et al. 2016). In our case, this issue is not so worrying since despite having overlapping generations, our Ne estimates are still high, giving an optimistic approximation about the conservation status of Iberian Branchinectella media.
Population extirpation in metapopulational systems may come from natural causes such as high levels of inbreeding or limited reproduction effectivity, which lead to the decrease of genetic diversity. The extent of the effect of these factors within populations depends on population size. Small populations usually tend to disappear randomly and faster than larger populations (Vrijenhoek 1994). Most species of Anostraca frequently occupy small, human-modified ponds (Horváth and Vad 2015; Sainz-Escudero et al. 2021), usually forming metapopulation systems, and therefore likely subjected to high population turnover. However, the case of Iberian B. media, in which habitat restrictions (saline ponds) determine extremely limited availability of possible population settling, population turnover or recolonization is highly unlikely. This fact together with the limited connectivity (low levels of genetic diversity) between the three studied genetic units would make short term local extinction a probable output for B. media populations. Only large population sizes can assure long-term population survival under such circumstances, as it is precisely the case observed in our sampling. According to our data, B. media diversified within Iberia at least from the late Pliocene (unpublished data), and therefore it was established in the territory at least since then. In addition, an historical record of B. media in Piñol (Brehm and Margalef 1948) also assures the continuity of the population from, at least, fifty years ago. Following these considerations, we put forward that other species of Anostraca whose populations are likely subjected to genetic isolation, such as those of Branchipodopsis in southern Africa (Brendonck et al. 2000a, b), may be relying on their large Ne to persist through time despite genetic diversity losses due to stochasticity. Indeed, there are other invertebrates adapted to temporary aquatic ecosystems which also display cases of strong genetic differentiation such as Cladocera, Copepoda and Ostracoda (Haag et al. 2006; Rossi et al. 1994; Scheihing et al. 2011). In order to recognize threatened units of species living in temporary aquatic ecosystems and with limited dispersal capacity, conservation studies should include the estimation of effective sizes, accompanied, if possible, by an approximate observation of the total size of the population as an additional support (Wang & Rogers 2018).
Conservation implications
At present, Iberian lineages of Branchinectella media would not require specific nor urgent conservation actions. Large effective population sizes in B. media populations, together with current local strict protection of their inhabited and potential salt lakes guarantee its short-term persistence. However, our optimistic Ne estimates should not preclude this species from being monitored in the future. Continental aquatic ecosystems are currently threatened by direct and indirect anthropogenic disturbances (Dudgeon et al. 2006; Reid et al. 2019). Among them, temporary ecosystems are especially vulnerable (Zacharias and Zamparas 2010). Climatic change and the resulting unpredictable rainfall distribution threaten the fill of endorheic basins, halting the formation of the necessary aquatic habitats for development of anostracans (Asem et al. 2019; Rautio et al. 2011) and other invertebrates (Williams 1997). Similarly, changes in light and temperature alter hatching success in Anostraca (Tladi et al. 2020). Besides that, agricultural and farming development causes aquifer collapse due to water overuse (Löffler 1993) and water pollution deriving of the use of pesticides, chemical fertilizers (Migliore et al. 1993; Sánchez-Bayo 2006) and veterinary pharmaceuticals (Udebuani et al. 2021). In addition, modifications of the temporary nature of lakes with artificial water inputs (Brock et al. 1999) impede fairy shrimps and other organisms adapted to habitat filling-drying conditions to complete their life cycle. The persistence of temporary habitats is thus compromised by anthropogenic disturbances, which may condition the survival of B. media despite the evolutionary efforts (high Ne) nature is making for its persistence.
Data availability
The datasets generated and analysed during the current study are available on NCBI repository. Sequences read archives are stored in the BioProject PRJNA973723 (https://www.ncbi.nlm.nih.gov/sra/PRJNA973723.
References
Ada A (2021) Population genetic delineation of conservation units of the endangered California freshwater shrimp (Syncaris pacifica). Thesis Dissertation, San Diego State University.
