
Abstract
Fragmentation of isolated populations increases the risk of inbreeding and loss of genetic diversity. The endemic Saimaa ringed seal (Pusa hispida saimensis) is one of the most endangered pinnipeds in the world with a population of only ~ 400 individuals. The current genetic diversity of this subspecies, isolated in Lake Saimaa in Finland for ca. 1000 generations, is alarmingly low. We performed whole-genome sequencing on Saimaa ringed seals (N = 30) and analyzed the level of homozygosity and genetic composition across the individual genomes. Our results show that the Saimaa ringed seal population has a high number of runs of homozygosity (RoH) compared with the neighboring Baltic ringed seal (Pusa hispida botnica) reference population (p < 0.001). There is also a tendency for stillborn seal pups to have more pronounced RoH. Since the population is divided into semi-isolated subpopulations within the Lake Saimaa exposing the population to deleterious genomic effects, our results support augmented gene flow as a genetic conservation action. Based on our results suggesting inbreeding depression in the population, we recommend Pihlajavesi as a potential source and Southern Saimaa as a potential recipient subpopulation for translocating individuals. The Saimaa ringed seal is a recognized subspecies and therefore translocations should be considered only within the lake to avoid an unpredictable risk of disease, the introduction of deleterious alleles, and severe ecological issues for the population.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Preservation of genetic diversity has been recognized as essential to the conservation efforts of endangered species (Frankel 1974). High genetic diversity is positively linked to the viability and evolutionary potential of a population but is often compromised in endangered species. Decreasing population size and increasing isolation reduce fitness both at individual and population level via inbreeding depression (e.g., Frankham 1996, 2010, 2015; O’Grady et al. 2006; Hedrick and Garcia-Dorado 2016; Kardos et al. 2016, 2018; Weiser et al. 2016; Frankham et al. 2019; Harrisson et al. 2019; Gkafas et al. 2020; Kyriazis et al. 2021; Ning et al. 2021). Genetic rescue aims to restore, preserve, and increase current genetic diversity in semi-isolated subpopulations and thus reduce the extinction risk of endangered populations. It is largely implemented through translocations, which aim to increase the diversity by adding individuals to areas with impoverished genetic variation. The International Union for Conservation of Nature defines translocation as deliberate and mediated movement of wild individuals or populations from one part of their range to another (IUCN/SSC 2013).
The Saimaa ringed seal (Pusa hispida saimensis), occupying a 4400 km2 freshwater lake in Finland, is one of the most endangered pinnipeds in the world (Kovacs et al. 2012). As a landlocked subspecies, it has suffered strongly from a variety of human-induced factors such as hunting, habitat loss, environmental toxins, incidental bycatch mortality, and wintertime water-level fluctuations related to hydropower production. Due to systematic conservation efforts since the early 1980s, the population has slowly grown from ~ 100 individuals in 1980 to ~ 400 individuals in 2020 (Kunnasranta et al. 2021). However, this endemic population is increasingly threatened by climate change, reducing the ice and snow cover, crucial for ringed seals giving birth in snow lairs (Auttila et al. 2014). To protect this unique environment, main Saimaa ringed seal habitats in the Lake Saimaa archipelago are being proposed as a new natural site on UNESCO’s list of World Heritage sites (Ministry of the Environment 2020).
The deglaciation of Fennoscandia and subsequent isolation of Lake Saimaa from the Baltic Ice Lake (a proto-Baltic Sea) approximately 9500–11,000 years ago (Stroeven et al. 2016) set the stage for the founding of the Saimaa ringed seal population and for the ongoing total genetic isolation from other ringed seals. Not surprisingly, the genetic variation, as shown both by nuclear microsatellites and mitochondrial DNA markers of the current Saimaa ringed seal population is among the lowest detected in pinnipeds (Valtonen et al. 2012; Nyman et al. 2014; Stoffel et al. 2018; Peart et al. 2020). For instance, the heterozygosity at microsatellite loci (HE = 0.34) is as low as observed in other endangered or genetically depauperate pinnipeds such as the Mediterranean monk seal (Monachus monachus, HE ≈ 0.37, Pastor et al. 2004; Karamanlidis et al. 2021) or northern elephant seal (Mirounga angustirostris HE ≈ 0.37, Abadía-Cardoso et al. 2017), and is notably lower than in the marine ringed seals (HE ≥ 0.80; Davis et al. 2008; Martinez-Bakker et al. 2013; Nyman et al. 2014). In addition to the impoverished genetic diversity, genetic studies indicate that the Saimaa ringed seal population is divided into semi-isolated subpopulations across the lake (Valtonen et al. 2012, 2014, 2015). This division into units with only restricted gene flow among them can make the already small population even more vulnerable to stochastic environmental and genetic effects (e.g., Lacy 1987; Reed and Frankham 2003; Kardos et al. 2018; Kyriazis et al. 2021).
In addition to diversity-level estimations, modern genomic analyses have offered new tools to assess inbreeding in natural populations. Genome-level nucleotide differences between individuals are commonly known as single-nucleotide polymorphisms (SNPs). SNPs are single-nucleotide differences in genome sequence between the alleles of an individual or between two or more individuals, which implicate the genetic variability in a population. They can be used to observe slight genetic differences within a population and predict an individual’s ancestry. An alternative estimator for the level of inbreeding is provided by the analysis of SNP-based contiguous lengths of homozygous areas, i.e., runs of homozygosity (RoH, Broman and Weber 1999; Ceballos et al. 2018), across the whole genome (Lencz et al. 2007; Keller et al. 2011; Pemberton et al. 2012; Grossen et al. 2017). RoH correlate positively with inbreeding and extensive RoH indicate close relatedness of individuals. RoH length also indicates how recent the observed inbreeding is: the longer the RoH the more recent the observed inbreeding (Browning and Browning 2012). The longest RoH in mammal genomes may exceed 10 million base pairs (Mbp) (Purfield et al. 2012; Grossen et al. 2017). In the Saimaa ringed seal population, a genome-wide SNP and RoH analysis in comparison to the neighboring Arctic (P. h. hispida), Baltic (P. h. botnica) and Ladoga (P. h. ladogensis) subspecies shows evidence of lost genetic diversity and contemporary inbreeding (Löytynoja et al. 2022).
