
Abstract
Intraspecific genetic variation can have similar effects as species diversity on ecosystem function; understanding such variation is important, particularly for ecological key species. The brown trout plays central roles in many northern freshwater ecosystems, and several cases of sympatric brown trout populations have been detected in freshwater lakes based on apparent morphological differences. In some rare cases, sympatric, genetically distinct populations lacking visible phenotypic differences have been detected based on genetic data alone. Detecting such “cryptic” sympatric populations without prior grouping of individuals based on phenotypic characteristics is more difficult statistically, though. The aim of the present study is to delineate the spatial connectivity of two cryptic, sympatric genetic clusters of brown trout discovered in two interconnected, tiny subarctic Swedish lakes. The structures were detected using allozyme markers, and have been monitored over time. Here, we confirm their existence for almost three decades and report that these cryptic, sympatric populations exhibit very different connectivity patterns to brown trout of nearby lakes. One of the clusters is relatively isolated while the other one shows high genetic similarity to downstream populations. There are indications of different spawning sites as reflected in genetic structuring among parr from different creeks. We used >3000 SNPs on a subsample and find that the SNPs largely confirm the allozyme pattern but give considerably lower F ST values, and potentially indicate further structuring within populations. This type of complex genetic substructuring over microgeographical scales might be more common than anticipated and needs to be considered in conservation management.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Identifying and understanding patterns of population genetic structure is an important task in evolutionary and conservation biology, because such intraspecific, genetic biodiversity represents results of evolutionary processes that provide the basic units for future adaptation processes (Allendorf et al. 2013). Research over the past decade has reported that genetic biodiversity can affect species productivity, diversity and viability (Reusch et al. 2005; Lindley et al. 2009), resilience to environmental stressors (Frankham 2005; Hajjar et al. 2008; Hellmair and Kinziger 2014) and adaptation to changing environmental features including climate change (McGinnity et al. 2009; Barshis et al. 2013). This knowledge is of key importance for sustainable management and is recognised in the Convention on Biological Diversity (CBD; http://www.cbd.int), and in the Strategic Plan for 2011–2020 where increasing efforts to protect genetic diversity is called for (cf. Aichi Biodiversity Target 13; CBD COP Dec http://www.cbd.int/sp/targets).
Genetic biodiversity has been suggested to be of particular importance in species-poor environments such as the boreal forest biome (Pastor 1996; Jetz and Fine 2012), including its fresh and marine water systems (Laikre et al. 2008; Johannesson et al. 2011). This is because genetically divergent populations within species can have similar effects for ecosystem function, resilience and services as species diversity does (Hughes et al. 1997), as empirically demonstrated in some biological systems (e.g. Cook-Patton et al. 2011; Yang et al. 2015). In salmonids, indications of genetically divergent populations contributing to different ecosystem roles include the portfolio effect documented in sockeye salmon (Schindler et al. 2010), where intraspecific life history variation stabilises ecosystem services, as well as in lake whitefish, in which different ecotypes take on different ecological roles as they occupy separate trophic niches (Rogers and Bernatchez 2007).
The brown trout (Salmo trutta) is an important salmonid species in freshwater lakes and streams in the northern hemisphere, particularly in the mountainous areas of Scandinavia. It is often one of a few occurring fish species, or the only one present, and thus of obvious ecological impact (Laikre 1999). Also, the brown trout represents a high socio-economic value as target for both sports and commercial fisheries (Marco-Rius et al. 2013). At the same time, rapid climate change is expected to be particularly severe for freshwater habitats such as the ones studied in the present paper (Prowse et al. 2006; Ficke et al. 2007). Taking all these aspects together, the brown trout represents a typical species to which the Aichi Target 13 of the CBD Strategic Plan 2011–2020 applies and for which increased efforts are needed to understand and safeguard genetic biodiversity.
The brown trout typically shows high genetic structuring (Laikre 1999). In some freshwater lakes multiple and genetically distinct, sympatric populations that also differ in morphology, diet and life history traits have been documented (Ferguson and Mason 1981; Ferguson and Taggart 1991; Prodöhl et al. 1992; Duguid et al. 2006), indicating different ecological roles of intraspecific groupings also in this species. Likewise, such genetically, phenotypically and ecologically divergent sympatric populations have been reported in other freshwater fish species including perch, walleye and rainbow smelt (Pigeon et al. 1998; Bergek and Björklund 2007; Dupont et al. 2007), as well as in other salmonids such as Arctic char and whitefish (Power et al. 2009; Gowell et al. 2012; Præbel et al. 2013; May-McNally et al. 2015). In contrast, sympatric populations that are cryptic, i.e. without visible phenotypic differences and where genetic data have been needed to detect the structures—have rarely been documented. As far as we have been able to find, only two cases in each of the species Arctic char (Wilson et al. 2004) and brown trout (Ryman et al. 1979; Palmé et al. 2013) have been documented, including the one that is the focus of this study.