Alcorlo P (2004) Las redes tróficas en las lagunas salinas temporales de Los Monegros (Zaragoza, España). Ecosistemas 13:37–51
Alcorlo P, Baltanás A (1999) Limnología de las lagunas salinas de Los Monegros y caracterización de sus comunidades animales. Manifiesto Científico por Los Monegros. Bol Soc Entomol Aragonesa 24:113–120
Alcorlo P, Díaz P, Lacalle J, Baltanás J, Florín A, Guerrero MC, Montes C (1997) Sediment features, primary producers and food web structure in two shallow temporary lakes (Monegros, Spain). Wat Air Soil Poll 99:681–688
Alonso M (1985) A survey of the Spanish Euphyllopoda. Miscellanea Zool 9:179–208
Alonso M (1998) Las lagunas de la España peninsular. Limnetica 15:1–176
Alonso M (2010) Branchiopoda and Copepoda (Crustacea) in Mongolian Saline Lakes. Mong J Biol Sci 8(1):9–16. https://doi.org/10.22353/mjbs.2010.08.02
Amat JA, Rendón MA, Rendón-Martos M, Garrido A, Ramírez JM (2005) Ranging behaviour of greater flamingos during the breeding and post-breeding periods: Linking connectivity to biological processes. Biol Conserv 125(2):183–192. https://doi.org/10.1016/j.biocon.2005.02.018
Anagnostou C, Schubart CD (2017) Evidence for a single panmictic and genetically diverse population of the coconut crab Birgus latro (Decapoda: Anomura: Coenobitidae) on Christmas Island in the Indian Ocean. Mar Freshw Res 68(6):1165–1177. https://doi.org/10.1071/MF16031
Asem A, Eimanifar A, Van Stappen G, Sun SC (2019) The impact of one-decade ecological disturbance on genetic changes: a study on the brine shrimp Artemia urmiana from Urmia Lake. Iran. Peerj 7:e7190. https://doi.org/10.7717/peerj.7190
Beladjal L, Mertens J (2009) Diaspore dispersal of Anostraca by flying insects. J Crustac Biol 29(2):266–268. https://doi.org/10.1651/08-3059R.1
Belk D (1991) Anostracan mating behavior: a case of scramble-competition polygyny. In: Bauer RT, Martin JW (eds) Crustacean sexual biology. Columbia University Press, New York Chichester, pp 111–125
Bellin N, Groppi M, Rossi V (2020) A model of egg bank dynamics in ephemeral ponds. Ecol Modell 430:109–126. https://doi.org/10.1016/j.ecolmodel.2020.109126
Benestan LM, Ferchaud AL, Hohenlohe PA, Garner BA, Naylor GJP, Baums IB, Schwartz MK, Kelley JL, Luikart G (2016) Conservation genomics of natural and managed populations: Building a conceptual and practical framework. Mol Ecol 25(13):2967–2977. https://doi.org/10.1111/mec.13647
Benito-Calvo A, Pérez-González A, Magri O, Meza P (2009) Assessing regional geodiversity: the Iberian Peninsula. Earth Surf Process Landforms 34:1433–1445. https://doi.org/10.1002/esp.1840
Bohonak AJ (1998) Genetic population structure of the fairy shrimp Branchinecta coloradensis (Anostraca) in the Rocky Mountains of Colorado. Can J Zool 76(11):2049–2057. https://doi.org/10.1139/z98-170
Boileau MG, Hebert PDN, Schwartz SS (1992) Non-equilibrium gene frequency divergence: persistent founder effects in natural populations. J Evol Biol 5(1):25–39. https://doi.org/10.1046/j.1420-9101.1992.5010025.x
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Boronat-Chirivella D (2003) Distribución de los microcrustáceos en lagunas de Castilla La Mancha. Ciclos estacionales y migración vertical en lagunas cársticas estratificadas. Thesis Dissertation, Universitat de València, Comunitat Valenciana.
BrehmV V, Margalef R (1948) Eucypris aragonica nov. sp., nuevo ostrácodo de una laguna salada de Los Monegros. Ins Biol Apl 5:5–9
Brendonck L, Hamer ML, Riddoch BJ, Seaman MT (2000a) Branchipodopsis species—specialists of ephemeral rock pools. Afr J Aquat Sci 25(1):98–104. https://doi.org/10.2989/160859100780177668
Brendonck L, De Meester L, Riddoch BJ (2000b) Regional structuring of genetic variation in short-lived rock pool populations of Branchipodopsis wolfi (Crustacea: Anostraca). Oecologia 123(4):506–515. https://doi.org/10.1007/s004420000344
Brock MA, Smith RGB, Jarman PJ (1999) Drain it, dam it: alteration of water regime in shallow wetlands on the New England Tableland of New South Wales. Australia Wetl Ecol Manag 7(1–2):37–46. https://doi.org/10.1023/A:1008416925403
Caballero A (1994) Developments in the prediction of effective population size. Heredity 73(6):657–679. https://doi.org/10.1038/hdy.1994.174
Caballero A (2020) Quantitative genetics. Cambridge University Press
Caballero A, Bravo I, Wang J (2017) Inbreeding load and purging: implications for the short-term survival and the conservation management of small populations. Heredity 118(2):177–185. https://doi.org/10.1038/hdy.2016.80
Cabezas P, Alda F, Macpherson E, Machordom A (2012) Genetic characterization of the endangered and endemic anchialine squat lobster Munidopsis polymorpha from Lanzarote (Canary Islands): management implications. ICES Mar Sci Symp 69:1030–1037. https://doi.org/10.1093/icesjms/fss062
Carreras C, Ordóñez V, Zane L, Kruschel C, Nasto I, MacPherson E, Pascual M (2017) Population genomics of an endemic Mediterranean fish: differentiation by fine scale dispersal and adaptation. Sci Rep 7:1–12. https://doi.org/10.1038/srep43417
Catchen JM, Amores A, Hohenlohe P, Cresko W, Postlethwait JH (2011) Stacks: building and genotyping loci de novo from short-read sequences. G3: Genes, Genomes, Genetics 1(3):171–182. https://doi.org/10.1534/g3.111.000240
Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA (2013) Stacks: an analysis tool set for population genomics. Mol Ecol 22(11):3124–3140. https://doi.org/10.1111/mec.12354
Ceballos G, Ehrlich PR, Dirzo R (2017) Biological annihilation via the ongoing sixth mass extinction signaled by vertebrate population losses and declines. Proc Natl Acad Sci USA 114(30):E6089–E6096. https://doi.org/10.1073/pnas.1704949114
Cesari M, Luchetti A, Scanabissi F, Mantovani B (2007) Genetic variability in European Leptestheria dahalacensis (Rüppel, 1837) (Crustacea, Branchiopoda, Spinicaudata). Hydrobiologia 586(1):249–260. https://doi.org/10.1007/s10750-007-0645-2
Coughlan NE, Kelly TC, Davenport J, Jansen MAK (2017) Up, up and away: bird-mediated ectozoochorous dispersal between aquatic environments. Freshw Bio 62(4):631–648. https://doi.org/10.1111/fwb.12894
Crnokrak P, Roff DA (1999) Inbreeding depression in the wild. Heredity 83(3):260–270. https://doi.org/10.1038/sj.hdy.6885530
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth G, Sherry ST, McVean G, Durbin R, 1000 Genomes Project Analysis Group (2011) The variant call format and VCFtools. Bioinformatics 27(15):2156–2158. https://doi.org/10.1093/bioinformatics/btr330
Davies CP, Simovich MA, Hathaway SA (1997) Population genetic structure of a California endemic branchiopod, Branchinecta sandiegonensis. Hydrobiologia 359:149–158
Do C, Waples RS, Peel D, Macbeth GM, Tillett BJ, Ovenden JR (2014) NeEstimator v2: Re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol Ecol Res 14(1):209–214. https://doi.org/10.1111/1755-0998.12157
Dudgeon D, Arthington AH, Gessner MO, Kawabata ZI, Knowler DJ, Lévêque C, Naiman RJ, Prieur-Richard AH, Soto D, Stiassny ML, Sullivan CA (2006) Freshwater biodiversity: Importance, threats, status and conservation challenges. Biol Rev Biol Proc Camb Philos Soc 81(2):163–182. https://doi.org/10.1017/S1464793105006950
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14(8):2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Ewels P, Magnusson M, Lundin S, Käller M (2016) MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32(19):3047–3048. https://doi.org/10.1093/bioinformatics/btw354
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164(4):1567–1587. https://doi.org/10.1093/genetics/164.4.1567
Figuerola J, Green AJ, Santamaría L (2003) Passive internal transport of aquatic organisms by waterfowl in Doñana, south-west Spain. Glob Ecol Biogeogr 12(5):427–436. https://doi.org/10.1046/j.1466-822X.2003.00043.x
Foll M (2012) BayeScan v2. 1 user manual. Ecol 20(10).
Foll M, Gaggiotti O (2008) A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180(2):977–993. https://doi.org/10.1534/genetics.108.092221
Frankham R (1995) Effective population size/adult population size ratios in wildlife: a review. Genet Res 66(2):95–107. https://doi.org/10.1017/S0016672300034455
Frankham R (1996) Relationship of genetic variation to population size in wildlife. Biol Conserv 10(6):1500–1508. https://doi.org/10.1046/j.1523-1739.1996.10061500.x
Franklin IR (1980) Evolutionary change in small populations. In: Soule ME, Wilcox BA (eds) Conservation biology—an evolutionary ecological perspective. Sinauer, Sunderland, pp 135–149
Franklin IR, Frankham R (1998) How large must populations be to retain evolutionary potential? Anim Conserv Forum 1(1):69–70. https://doi.org/10.1111/j.1469-1795.1998.tb00228.x
García CM, García-Ruiz R, Rendón M, Niell FX, Lucena J (1997) Hydrological cycle and interannual variability of the aquatic community in a temporary saline lake (Fuente de Piedra, southern Spain). Hydrobiologia 345:131–141. https://doi.org/10.1023/A:1002983723725
García-Marín JL, Utter FM, Carles P (1999) Postglacial colonization of brown trout in Europe based on distribution of allozyme variants. Heredit 82(1):46–56. https://doi.org/10.1038/sj.hdy.6884470
García de Lomas J, Sala J, García CM, Alonso M (2015) Orden Anostraca. Revista IDE@-SEA, 67, 1–12. http://sea-entomologia.org/IDE@/revista_67. pdf
García de Lomas J, Sala J, Barrios V, Prunier F, Camacho A, Machado M, Alonso M, Korn M, Boix D, Hortas F, García CM, Serrano L, Muñoz G (2017) How threatened are large branchiopods (Crustacea, Branchiopoda) in the Iberian Peninsula? Hydrobiologia 801(1):99–116. https://doi.org/10.1007/s10750-017-3322-0
Gergs R, Gemmer I, Koester M, Rothhaupt KO, Behrmann-Godel J (2019) No evidence for a genetic impoverishment of the indigenous amphipod Gammarus roeselii (Gervais, 1835) due to the invasion of Dikerogammarus villosus (Sowinsky, 1894) in Lake Constance. Hydrobiologia 829(1):189–204. https://doi.org/10.1007/s10750-018-3831-5
Giesel JT (1971) The relations between population structure and rate of inbreeding. Evolutions. 25:491–496
Goudet J (2005) Hierfstat, a package for R to compute and test hierarchical F-statistics. Mol Ecol Not 5(1):184–186. https://doi.org/10.1111/j.1471-8286.2004.00828.x
Graffelman J (2015) Exploring diallelic genetic markers: the HardyWeinberg package. J Stat Softw 64:1–23. https://doi.org/10.18637/jss.v064.i03