In this study, we further examine the most recent genomic data of the Saimaa ringed seals to better understand the diversity and the level of inbreeding of this small, isolated, and fragmented population as well as to assess the potential benefit of translocations within Lake Saimaa. This is the first study to combine samples from various age groups in different Saimaa ringed seal subpopulations including samples from stillborn individuals and young pups. We analyze all single-nucleotide differences in the individual genomes and summarize the RoH and SNP compositions. We compare the homozygosity levels observed in the genomes of Saimaa ringed seals against the levels observed in the Baltic ringed seals and between the Lake Saimaa subpopulations. The specific objectives of this study are to (1) investigate differences in the RoH levels between the Baltic and Saimaa populations and between the Saimaa subpopulations, (2) compare the SNP composition between subpopulations, and (3) identify potential source and recipient subpopulations for translocating individuals within Lake Saimaa based on the RoH levels and SNP composition. The results will be used to make recommendations for an improved genetic conservation protocol for the Saimaa ringed seal. These recommendations may be valuable in the conservation of other endangered species.
Materials and methods
Sample material
We analyzed the genomes of Saimaa ringed seal pups that were stillborn, died before weaning, or died at less than a year old (N = 30, Fig. 1, Appendix S1). We selected this rare and high-quality sample material due to its accessibility and non-invasive nature. Two of the samples were from weaned pups (To-20/A1702 and Sa-20) that were found in poor condition and taken into care in 2020. To-20 died soon in captivity, but Sa-20 was released back into the wild the same summer without further records of its existence. The samples were collected and examined under the Centre for Economic Development, Transport and the Environment permits VARELY/3480/2016 and VARELY/2497/2018 during 1995–2020.

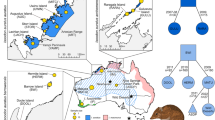
Map of Lake Saimaa showing the locations of seal samples used in this study
As reference data, we used published whole-genome sequencing data from Saimaa ringed seal samples (N = 11) collected during 1995–2017 (Fig. 1, Appendix S1) and Baltic ringed seal samples (N = 9) collected during 2007–2015 (Savriama et al. 2018). Baltic ringed seals were chosen as the reference group for its neighboring northern location, population size (N ≈ 13,000, Sundqvist et al. 2012), and available high-quality whole-genome data. The Saimaa samples consisted of individuals that survived beyond weaning (9 pups, one 1-year-old and one adult: Appendix S1). The Saimaa individuals had genomic coverages between 4.2X and 21.6X and those of Baltic individuals between 9.3X and 28.9X (Appendix S1). The Baltic ringed seal samples were from the Bothnian Bay; no detailed location information for these samples was available. We assigned each Saimaa individual into five groups representing the subpopulations: Main Haukivesi (n = 10), Pihlajavesi (n = 20), Kolovesi (n = 3), Northern Saimaa (n = 2), and Southern Saimaa (n = 6), according to the sample collection basin (Fig. 1).
Sequencing
We sequenced all samples with either the Illumina NextSeq500 or the MGI Tech Co. DNBseq-G400 sequencing platform (Appendix S1). The working principle of all platforms is essentially the same: base incorporation light color detection with a high-density camera. The main exception is that the Illumina NextSeq500 uses a bridging polymerase chain reaction (PCR) protocol to amplify the sample material whereas the DNBseq platform does not use PCR. We prepared the sequencing libraries with the standard library preparation protocols provided by Illumina and MGI.
Secondary analysis
We filtered the sequencing reads with Cutadapt (Martin 2011) version 1.9.1 and used a minimum read length of 50 base pairs and a sequencing quality threshold value of 25. This corresponds to a minimum base call correctness of 99.5% in the reads that pass the minimum length criterion. Cutadapt removed the residual sequencing library adapter sequences that may sometimes be introduced to raw sequencing reads due to the sequencing technology. We mapped the reads with the Burrows-Wheeler Aligner software (Li and Durbin 2009) against a draft reference genome produced by the Saimaa Ringed Seal Genome Project (https://www.saimaaringedseal.org/). The reference genome was built by scaffolding the 2183 contiguous sequences of the original draft assembly (Löytynoja et al. 2022) with a novel linkage disequilibrium (LD) -based method (Kivikoski et al. 2021) into 41 super scaffolds totaling 2,33 giga base pairs (Gbp). We then performed a joint variant calling pipeline of the Genome Analysis Toolkit (Van der Auwera and O’Connor 2020), which produced Variant Call Format (VCF) files for the next stage of the analysis. The secondary analysis statistics per individual are presented as supplementary material (Appendix S1).
Analysis of runs of homozygosity
RoH in the genomes were analyzed using the BCFtools/RoH pipeline (Narasimhan et al. 2016) which detects RoH based on the emission probabilities of a hidden Markov model. BCFtools is an open-source genome analysis software toolkit that is part of the SAMtools software (Li et al. 2009). It is common to use a cutoff length that discards short homozygous areas that appear quite frequently in animal genomes, for example, due to linkage disequilibrium (Purfield et al. 2012). Accordingly, we used a cutoff length of 500,000 base pairs (500 kbp) for RoH. We defined RoH exceeding 2 Mbp as long and RoH between 500 kbp and 2 Mbp as short. We used full coverage read data up to 28.9X to analyze the subpopulation level differences in RoH lengths. To investigate individual differences in long RoH, we also downsampled the mapped read data to 5X for all individuals to balance the sequencing coverages. We compared RoH longer than 3Mbp, 4Mbp and 5Mbp between stillborn individuals and those that died later.