Green AJ, Sánchez MI, Amat F, Figuerola J, Hontoria F, Ruiz O, Hortas F (2005) Dispersal of invasive and native brine shrimps Artemia (Anostraca) via waterbirds. Limnol Oceanogr 50(2):737–742. https://doi.org/10.4319/lo.2005.50.2.0737
Guzik MT, Adams MA, Murphy NP, Cooper SJB, Austin AD (2012) Desert springs: Deep phylogeographic structure in an ancient endemic crustacean (Phreatomerus latipes). PLoS ONE 7(7):e37642. https://doi.org/10.1371/journal.pone.0037642
Haag CR, Riek M, Hottinger JW, Pajunen VI, Ebert D (2006) Founder events as determinants of within-island and among-island genetic structure of Daphnia metapopulations. Heredity 96:150–156. https://doi.org/10.1038/sj.hdy.6800774
Hanski I, Simberloff D (1997) The metapopulation approach, its history, conceptual domain, and application to conservation. Metapopulation Biol. https://doi.org/10.1016/B978-012323445-2/50003-1
Hedrick PW, Kalinowski ST (2000) Inbreeding depression in conservation biology. Annu Rev Ecol Evol Syst 31:139–162
Heras S, Planella L, García-Marín JL, Vera M, Roldán MI (2019) Genetic structure and population connectivity of the blue and red shrimp Aristeus antennatus. Sci Rep 9(1):1–11. https://doi.org/10.1038/s41598-019-49958-5
Hohenlohe PA, Funk WC, Rajora OP (2021) Population genomics for wildlife conservation and management. Mol Ecol 30(1):62–82. https://doi.org/10.1111/mec.15720
Horn RL, Kuehn R, Drechsel V, Cowley DE (2014) Discriminating between the effects of founding events and reproductive mode on the genetic structure of Triops populations (Branchiopoda: Notostraca). PLoS ONE 9(5):1–10. https://doi.org/10.1371/journal.pone.0097473
Horváth Z, Vad CF (2015) Life history and current distribution of the fairy shrimp Chirocephalus carnuntanus (Brauer, 1877) (Crustacea: Anostraca). North-West J Zool 11(1):102–109
Hotelling H (1933) Analysis of a complex of statistical variables into principal components. J Educ Psychol 24(6):417
Jangjoo M, Matter SF, Roland J, Keyghobadi N (2016) Connectivity rescues genetic diversity after a demographic bottleneck in a butterfly population network. Proc Natl Acad Sci USA 113(39):10914–10919. https://doi.org/10.1073/pnas.1600865113
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24(11):1403–1405. https://doi.org/10.1093/bioinformatics/btn129
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Gen 11(1):1–15. https://doi.org/10.1186/1471-2156-11-94
Kappas I, Mura G, Synefiaridou D, Marrone F, Alfonso G, Alonso M, Abatzopoulos TJ (2017) Molecular and morphological data suggest weak phylogeographic structure in the fairy shrimp Streptocephalus torvicornis (Branchiopoda, Anostraca). Hydrobiologia 801(1):21–32. https://doi.org/10.1007/s10750-017-3203-6
Keller I, Largiadèr CR (2003) Recent habitat fragmentation caused by major roads leads to reduction of gene flow and loss of genetic variability in ground beetles. Proc R Soc B 270(1513):417–423. https://doi.org/10.1098/rspb.2002.2247
Keller LF, Waller DM (2002) Inbreeding effects in wild populations. Trends Ecol Evol 17(5):230–241. https://doi.org/10.1016/S0169-5347(02)02489-8
Ketmaier V, Pirollo D, De Matthaeis E, Tiedemann R, Mura G (2008) Large-scale mitochondrial phylogeography in the halophilic fairy shrimp Phallocryptus spinosa (Milne-Edwards, 1840) (Branchiopoda: Anostraca). Aquat Sci 70(1):65–76. https://doi.org/10.1007/s00027-007-7028-7
Ketmaier V, Marrone F, Alfonso G, Paulus K, Wiemann A, Tiedemann R, Mura G (2012) Mitochondrial DNA regionalism and historical demography in the extant populations of Chirocephalus kerkyrensis (Branchiopoda: Anostraca). PLoS ONE 7(2):19–22. https://doi.org/10.1371/journal.pone.0030082
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Res 15(5):1179–1191
Kořínková T, Godyn B (2011) Karyotypes and sex ratios in populations of Eubranchipus grubii (Dybowski, 1860) and Branchipus schaefferi Fischer, 1834 (Branchiopoda, Anostraca) from Poland. Crustaceana 84(5–6):707–720. https://doi.org/10.1163/001121611X577918
Lischer HE, Excoffier L (2012) PGDSpider: an automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 28(2):298–299. https://doi.org/10.1093/bioinformatics/btr642
Löffler H (1993) Anostraca, Notostraca, Laevicaudata and Spinicaudata of Pannonian region and in its Austrian area. Hydrobiologia 264(3):169–174. https://doi.org/10.1007/BF00007287
Lukić D, Waterkeyn A, Rabet N, Mioduchowska M, Geudens B, Vanschoenwinkel B, Brendonck L, Pinceel T (2019) High genetic variation and phylogeographic relations among Palearctic fairy shrimp populations reflect persistence in multiple southern refugia during Pleistocene ice ages and postglacial colonisation. Freshw Biol 64(11):1896–1907. https://doi.org/10.1111/fwb.13380
Makino W, Tanabe AS (2009) Extreme population genetic differentiation and secondary contact in the freshwater copepod Acanthodiaptomus pacificus in the Japanese Archipelago. Mol Ecol 18(17):3699–3713. https://doi.org/10.1111/j.1365-294X.2009.04307.x
Margalef R (1948) Estudios sobre la vida en las aguas continentales de la región endorréica manchega. Inst Biol Apl 4:5–51
Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet J 17(1):10–12. https://doi.org/10.14806/ej.17.1.200
Martino P (1988) Limnología de las lagunas salinas españolas. Thesis Dissertation, Universidad Autónoma de Madrid.
Méndez M, Tella JL, Godoy JA (2011) Restricted gene flow and genetic drift in recently fragmented populations of an endangered steppe bird. Biol Cons 144(11):2615–2622. https://doi.org/10.1016/j.biocon.2011.07.011
Migliore L, Brambilla G, Grassitellis A, Delupis GD (1993) Toxicity and bioaccumulation of sulphadimethoxine. Int J Salt Lake Res 2(2):141–152. https://doi.org/10.1007/BF02905906
Milligan BG (2003) Maximum-likelihood estimation of relatedness. Genetics 163(3):1153–1167. https://doi.org/10.1093/genetics/163.3.1153
Mills S, Lunt DH, Gómez A (2007) Global isolation by distance despite strong regional phylogeography in a small metazoan. BMC Evol Biol 7(1):1–10. https://doi.org/10.1186/1471-2148-7-225
Ministerio para la Transición Ecológica y el Reto Demográfico (MITECO) (2018). Cartografía y SIG. https://www.miteco.gob.es/es/cartografia-ysig/ide/descargas/default.aspx
Montero-Pau J, Serra M, Gómez A (2017) Diapausing egg banks, lake size, and genetic diversity in the rotifer Brachionus plicatilis Müller (Rotifera, Monogononta). Hydrobiologia 796(1):77–91. https://doi.org/10.1007/s10750-016-2833-4
Muñoz J, Gómez A, Green AJ, Figuerola J, Amat F, Rico C (2008) Phylogeography and local endemism of the native Mediterranean brine shrimp Artemia salina (Branchiopoda: Anostraca). Mol Ecol 17(13):3160–3177. https://doi.org/10.1111/j.1365-294X.2008.03818.x
Muñoz J, Green AJ, Figuerola J, Amat F, Rico C (2009) Characterization of polymorphic microsatellite markers in the brine shrimp Artemia (Branchiopoda, Anostraca). Mol Ecol Res 9(2):547–550. https://doi.org/10.1111/j.1755-0998.2008.02360.x
Muñoz J, Chaturvedi A, De Meester L, Weider LJ (2016) Characterization of genome-wide SNPs for the water flea Daphnia pulicaria generated by genotyping-by-sequencing (GBS). Sci Rep 6:28569
Newman D, Pilson D (1997) Increased probability of extinction due to decreased genetic effective population size: experimental populations of Clarkia pulchella. Evol 51(2):354–362. https://doi.org/10.1111/j.1558-5646.1997.tb02422.x
Nunney L, Elam DR (1994) Estimating the effective population size of conserved populations. Cons Biol 8(1):175–184. https://doi.org/10.1046/j.15231739.1994.08010175.x
O’Leary SJ, Puritz JB, Willis SC, Hollenbeck CM, Portnoy DS (2018) These aren’t the loci you’e looking for: Principles of effective SNP filtering for molecular ecologists. Mol Ecol 27(16):3193–3206. https://doi.org/10.1111/mec.14792
Ortego J, Aguirre MP, Cordero PJ (2010) Population genetics of Mioscirtus wagneri, a grasshopper showing a highly fragmented distribution. Mol Ecol 19(3):472–483. https://doi.org/10.1111/j.1365-294X.2009.04512.x
Palstra FP, Ruzzante DE (2008) Genetic estimates of contemporary effective population size: what can they tell us about the importance of genetic stochasticity for wild population persistence? Mol Ecol 17(15):3428–3447. https://doi.org/10.1111/j.1365-294X.2008.03842.x
Parekh PA, Paetkau MJ, Gosselin LA (2014) Historical frequency of wind dispersal events and role of topography in the dispersal of anostracan cysts in a semi-arid environment. Hydrobiologia 740(1):51–59. https://doi.org/10.1007/s10750-014-1936-z
Pearman WS, Urban L, Alexander A (2021) Commonly used Hardy-Weinberg equilibrium filtering schemes impact population structure inferences using RADseq data. Mol Ecol Res 22(7):2599–2613. https://doi.org/10.1111/1755-0998.13646
Peel D, Waples RS, Macbeth GM, Do C, Ovenden JR (2013) Accounting for missing data in the estimation of contemporary genetic effective population size (Ne). Mol Ecol Res 13:243–253
Pembleton LW, Cogan NO, Forster JW (2013) StAMPP: an R package for calculation of genetic differentiation and structure of mixed-ploidy level populations. Mol Ecol Res 13(5):946–952. https://doi.org/10.1111/1755-0998.12129
Plough LV (2017) Population genomic analysis of the blue crab Callinectes sapidus using genotyping-by-sequencing. J Shellfish Res 36(1):249–261. https://doi.org/10.2983/035.036.0128
Pons P, Gonçalves MSS, Gil-Delgado JA, Ortells R (2018) Spatial distribution of Branchinectella media (Crustacea, Branchiopoda) in a saline pond from “La Mancha Húmeda”: a case of habitat selection? Limnetica 37:69–83. https://doi.org/10.23818/limn.37.07
Pormehr N, Van Stappen G, Agh N, Atashbar B, Beladjal L (2019) Irregular hatching patterns of Branchinecta orientalis Sars, 1901 (Branchiopoda: Anostraca) in response to parental food, brood order, and pre-inundation conditions. J Crustac Biol 39(4):500–508. https://doi.org/10.1093/jcbiol/ruz020
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155(2):945–959. https://doi.org/10.1093/genetics/155.2.945
Puritz JB, Hollenbeck CM, Gold JR (2014) dDocent: A RADseq, variant-calling pipeline designed for population genomics of non-model organisms. PeerJ 2(1):e431. https://doi.org/10.7717/peerj.431
Rautio M, Dufresne F, Laurion I, Bonilla S, Vincent WF, Christoffersen KS (2011) Shallow freshwater ecosystems of the circumpolar Arctic. Ecoscience 18(3):204–222. https://doi.org/10.2980/18-3-3463