Analysis of genetic composition
The number of SNPs and the SNP density across the genome (e.g., per 1 Mbp) are indicators of genetic diversity. SNP information of the samples was acquired from the VCF files produced in the secondary analysis. First, we identified the number of raw SNPs and the SNP density per individual (Appendix S1). Second, we filtered the raw VCF data using a minimum genomic position sample depth of 10, i.e., at least 10 out of 50 samples must have a SNP in the genomic position for the position to pass the filter. Third, we required a minor alternate variant frequency (= minor allele frequency) of 0.05 for the variant in the position and a minimum variant call quality, Q = 20, which translates to a 99% probability that the variant call in the position is correct. Finally, we counted the numbers of group-specific and shared positions with single-nucleotide variance between the Lake Saimaa groups.
We performed a Mann–Whitney U test for the selected indices of diversity: the (1) number of RoH, (2) number of long RoH, (3) percentage of RoH, and (4) median RoH length across the individual genomes. First, we tested the differences in the observed diversity between the within-lake groups and the Saimaa reference group. The observed non-significant differences between the within-lake and reference individuals (Appendix S2) allowed us to pool all Saimaa individuals into their respective groups within Lake Saimaa. Second, we tested the differences in observed diversity between the Saimaa (n = 41) and Baltic ringed seal reference group (n = 9) individuals. We then performed a Kruskal–Wallis analysis of variance (ANOVA) test for the Lake Saimaa groups to test whether the areas within Lake Saimaa were differentiated. Finally, we performed a pairwise Mann–Whitney U test for the Lake Saimaa groups with Bonferroni adjusted p-values.
Results
We discovered considerable differences in the levels of homozygosity between Saimaa and Baltic individuals (Fig. 2, Appendices S2–S5). The Saimaa individuals had a significantly higher number and percentage of RoH compared with the Baltic ringed seals (p < 0.001, Fig. 2a–c and Appendix S2). The median RoH length was short, less than 1,000,000 base pairs in all individuals (Figs. 2d and 3d and Appendix S5). There were no significant differences in the median RoH lengths between Saimaa and Baltic individuals (p > 0.1, Fig. 2d, Appendix S2). Within Lake Saimaa, Pihlajavesi individuals showed the highest variation in their RoH levels (Fig. 3).

Distributions of a number of runs of homozygosity (RoH), b number of long RoH, c percentage of RoH and d median RoH length in kilobases per sample group. The IDs are: SAI = Saimaa, BAL = Baltic reference. The boxplots represent the first quartile (box base), the median (horizontal black line at the box center), and the third quartile (box top) in the sample. The box whiskers extend to 1.5*interquartile range. Round dots are considered outliers with respect to the rest of the data points in the dataset

Distributions of a number of runs of homozygosity (RoH), b number of long RoH, c percentage of RoH and d median RoH length in kilobases, per group within Lake Saimaa. The group IDs are: MHV Main Haukivesi, PV Pihlajavesi, SS Southern Saimaa, KV Kolovesi, NS Northern Saimaa
Of the stillborn samples, individual 2402 (Pihlajavesi) had the highest number of RoH of all samples, 907 (Appendix S3). Individuals 2377 (Main Haukivesi) and 2402 had the highest overall percentage of RoH across the genome, 39.7% and 37.1%, respectively (Appendix S4).
Overall, the number of RoH was highly variable among different individuals (Fig. 4a). When comparing the stillborns with pups that died under the age of 12 months, the difference was statistically significant (p = 0.0496, Fig. 4b, Appendix S2). However, as the sequencing coverage can affect RoH lengths and number, we made a more detailed comparison of RoH lengths after downsampling the data to 5X coverage. Differences in sequencing depth could be a concern in our data since stillborn samples tended to have lower quality DNA and sequencing coverage (Appendix S1). After downsampling, stillborns showed tendency for longer RoH with 58% (7/12) of stillborn and 28% (8/29) of pups born alive having RoH lengths exceeding 4 Mbp (Fig. 4c). In addition, the number of long RoH tended to be higher in the stillborn samples (Fig. 4d), but the differences were not significant (smallest p = 0.088 for the RoH greater than 3 Mbp, Mann–Whitney U test). We note that the individual having the largest number of long RoH in the downsampled data was the seal pup To-20 that was found in poor condition, and which then died in captivity (triangle in Fig. 4d). The high level of RoH could have contributed to the death of To-20. The use of DNBseq-G400 and high sequencing coverage for this individual may also have exaggerated the RoH values in the downsampled data. If To-20 is excluded from the analyses, the number of long RoH in stillborns is significantly greater compared to pups born alive (p = 0.035 for the RoH greater than 3 Mbp). Although these results should be considered tentative because of the limited sample size, they are suggestive of inbreeding depression in the Saimaa ringed seals shown especially in the stillborn individuals.

Examining inbreeding depression in Saimaa ringed seals between stillborns and pups aged under 12 months. Distributions of a number of RoH per individual across subpopulations, b distributions of the number of long RoH, c percentages of individuals with RoH exceeding 2–5 megabase-pairs (Mbp) and d distributions of the number of long RoH. In subplots b-d, the boxplots represent the first quartile (box base), the median (horizontal black line at the box center), and the third quartile (box top) in the sample. The box whiskers extend to 1.5*interquartile range. The black dots are data points, and black triangles mark the To-20/A1702 seal pup that was found in poor condition and died in captivity
Of the Lake Saimaa groups, Southern Saimaa showed the highest and Northern Saimaa the lowest median level of homozygosity (Fig. 3). The Kruskal–Wallis ANOVA test of the groups indicated significant differences in number, percentage, and median length of RoH within Lake Saimaa (p < 0.05 for all diversity indices, Appendix S2). While this is a general signal of significant within-lake differentiation, the pairwise Mann–Whitney U test for the within-lake groups did not find significant differences between the subpopulations for any of the four diversity indices after applying Bonferroni correction (Appendix S2).