Recuerda M, Illera JC, Blanco G, Zardoya R, Milá B (2021) Sequential colonization of oceanic archipelagos led to a species-level radiation in the common chaffinch complex (Aves: Fringilla coelebs). Mol Phylogenet Evol. https://doi.org/10.1016/j.ympev.2021.107291
Reed DH (2007) Selection and the rate of loss of genetic variation: natural selection and genetic diversity. Heredity 99(1):1–2. https://doi.org/10.1038/sj.hdy.6800990
Reid AJ, Carlson AK, Creed IF, Eliason EJ, Gell PA, Johnson PTJ, Kidd KA, MacCormack TJ, Olden JD, Ormerod SJ, Smol JP, Taylor WW, Tockner K, Vermaire JC, Dudgeon D, Cooke SJ (2019) Emerging threats and persistent conservation challenges for freshwater biodiversity. Biol Rev 94(3):849–873. https://doi.org/10.1111/brv.12480
Rey-Iglesia A, Gopalakrishan S, Carøe C, Alquezar-Planas DE, Ahlmann-Nielsen A, Röder T, Pedersen LB, Næsborg-Nielsen C, Sinding MHS, Rath MF, Li Z, Petersen B, Thomas M, Gilbert P, Bunce M, Mourier T, Hansen AJ (2019) MobiSeq: De novo SNP discovery in model and non-model species through sequencing the flanking region of transposable elements. Mol Ecol Res 19(2):512–525. https://doi.org/10.1111/1755-0998.12984
Rich SS, Bell AE, Wilson SP (1979) Genetic drift in small populations of Tribolium. Evol 33(2):579–584. https://doi.org/10.2307/2407781
Rodríguez-Flores PC, Sánchez-Vialas A, García-París M (2016) Muestreos taxonómicos en charcos estacionales: una herramienta imprescindible para el conocimiento de la distribución geografica de Anostraca (Crustacea: Branchiopoda) en el centro de la Peninsula Ibérica. Heteropterus Rev Entomol 16(1):29–52
Rodríguez-Flores PC, Jiménez-Ruiz Y, Forró L, Vörös J, García-París M (2017) Non-congruent geographic patterns of genetic divergence across European species of Branchinecta (Anostraca: Branchinectidae). Hydrobiologia 801(1):47–57. https://doi.org/10.1007/s10750-017-3266-4
Rodríguez-Flores PC, Recuero E, Jiménez-Ruiz Y, García-París M (2019) Limited long-distance dispersal success in a western European fairy shrimp evidenced by nuclear and mitochondrial lineage structuring. Curr Zool 66(3):1–11. https://doi.org/10.1093/cz/zoz054
Rogers DC (2014a) Larger hatching fractions in avian dispersed anostracan eggs (Branchiopoda). J Crustac Biol 34(2):135–143. https://doi.org/10.1163/1937240X-00002220
Rogers DC (2014b) Anostracan (Crustacea: Branchiopoda) zoogeography II: Relating distribution to geochemical substrate properties in the USA. Zootaxa 3856(1):1–49
Rogers DC (2015) A conceptual model for anostracan biogeography. J Crustac Biol 35(5):686–699. https://doi.org/10.1163/1937240X-00002369
Rossi V, Paris G, Menozzi P (1994) Genetic variability in Mixodiaptomus kupelwieseri (Crustacea, Copepoda). Verh Lnternat Verein Limnol 25:2436–2438. https://doi.org/10.1080/03680770.1992.11900662
Rowe CL, Adamowicz SJ, Hebert PDN (2007) Three new cryptic species of the freshwater zooplankton genus Holopedium (Crustacea: Branchiopoda: Ctenopoda), revealed by genetic methods. Zootaxa 1656:1–49. https://doi.org/10.11646/zootaxa.1656.1.1
Sainz-Escudero L, Stănescu FS, Rodríguez-Flores PC, García-París M (2019) First record of Branchinectella media (Schmankewitsch, 1873) (Branchiopoda, Anostraca) in Romania. Crustaceana 92(10):1231–1247. https://doi.org/10.1163/15685403-00003936
Sainz-Escudero L, García-París M, Sánchez-Vialas A, Rosas-Ramos N, Rodríguez-Flores PC (2021) Nuevas localizaciones de Anostraca (Crustacea: Branchiopoda ) en el oeste de la Península Ibérica y consideraciones sobre su estado de conservación. Heteropterus Rev Entomol 21(1):55–77
Sainz-Escudero L, López-Estrada EK, Rodríguez-Flores PC, García-París M (2022) Brine shrimps adrift: historical species turnover in Western Mediterranean Artemia (Anostraca). Biol Inv 24:2477–2498. https://doi.org/10.1007/s10530-022-02779-6
Sala J, Gascón S, Boix D (2005) Nueva localidad para Branchinectella media (Crustacea: Anostraca) en Los Monegros. Bol Soc Entomol Aragonesa 37:164
Sánchez-Bayo F (2006) Comparative acute toxicity of organic pollutants and reference values for crustaceans: I—Branchiopoda, Copepoda and Ostracoda. Environ Pollut 139(3):385–420. https://doi.org/10.1016/j.envpol.2005.06.016
Sánchez-Vialas A, Recuero E, Jiménez-Ruiz Y, Ruiz JL, Marí-Mena N, García-París M (2021) Phylogeny of Meloini blister beetles (Coleoptera, Meloidae) and patterns of island colonization in the Western Palaearctic. Zool Scr 50(3):358–375. https://doi.org/10.1111/zsc.12474
Saunders DA, Hobbs RJ, Margules CR (1991) Biological consequences of ecosystem fragmentation: a review. Soc Cons Biol 5(1):18–32. https://doi.org/10.1111/j.1523-1739.1991.tb00384.x
Scheihing R, Cardenas L, Labarca P, Nespolo RF (2011) Spatial and temporal analysis of the genetic diversity and population structure of a freshwater ostracod from the high Andean plateau. Evol Ecol Res 13:91–104
Schmankewitsch W (1873) Fakty, imeyushchiye otnosheniye k vliyaniyu okruzhayushchey sredy na fiziologicheskiye funktsii i organizatsiyu zhivotnykh [Facts relevant to the influence of the environment on the physiological functions and organization of animals]. Trudy Tret’ego Syezda Russkikh Estestvo Ispitateley (Zoologiya, Anatomiya i Fiziologiya) 65: 89–92.