After filtering, there were 731,209 genomic positions with single-nucleotide variance at the population level. Altogether, 292,302 (40.0%) of the positions were shared between all Lake Saimaa groups (Fig. 5). Notably, there were a total of 39,986 group-specific positions between the groups. Pihlajavesi had the highest number of these, 19,943, i.e., 2.7% of all positions and 49.9% of group-specific positions found between the groups (Fig. 5).

Overlaps of the 731,209 genomic positions with single-nucleotide variance between the Lake Saimaa groups. The group IDs are: MHV Main Haukivesi (n = 10), PV Pihlajavesi (n = 20), KV Kolovesi (n = 3), SS Southern Saimaa (n = 6), NS Northern Saimaa (n = 2)
Discussion
This is the first study to investigate RoH and single-nucleotide variation at whole-genome level on the Saimaa ringed seal, a small landlocked population that has lived in total isolation for ca. 1000 generations. Our results are in line with earlier studies showing low genetic diversity in the population (Palo et al. 2003; Valtonen et al. 2012; Martinez-Bakker et al. 2013; Nyman et al. 2014; Savriama et al. 2018; Stoffel et al. 2018; Peart et al. 2020; Löytynoja et al. 2022). The diversity loss has been attributed to either a founder event after the colonization bottleneck, gradual erosion of diversity over a long isolation, or a more recent anthropogenic bottleneck caused by targeted hunting in the 1900s (Palo et al. 2003; Nyman et al. 2014; Stoffel et al. 2018; Peart et al. 2020). Populations experiencing an abrupt loss of diversity and increase of inbreeding suffer inbreeding depression more acutely (Hedrick and Kalinowski 2000; Kardos at al. 2018; Kyriazis et al. 2021). Nyman et al. (2014) suggested that low diversity of the Saimaa ringed seal largely results from a founder event by a small number of colonizers. However, Valtonen et al. (2014) found evidence for a slow decline in individual heterozygosity occurring over the past 50 years, and recent analyses by Stoffel et al. (2018) and Peart et al. (2020) likewise suggest that the human-induced population crash in the latter half of the 20th century (Sipilä 2003; Kunnasranta et al. 2021) has played a role in shaping the genetic variation of the Saimaa ringed seals.
Differences in runs of homozygosity levels between Baltic and Saimaa populations and between Saimaa subpopulations
The higher number and percentage of RoH in Saimaa ringed seals compared to the Baltic population suggest high and relatively recent inbreeding in the Saimaa ringed seals, which varies among the subpopulations in Lake Saimaa. Furthermore, some stillborn seals showed the highest percentage and number of long RoH (Appendices S4 and S5). Although limited by sample size, we observed a tendency for stillborn individuals to have higher numbers of long RoH when compared with pups that died of other causes. The difference was further emphasized when comparing RoH with lengths exceeding several megabase-pairs. While obviously only individual cases, they suggest that inbreeding in Saimaa ringed seals directly affects fitness in individuals. The long RoH results further indicate that the parents of the stillborn individuals are more likely to be closely related.
The results are in line with several previous studies documenting reduced genetic diversity and significant population sub structuring in Saimaa ringed seals (Palo et al. 2003; Valtonen et al. 2012, 2014; Löytynoja et al. 2022). Mitochondrial diversity patterns in the Saimaa population suggest that female seals are highly philopatric, staying and reproducing in their natal regions (Valtonen et al. 2012). Furthermore, nuclear markers show highly significant population structure (Valtonen et al. 2014; Löytynoja et al. 2022), indicating that sporadic trips made by single individuals between water basins (Biard et al. 2022) do not translate to effective gene flow (cf. Virrueta Herrera et al. 2022).
Single-nucleotide polymorphism composition between subpopulations
Our results show that the SNPs are unevenly distributed in the Saimaa ringed seal population. Thousands of group-specific positions of genomic single-nucleotide variance were found in all within-lake groups, the Pihlajavesi group alone containing half of the positions. The Southern Saimaa group had the second highest number of unique positions, but it also had the highest number of overlapping positions with all the other Lake Saimaa groups. Investigating the consequences, for example, whether the variants cause amino-acid changes or how the variants are distributed between the individuals in the group, will be the target of future research.
Preserving genetic variation is one way to increase the long-term survival of a population. Translocating individuals between subpopulations decreases the risk of losing useful SNPs, and in some cases, the whole population. SNPs that are unique will disappear if any of the subpopulations becomes extinct. While the significance of individual SNPs is often impossible to determine, in the case of a genetically depauperate population, such as the Saimaa ringed seals, all measures safeguarding the existing variation can be considered valuable.
Identifying potential source and recipient subpopulations
The results obtained in this study, combined with ecological information regarding the subpopulations, can be used to identify potential source and recipient regions in Lake Saimaa. Pihlajavesi and Main Haukivesi individuals showed the most variable RoH levels among the Lake Saimaa subpopulations. This is an expected result: the Pihlajavesi basin is estimated to have around 130 individuals and the Main Haukivesi basin 90 (Metsähallitus 2021), i.e., more than half of the whole population.
We observed a lower level of homozygosity in the Northern Saimaa subpopulation compared with the other Lake Saimaa subpopulations. The difference was not statistically significant but was biased toward the lower end for the RoH number, RoH percentage, and median RoH length. However, due to a small sample size (n = 2) we cannot be conclusive on the level of inbreeding in the Northern Saimaa population. Moreover, with a total population of only 20–30 individuals, Northern Saimaa looks less suitable as a source population for translocations. Conversely, at least the Southern Saimaa subpopulation and possibly Main Haukivesi subpopulation show relatively high numbers, percentages, and median lengths of RoH (Fig. 3). Despite the high variation in number, percentage, and median length of RoH, Pihlajavesi shows low median values of the diversity indices and has the highest subpopulation size, making it the most suitable source population.
Although the differences in the pairwise diversity indices within Lake Saimaa were found to be non-significant, we suggest Southern Saimaa as the potential recipient subpopulation for translocating individuals. We further identify the Kolovesi basin as a region of high concern due to its genetic isolation from the other Saimaa basins (Valtonen et al. 2014) and reduced pup production (Metsähallitus 2021). Since 2005, the population has decreased from 35 individuals to 12. The Kolovesi basin has conservation status as a national park and therefore the habitat would be favorable for translocation due to strict protection restrictions.
Saimaa ringed seal conservation perspectives
Ongoing climate change, which is estimated to be extreme in northern latitudes, increases the probability of winters without ice cover on Lake Saimaa. This would be highly detrimental to pup production as females give birth in snow lairs on icy lakeshores. Reducing the losses caused by climate change and other human-induced effects is vital for reaching favorable population size and strengthening the genetic diversity of the Saimaa ringed seal population. In addition to translocations, several actions are being carried out to improve seal survival into adulthood: fishing restrictions, water-level regulation, land use planning, building artificial nest boxes, and improving snow conditions by piling snow, are key conservation elements (see e.g., Kunnasranta et al. 2021, 2022).
A common concern with genetic rescue is that augmented gene flow may also lead to outbreeding depression and decrease fitness when source and recipient populations are genetically distant (Bell et al. 2019; Robinson et al. 2021). Within Saimaa, this risk can be ignored as the source and recipient populations originate from the same lake and have low population divergence, but the risk of outbreeding depression prevents translocations from other ringed seal populations e.g., from Like Ladoga. One within-lake translocation was successfully piloted in 1992 with an adult female which, based on the photo-identification database, was still alive 25 years later in the same region (Kunnasranta et al. 2021). Captive-reared individuals are used in many translocations (Griffiths and Pavajeau 2008; Attard et al. 2016; Landa et al. 2017), but actions are known to be more successful when wild-born individuals are used (Griffith et al. 1989).
Currently, the population size of the Saimaa ringed seals is highly dependent on the direct and indirect consequences of human activity. Since the alarmingly low numbers of 100–150 individuals in the 1980s, the population has slowly grown to ~ 400 individuals. Despite the gradual increase in population size, a few consecutive years with poor reproductive success could cause the population size to decline again. Based on a previous study on breeding habitat requirements of Saimaa ringed seals, it has been estimated that the carrying capacity of Lake Saimaa could be as high as 4,000 seals in favorable conditions (Niemi et al. 2019).
Our study shows that genomic approaches involving the analysis of RoH and SNPs are powerful in detecting differences in the genetic composition between populations, subpopulations, and individuals. Based on our results, we argue that within-lake translocations would have a positive effect on preserving the genetic diversity and population dynamics of the Saimaa ringed seal. Nevertheless, the effect of translocations will only be beneficial when performed along with other conservation actions.
Data availability
The sequencing reads will be made available in the Sequencing Read Archive of the U.S. National Library of Medicine’s National Center for Biotechnology Information (https://ncbi.nlm.nih.gov/SRA) under the accession SUB10800404 and embargoed therein until 31 Dec 2022 or until the publication of this manuscript. The Saimaa ringed seal (Pusa hispida saimensis) reference genome cannot be made publicly available before the actual publication of the genome (scheduled 2023). Before this, however, the data are limitedly available from the Saimaa Ringed Seal Genome Project (SRSGP; https://www.saimaaringedseal.org/genome.html) for researchers who meet the criteria for accessing confidential data. Interested researchers can contact Juhana Kammonen, Jukka Jernvall or Petri Auvinen of the SRSGP (ge-norppa@helsinki.fi).
References
Abadía-Cardoso A, Freimer NB, Deiner K, Garza JC (2017) Molecular population genetics of the northern elephant seal Mirounga angustirostris. J Hered 108:618–627. https://doi.org/10.1093/jhered/esx053
Attard CRM, Möller LM, Sasaki M, Hammer MP, Bice CM, Brauer CJ, Carvalho DC, Harris JO, Beheregaray LB (2016) A novel holistic framework for genetic-based captive-breeding and reintroduction programs. Conserv Biol 30:1060–1069. https://doi.org/10.1111/cobi.12699
Auttila M, Niemi M, Skrzypczak T, Viljanen M, Kunnasranta M (2014) Estimating and mitigating perinatal mortality of the endangered saimaa ringed seal (Phoca hispida saimensis) in a changing climate. Ann Zool Fenn 51:526–534. https://doi.org/10.5735/086.051.0601
Bell DA, Robinson ZL, Funk WC, Fitzpatrick SW, Allendorf FW, Tallmon DA, Whiteley AR (2019) The exciting potential and remaining uncertainties of genetic rescue. Trends Ecol Evol 34:1070–1079. https://doi.org/10.1016/j.tree.2019.06.006
Biard V, Nykänen M, Niemi M, Kunnasranta M (2022) Extreme moulting site fidelity of the Saimaa ringed seal. Mamm Biol. https://doi.org/10.1007/s42991-021-00209-z
Broman KW, Weber JL (1999) Long homozygous chromosomal segments in reference families from the centre d’Etude du polymorphisme humain. Am J Hum Genet 65:1493–1500. https://doi.org/10.1086/302661
Browning SR, Browning BL (2012) Identity by descent between distant relatives: detection and applications. Annu Rev Genet 46:617–633. https://doi.org/10.1146/annurev-genet-110711-155534