Schwentner M, Giribert G, Combosch DJ, Timms BV (2018) Genetic differentiation in mountain-dwelling clam shrimp, Paralimnadia (Crustacea: Branchiopoda: Spinicaudata), in eastern Australia. Invertebr Syst 34(1):88–100. https://doi.org/10.1071/IS19027
Servicio de Cartografía Universidad Autónoma de Madrid (SCUAM) (1987) http://guiadigital.uam.es/SCUAM/index.php
Star B, Spencer HG (2013) Effects of genetic drift and gene flow on the selective maintenance of genetic variation. Genetics 194(1):235–244. https://doi.org/10.1534/genetics.113.149781
Templeton A, Shaw K, Routman E, Davis S (1990) The genetic consequences of habitat fragmentation. Ann Missouri Bot Gard 77(1):13–27. https://doi.org/10.2307/2399621
Timms BV (2006) The large branchiopods (Crustacea: Branchiopoda) of gnammas (rock holes) in Australia. J R Soc West Aust 89(4):163–173
Tladi M, Nyamukondiwa C, Cuthbert RN, Wasserman RJ (2020) Emergent effects of light and temperature on hatching success of Streptocephalus cafer (Branchiopoda: Anostraca) resting eggs. Austral Ecol 45(8):1062–1066. https://doi.org/10.1111/aec.12934
Udebuani AC, Pereao O, Akharame MO, Fatoki OS, Opeolu BO (2021) Acute toxicity of piggery effluent and veterinary pharmaceutical cocktail on freshwater organisms. Environ Monit Assess 193(5):1–13. https://doi.org/10.1007/s10661-021-09085-z
Unal E, Bucklin A (2010) Basin-scale population genetic structure of the planktonic copepod Calanus finmarchicus in the North Atlantic Ocean. Prog Oceanog 87(1–4):175–185. https://doi.org/10.1016/j.pocean.2010.09.017
Vanschoenwinkel B, Waterkeyn A, Vandecaetsbeek T, Pineau O, Grillas P, Brendonck L (2008) Dispersal of freshwater invertebrates by large terrestrial mammals: a case study with wild boar (Sus scrofa) in Mediterranean wetlands. Freshw Biol 53(11):2264–2273. https://doi.org/10.1111/j.1365-2427.2008.02071.x
Vanschoenwinkel B, Seaman M, Brendonck L (2010) Hatching phenology, life history and egg bank size of fairy shrimp Branchipodopsis spp. (Branchiopoda, Crustacea) in relation to the ephemerality of their rock pool habitat. Aquat Ecol 44:771–780. https://doi.org/10.1007/s10452-010-9315-y
Vanschoenwinkel B, Brendonck L, Pincee T, Dupriez P, Waterkeyn A (2013) Rediscovery of Branchipus schaefferi (Branchiopoda: Anostraca) in Belgium—Notes on habitat requirements and conservation management. Belg J Zool 143(1):3–14
Velonà A, Luchetti A, Scanabissi F, Mantovani B (2009) Genetic variability and reproductive modalities in European populations of Triops cancriformis (Crustacea, Branchiopoda, Notostraca). Ital J Zool 76(4):366–375. https://doi.org/10.1080/11250000902785314
Vilà C, Sundqvist AK, Flagstad Ø, Seddon J, Björnerfeldt S, Kojola I, Casullo A, Sand H, Wabakken P, Ellegren H (2003) Rescue of a severely bottlenecked wolf (Canis lupus) population by a single immigrant. Proc Royal Soc B 270(1510):91–97. https://doi.org/10.1098/rspb.2002.2184
Vrijenhoek RC (1994) Genetic diversity and fitness in small populations. In: Loeschcke V, Jain SK, Tomiuk J (eds) Conservation genetics. Birkhäuser, Basel, pp 37–53
Wahlund S (1928) Zusammensetzung von Populationen und Korrelationerscheinungen vom Standpunkt der Vererbungslehre aus betrachtet. Hereditas 11:65–106
Wang J (2007) Triadic IBD coefficients and applications to estimating pairwise relatedness. Genet Res 89(3):135–153. https://doi.org/10.1017/S0016672307008798
Wang J (2011) Coancestry: A program for simulating, estimating and analysing relatedness and inbreeding coefficients. Mol Ecol Res 11(1):141–145. https://doi.org/10.1111/j.1755-0998.2010.02885.x
Wang CC, Rogers DC (2018) Bet hedging in stochastic habitats: an approach through large branchiopods in a temporary wetland. Oecologia 188(4):1081–1093
Wang J, Santiago E, Caballero A (2016) Prediction and estimation of effective population size. Heredity 117(4):193–206. https://doi.org/10.1038/hdy.2016.43
Waples RS (2006) A bias correction for estimates of effective population size based on linkage disequilibrium at unlinked gene loci. Conserv Genet 7(2):167–184. https://doi.org/10.1007/s10592-005-9100-y
Waples RS (2015) Testing for Hardy-Weinberg proportions: have we lost the plot? J Hered 106(1):1–19. https://doi.org/10.1093/jhered/esu062
Waples RS, Do C (2008) LDNE: a program for estimating effective population size from data on linkage disequilibrium. Mol Ecol Res 8(4):753–756. https://doi.org/10.1111/j.1755-0998.2007.02061.x
Waples RS, Do C (2010) Linkage disequilibrium estimates of contemporary Ne using highly variable genetic markers: a largely untapped resource for applied conservation and evolution. Evol Appl 3(3):244–262. https://doi.org/10.1111/j.1752-4571.2009.00104.x
Waples RS, Antao T, Luikart G (2014) Effects of overlapping generations on linkage disequilibrium estimates of effective population size. Genetics 197(2):769–780
Waples RK, Larson WA, Waples RS (2016) Estimating contemporary effective population size in non-model species using linkage disequilibrium across thousands of loci. Heredity 117(4):233–240. https://doi.org/10.1038/hdy.2016.60
Wei T, Simko VR, Levy M (2021) R package ‘corrplot’: Visualization of a Correlation Matrix (Version 0.92). https://github.com/taiyun/corrplot
Williams DD (1997) Temporary ponds and their invertebrate communities. Aquat Conserv Mar Freshw Ecosyst 7:105–107
Wright S (1931) Evolution in mendelian populations. Bull Math Biol 52(1–2):241–295. https://doi.org/10.1007/BF02459575
Wright S (1938) Size of population and breeding structure in relation to evolution. Sci 87:430–431
Wright S (1951) The genetical structure of populations. Ann Eugen 15:323–354
Xu S, Hebert PDN, Kotov AA, Cristescu ME (2009) The noncosmopolitanism paradigm of freshwater zooplankton: insights from the global phylogeography of the predatory cladoceran Polyphemus pediculus (Linnaeus, 1761) (Crustacea, Onychopoda). Mol Ecol 18(24):5161–5179. https://doi.org/10.1111/j.1365-94X.2009.04422.x
Zacharias I, Zamparas M (2010) Mediterranean temporary ponds: a disappearing ecosyatem. Biodivers Conserv 19(14):3827–3834. https://doi.org/10.1007/s10531-010-9933-7
Acknowledgements
We thank Robin Waples, Ryan Waples, and Jinliang Wang for kindly and thoroughly answering several questions about their methods to estimate Ne and relatedness. We kindly appreciate the valuable comments and suggestion received by Armando Caballero,Horacio Naveira, Michael Ford, and Christopher Rogers. We also thank Carlos Fernandez-Lozano, Christian Ponte-Fernández, E. Karen López-Estrada, Laura Torrado-Blanco and María Recuerda for their great help with some bioinformatic issues. We are grateful to Alberto Sánchez, Natalia Rojas and Paula C. Rodríguez for their help during sampling as well as to Begoña Sánchez (curator of the Arthropod Collection of the Museo Nacional de Ciencias Naturales) and Beatriz Álvarez (from the Tissue and DNA Collection) for all the resources provided. Finally, we thank the Comunidad Autónoma de Madrid (Spain), for the concession of the “Doctorado Industrial” grant (IND2018/AMB9692), through Fundación Global Nature, with support of MITECO (Ministerio para la Transición Ecológica y el Reto Demográfico,) and the MNCN- CSIC for all facilities provided.
Funding
Open Access funding provided thanks to the CRUE-CSIC agreement with Springer Nature. This work was supported by the “Doctorado Industrial” grant ((IND2018/AMB9692). LS-E has received research support from Comunidad Autónoma de Madrid (Spain), Fundación Global Nature and MITECO (Ministerio para la Transición Ecológica y Reto Demográfico).
Author information
Authors and Affiliations
Contributions
LS-E and MG-P conceived the original idea and carried out the fieldwork. LS-E and SP generated and processed the molecular data. LS-E and MV designed and conducted the data analyses. LS-E wrote the first draft. LS-E, MV and MG-P wrote the final manuscript with comments from SP.
Corresponding authors
Ethics declarations
Competing interest
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sainz-Escudero, L., Vila, M., Perea, S. et al. Large effective size as determinant of population persistence in Anostraca (Crustacea: Branchiopoda). Conserv Genet 24, 675–692 (2023). https://doi.org/10.1007/s10592-023-01534-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-023-01534-1