Ceballos FC, Joshi PK, Clark DW, Ramsay M, Wilson J (2018) Runs of homozygosity: windows into population history and trait architecture. Nat Rev Genet 19(4):220–234. https://doi.org/10.1038/nrg.2017.109
Davis CS, Stirling I, Strobeck C, Coltman DW (2008) Population structure of ice-breeding seals. Mol Ecol 17:3078–3094. https://doi.org/10.1111/j.1365-294X.2008.03819.x
Frankel OH (1974) Genetic conservation: our evolutionary responsibility. Genetics 78:53–65
Frankham R (1996) Relationship of genetic variation to population size in wildlife. Conserv Biol 10:1500–1508
Frankham R (2010) Inbreeding in the wild really does matter. Heredity 104:124
Frankham R (2015) Genetic rescue of small inbred populations: meta-analysis reveals large and consistent benefits of gene flow. Mol Ecol 24:2610–2618. https://doi.org/10.1111/mec.13139
Frankham R, Ballou JD, Ralls K, Eldridge M, Dudash MR, Fenster CB, Lacy RC, Sunnucks P (2019) A practical guide for genetic management of fragmented animal and plant populations. Oxford University Press, Oxford
Gkafas GA, de Jong M, Exadactylos A, Raga JA, Aznar FJ, Hoelzel AR (2020) Sex-specific impact of inbreeding on pathogen load in the striped dolphin. Proc R Soc B 287:20200195. https://doi.org/10.1098/rspb.2020.0195
Griffith B, Scott JM, Carpenter JW, Reed C (1989) Translocation as a species conservation tool. Status Strategy Sci 245:477–480
Griffiths RA, Pavajeau L (2008) Captive breeding, reintroduction, and the conservation of Amphibians. Conserv Biol 22:852–861. https://doi.org/10.1111/j.1523-1739.2008.00967.x
Grossen C, Biebach I, Angelone-Alasaad S, Keller LF, Croll D (2017) Population genomics analyses of european ibex species show lower diversity and higher inbreeding in reintroduced populations. Evol Appl 11:123–139. https://doi.org/10.1111/eva.12490
Harrisson KA, Magrath MJL, Yen JDL, Pavlova A, Murray N, Quin B, Menkhorst P, Miller KA, Cartwright K, Sunnucks P (2019) Lifetime fitness costs of inbreeding and being inbred in a critically endangered bird. Curr Biol 29:2711–2717. https://doi.org/10.1016/j.cub.2019.06.064
Hedrick PW, Garcia-Dorado A (2016) Understanding inbreeding depression, purging, and genetic rescue. Trends Ecol Evol 31:940–952. https://doi.org/10.1016/j.tree.2016.09.005
Hedrick PW, Kalinowski ST (2000) Inbreeding depression in conservation biology. Annu Rev Ecol Syst 31:139–162. https://doi.org/10.1146/annurev.ecolsys.31.1.139
IUCN/SSC (2013) Guidelines for reintroductions and other conservation translocations. Gland: IUCN Species Survival Commission. https://portals.iucn.org/library/efiles/documents/2013-009.pdf. Accessed 21 Nov 2022
Karamanlidis AA, Skrbinšek T, Amato G, Dendrinos P, Gaughran S, Kasapidis P, Kopatz A, Stronen AV (2021) Genetic and demographic history define a conservation strategy for earth’s most endangered pinniped, the Mediterranean monk seal Monachus monachus. Sci Rep 11:1–9. https://doi.org/10.1038/s41598-020-79712-1
Kardos M, Taylor HR, Ellegren H, Luikart G, Allendorf FW (2016) Genomics advances the study of inbreeding depression in the wild. Evol Appl 9:1205–1218. https://doi.org/10.1111/eva.12414
Kardos M, Åkesson M, Fountain T, Flagstad Ø, Liberg O, Olason P, Sand H, Wabakken P, Wikenros C, Ellegren H (2018) Genomic consequences of intensive inbreeding in an isolated wolf population. Nat Ecol Evol 2:124–131. https://doi.org/10.1038/s41559-017-0375-4
Keller MC, Visscher PM, Goddard ME (2011) Quantification of inbreeding due to distant ancestors and its detection using dense single nucleotide polymorphism data. Genetics 189:237–249. https://doi.org/10.1534/genetics.111.130922
Kivikoski M, Rastas P, Löytynoja A, Merilä J (2021) Automated improvement of stickleback reference genome assemblies with Lep-Anchor software. Mol Ecol Resour 21:2166–2176. https://doi.org/10.1111/1755-0998.13404
Kovacs KM, Aguilar A, Aurioles D, Burkanov V, Campagna C, Gales N, Gelatt T, Goldsworthy SD, Goodman SJ, Hofmeyr GJG, Härkönen T, Lowry L, Lydersen C, Schipper J, Sipilä T, Southwell C, Stuart S, Thompson D, Trillmich F (2012) Global threats to pinnipeds. Mar Mammal Sci 28:414–436. https://doi.org/10.1111/j.1748-7692.2011.00479.x
Kunnasranta M, Niemi M, Auttila M, Valtonen M, Kammonen J, Nyman T (2021) Sealed in a lake—biology and conservation of the endangered Saimaa ringed seal: a review. Biol Conserv. https://doi.org/10.1016/j.biocon.2020.108908
Kunnasranta M, Niemi M, Auttila M (2022) Developing artificial nest boxes for a large aquatic mammal. Aquat Conserv: Mar Freshw Ecosyst 32(8):1365–1371. https://doi.org/10.1002/aqc.3851
Kyriazis CC, Wayne RK, Lohmueller KE (2021) Strongly deleterious mutations are a primary determinant of extinction risk due to inbreeding depression. Evol Lett 5(1):33–47. https://doi.org/10.1002/evl3.209
Lacy RC (1987) Loss of genetic diversity from managed populations: interacting effects of drift, mutation, immigration, selection, and population subdivision. Conserv Biol 1:143–158
Landa A, Flagstad Ø, Areskoug V, Linnell JDC, Strand O, Ulvund KR, Thierry AM, Rød-Eriksen L, Eide NE (2017) The endangered Arctic fox in Norway—the failure and success of captive breeding and reintroduction. Polar Res 36:1–14. https://doi.org/10.1080/17518369.2017.1325139
Lencz T, Lambert C, DeRosse P, Burdick KE, Morgan TV, Kane JM, Kucherlapati R, Malhotra AK (2007) Runs of homozygosity reveal highly penetrant recessive loci in schizophrenia. Proc Natl Acad Sci USA 104:19942–19947. https://doi.org/10.1073/pnas.0710021104
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25:1754–1760. https://doi.org/10.1093/bioinformatics/btp324
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R (2009) 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079. https://doi.org/10.1093/bioinformatics/btp352
Löytynoja A, Rastas P, Valtonen M, Kammonen J, Holm L, Paulin L, Jernvall J, Auvinen P (2022) Fragmented habitat compensates for the adverse effects of genetic bottleneck. bioRxiv. https://doi.org/10.1101/2022.04.05.487133
Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 1:10–12. https://doi.org/10.14806/ej.17.1.200
Martinez-Bakker ME, Sell SK, Swanson BJ, Kelly BP, Tallmon DA (2013) Combined Genetic and telemetry data reveal high rates of gene flow, migration, and long-distance dispersal potential in Arctic ringed seals (Pusa hispida). PLoS ONE 8:1–17. https://doi.org/10.1371/journal.pone.0077125
Metsähallitus (2021) Saimaannorpan suojelu. https://www.metsa.fi/luonto-ja-kulttuuriperinto/lajien-suojelu/saimaannorppa/ (in Finnish). Accessed 21 Nov 2022
Ministry of the Environment (2020) Saimaa ringed seal habitats proposed to UNESCO World Heritage tentative list. https://ym.fi/en/-/saimaa-ringed-seal-habitats-proposed-to-unesco-world-heritage-tentative-list. Accessed 21 Nov 2022
Narasimhan V, Danecek P, Scally A, Xue Y, Tyler-Smith C, Durbin R (2016) BCFtools/RoH: a hidden Markov model approach for detecting autozygosity from next-generation sequencing data. Bioinformatics 1:1749–1751. https://doi.org/10.1093/bioinformatics/btw044
Niemi M, Liukkonen L, Koivuniemi M, Auttila M, Rautio A, Kunnasranta M (2019) Winter behavior of Saimaa ringed seals: non-overlapping core areas as indicators of avoidance in breeding females. PLoS ONE 14:1–15. https://doi.org/10.1111/j.1748-7692.2011.00521.x
Ning Y, Roberts NJ, Qi J, Peng Z, Long Z, Zhou S, Gu J, Hou Z, Yang E, Ren Y, Lang J, Liang Z, Zhang M, Ma J, Jiang G (2021) Inbreeding status and implications for Amur tigers. Anim Conserv 25:521–531. https://doi.org/10.1111/acv.12761
Nyman T, Valtonen M, Aspi J, Ruokonen M, Kunnasranta M, Palo JU (2014) Demographic histories and genetic diversities of Fennoscandian marine and landlocked ringed seal subspecies. Ecol Evol 4:3420–3434. https://doi.org/10.1002/ece3.1193
O´Grady JJ, Brook BW, Reed DH, Ballou JD, Tonkyn DW, Frankham R (2006) Realistic levels of inbreeding depression strongly affect extinction risk in wild populations. Biol Conserv 133:42–51. https://doi.org/10.1016/j.biocon.2006.05.016
Palo JU, Hyvärinen H, Helle E, Mäkinen HS, Väinölä R (2003) Postglacial loss of microsatellite variation in the landlocked Lake Saimaa ringed seal. Conserv Genet 4:801–803. https://doi.org/10.1023/A:1023303109701
Pastor T, Garza JC, Allen P, Amos W, Aguilar A (2004) Low genetic variability in the highly endangered Mediterranean monk seal. J Hered 95:291–300. https://doi.org/10.1093/jhered/esh055
Peart CR, Tusso S, Pophaly SD, Botero-Castro F, Wu CC, Aurioles-Gamboa D, Baird AB, Bickham JW, Forcada J, Galimberti F, Gemmell NJ, Hoffman JI, Kovacs KM, Kunnasranta M, Lydersen C, Nyman T, de Rosa L, Orr AJ, Sanvito S, Valtonen M, Shafer ABA, Wolf JBW (2020) Determinants of genetic variation across eco-evolutionary scales in pinnipeds. Nat Ecol Evol 4:1095–1104. https://doi.org/10.1038/s41559-020-1215-5
Pemberton TJ, Absher D, Feldman MW, Myers RM, Rosenberg NA, Li JZ (2012) Genomic patterns of homozygosity in worldwide human populations. Am J Hum Genet 10:275–292. https://doi.org/10.1016/j.ajhg.2012.06.014
Purfield DC, Berry DP, McParland S, Bradley DG (2012) Runs of homozygosity and population history in cattle. BMC Genet 13:1–11. https://doi.org/10.1186/1471-2156-13-70
Reed DH, Frankham R (2003) Correlation between fitness and genetic diversity. Conserv Biol 17:230–237. https://doi.org/10.1046/j.1523-1739.2003.01236.x
Robinson ZL, Bell DA, Dhendup T, Luikart G, Whiteley AR, Kardos M (2021) Evaluating the outcomes of genetic rescue attempts. Conserv Biol 35:666–677. https://doi.org/10.1111/cobi.13596
Savriama Y, Valtonen M, Kammonen JI, Rastas P, Smolander O-P, Lyyski A, Häkkinen TJ, Corfe IJ, Gerber S, Salazar-Ciudad I, Paulin L, Holm L, Löytynoja A, Auvinen P, Jernvall J (2018) Bracketing phenogenotypic limits of mammalian hybridization. R Soc Open Sci 5:180903. https://doi.org/10.1098/rsos.180903
Sipilä T (2003) Conservation biology of Saimaa ringed seal (Phoca hispida saimensis) with reference to other European seal populations. PhD Thesis. University of Helsinki, Helsinki, Finland. https://helda.helsinki.fi/bitstream/handle/10138/22401/conserva.pdf?sequence=2
Stoffel MA, Humble E, Paijmans AJ, Acevedo-Whitehouse K, Chilvers BL, Dickerson B, Galimberti F, Gemmel NJ, Goldsworthy SD, Nichols HJ, Krüger O, Negro S, Osborne A, Pastor T, Robertson BC, Sanvito S, Schultz JK, Shafer ABA, Wolf JBW, Hoffman JI (2018) Demographic histories and genetic diversity across pinnipeds are shaped by human exploitation, ecology and life-history. Nat Commun 9:1–12. https://doi.org/10.1038/s41467-018-06695-z
Stroeven AP, Hättestrand C, Kleman J, Heyman J, Fabel D, Fredin O, Goodfellow BW, Harbor JM, Jansen JD, Olsen L, Caffee WM, Fink D, Lundqvist J, Rosqvist GC, Strömberg B, Jansson KN (2016) Deglaciation of Fennoscandia. Quat Sci Rev 147:91–121. https://doi.org/10.1016/j.quascirev.2015.09.016
Sundqvist L, Harkonen T, Svensson CJ, Harding KC (2012) Linking climate trends to population dynamics in the Baltic ringed seal: impacts of historical and future winter temperatures. Ambio 418:865–872. https://doi.org/10.1007/s13280-012-0334-x
Valtonen M, Palo J, Ruokonen M, Kunnasranta M, Nyman T (2012) Spatial and temporal variation in genetic diversity of an endangered freshwater seal. Conserv Genet 13:1231–1245. https://doi.org/10.1007/s10592-012-0367-5
Valtonen M, Palo JU, Aspi J, Ruokonen M, Kunnasranta M, Nyman T (2014) Causes and consequences of fine-scale population structure in a critically endangered freshwater seal. BMC Ecol 14:1–14. https://doi.org/10.1186/1472-6785-14-22
Valtonen M, Heino M, Aspi J, Buuri H, Kokkonen T, Kunnasranta M, Palo JU, Nyman T (2015) Genetic monitoring of a critically-endangered seal population based on field-collected placentas. Ann Zool Fenn 52:51–65. https://doi.org/10.5735/086.052.0205
Van der Auwera GA, O’Connor BD (2020) Genomics in the cloud: using docker, GATK, and WDL in Terra (1st Edition). O’Reilly Media
Virrueta Herrera S, Johnson KP, Sweet AD, Ylinen E, Kunnasranta M, Nyman T (2022) High levels of inbreeding with spatial and host-associated structure in lice of an endangered freshwater seal. Mol Ecol 31:4593–4606. https://doi.org/10.1111/mec.16569
Weiser EL, Grueber CE, Kennedy ES, Jamieson IG (2016) Unexpected positive and negative effects of continuing inbreeding in one of the world’s most inbred wild animals. Evolution 70:154–166. https://doi.org/10.1111/evo.12840
Acknowledgements
We are grateful for project funding received from the LIFE Programme of the European Commission (LIFE19NAT/FI/000832). The material reflects the views of the authors; neither the European Commission nor the EASME is responsible for any use that may be made of the information it contains. We also thank the Jane and Aatos Erkko Foundation for funding (JJ and PA). In addition, we thank Metsähallitus and Finnish Food Authority for co-operation and providing samples. We thank J. Laakkonen, H. Nihtilä, S. Sainmaa, N. Airas and P. Sarkanen for help in acquiring samples from To-20 and Sa-20. We thank the personnel of the DNA Sequencing and Genomics Laboratory (Institute of Biotechnology, Helsinki) for performing the NGS sequencing.
Funding
Open Access funding provided by University of Helsinki including Helsinki University Central Hospital. This work was supported by the LIFE Programme of the European Commission (Grant Number LIFE19NAT/FI/000832). Authors PA and JJ have received funding from the Jane and Aatos Erkko Foundation (Grant Numbers 4-2013 and 5-2017).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by all authors. The first draft of the manuscript was written by Tarja Sundell and Juhana Kammonen and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors have no relevant financial or non-financial interests to disclosure.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary material 1 (XLSX 17.8 kb)
Primary (sequencing coverage) and secondary (single nucleotide polymorphism number and density) analysis statistics of the individuals in this study. Microsoft Excel Spreadsheet attached
Supplementary material 2 (XLSX 12.7 kb)
Results of non-parametric statistical tests for the diversity indices: 1) number of runs of homozygosity (RoH), 2) percentage of RoH, and 3) median RoH length. ANOVA = analysis of variance, SAI = Saimaa, BAL = Baltic reference, MHV = Main Haukivesi, PV = Pihlajavesi, KV = Kolovesi, SS = Southern Saimaa, NS = Northern Saimaa. Microsoft Excel Spreadsheet attached
Supplementary material 3 (TIF 298.2 kb)
Number of runs of homozygosity per individual (N = 50). The SR group consists of Saimaa ringed seal individuals that survived beyond weaning. BAL = Baltic, KV = Kolovesi, MHV = Main Haukivesi, PV = Pihlajavesi, SS = Southern Saimaa
Supplementary material 4 (TIF 280.3 kb)
Percentage of runs of homozygosity per individual (N = 50, genome size≈2.33Gbp). The SR group consists of Saimaa ringed seal individuals that survived beyond weaning. BAL = Baltic, KV = Kolovesi, MHV = Main Haukivesi, PV = Pihlajavesi, SS = Southern Saimaa
Supplementary material 5 (TIF 265.4 kb)
Distributions of runs of homozygosity (RoH) lengths per individual (N = 50, genomesize≈2.33 Gbp). The SR group consists of Saimaa ringed seal individuals that survived beyond weaning. The y-axis reference line at 2 megabases depicts the boundary between short and long RoHs. The boxplots represent the first quartile (box base), the median (horizontal black line at the box center), and the third quartile (box top) in the sample. The box whiskers extend to 1.5*interquartile range. The black dots are considered outliers with respect to the rest of the data points in the dataset. The boxplot ordering is based on the medians of the datasets of each sample in decreasing order. BAL = Baltic, KV = Kolovesi, MHV = Main Haukivesi, PV = Pihlajavesi, SS = Southern Saimaa
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sundell, T., Kammonen, J.I., Mustanoja, E. et al. Genomic evidence uncovers inbreeding and supports translocations in rescuing the genetic diversity of a landlocked seal population. Conserv Genet 24, 155–165 (2023). https://doi.org/10.1007/s10592-022-01497-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-022-01497-9