In the present case, what appears to be two sympatric brown trout populations have previously been documented to occur in stable sympatry over a minimum of 19 years (almost three generations) in two very small, interconnected mountain lakes in a remote area in the Hotagen Nature Reserve, County of Jämtland, central Sweden (Palmé et al. 2013; examining samples collected annually 1987–2005). The two lakes—collectively referred to as Lakes Trollsvattnen—are part of a long-term genetic monitoring study (Jorde and Ryman 1996; Charlier et al. 2012; Palmé et al. 2013) where the existence of the two genetic clusters was detected when consistent heterozygote deficiency as compared to Hardy–Weinberg proportions became apparent after several years of genetic monitoring using allozyme markers (Jorde and Ryman 1996). The population structures occur in stable sympatry at about the same frequency within both lakes and show no obvious morphological dichotomies (Palmé et al. 2013). An extensive screening for phenotypic differences of the two clusters detected only minor tendencies for morphological divergence, and no indication of trophic niche separation (Andersson et al. 2016), in spite of a high genetic differentiation (F ST ≈ 0.1; Palmé et al. 2013). Differences did occur between lakes, however, indicating the same pattern of plasticity with respect to diet and morphology within both genetic clusters (Andersson et al. 2016). This lack of trophic niche separation is in agreement with suggested food web limitations of nutrient-poor subarctic lake ecosystems (Karlsson et al. 2009).
The present study focuses on further exploration of these cryptic structures; delineating the spatial connectivity of them, including their genetic relationship to nearby populations beyond the two lakes where they were originally detected. We address the following questions: (a) can the sympatric populations detected using allozyme markers be identified with SNPs (single nucleotide polymorphisms)? (b) What is the spatial distribution and genetic relationships of the two populations with respect to brown trout in neighbouring lakes? (c) Are there indications of differences with respect to spawning site?
Materials and methods
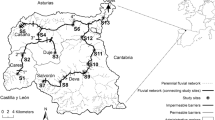
The study area is located in the Hotagen Nature Reserve in central Sweden and includes several small oligotrophic mountain lakes at an elevation of about 700 m (Fig. 1). The system belongs to the uppermost part of the River Indalsälven catchment, which is flowing into the Baltic Sea. The Lakes Trollsvattnen, where the sympatric clusters were detected, comprise the two interconnected Lake Östra Trollsvattnet (ÖT) and Lake Västra Trollsvattnet (VT). The lakes are tiny—0.10 km2 (ÖT) and 0.17 km2 (VT), with depths of 1–2 m for the major parts of the area; maximum depths are 5 and 6 m for ÖT and VT, respectively. They are included in a long-term genetic monitoring study, the Lakes Bävervattnen Project, which focuses on spatio-temporal genetic variability patterns in natural populations of brown trout in a pristine, conserved landscape with virtually no direct human manipulation of local fish populations (Jorde and Ryman 1996; Laikre et al. 1998; Palm and Ryman 1999; Palm et al. 2003; Charlier et al. 2011, 2012; Palmé et al. 2013). The study area is located c. 20 km from the nearest road and is reached only after a full day of hiking in partly harsh terrain.

Lakes and creeks sampled for the present study. The site is located in the Hotagen Nature Reserve, County of Jämtland, central Sweden. The sampled lakes are in dark grey; letters A–K indicate creek sampling locations. Small arrows show the direction of water flow
We collected about 100 fish annually during the past three decades from each of Lakes Östra and Västra Trollsvattnet within the monitoring project, and in recent years, we also sampled from selected neighbouring lakes. The trout used in the current study were collected in 1987–2014 (total n = 8078; Table 1), and represent samples from Lakes Trollsvattnen, VVT1, VVT2, Hästskotjärnen, Alfred Larsa Tjärnen, Trollsflyn, Häbbersflyn and Häbbersvattnet (Fig. 1). All sampling was conducted in late August–early September. The fish were caught using gillnets of various mesh sizes. Tissue samples of eye, muscle and liver were collected for genetic analysis. Individual data on length, weight, sex, reproductive status (mature to spawn or not) were also recorded, and otoliths were prepared for subsequent age determination.
In addition to the above sampling from the lakes we also collected young-of-the-year parr in the in- and outflows of Lakes Östra and Västra Trollsvattnet in 2013 and 2014 (total n = 888; Fig. 1). This sampling from creeks and streams was aimed to target potential spawning sites of the two populations and was done by single sweep electrofishing. We sampled a stretch of about 50 m at seven of the ten localities, and about 75, 100 and 150 m at the three remaining ones, covering the whole width of the separate creeks.
Genetic data
When the long-term genetic monitoring study of Lakes Bävervattnen Project started in the 1980s allozymes were the only genetic markers available for large-scale screening of natural populations, and we have therefore continued to use allozymes to provide consistency throughout the project. All fish were genotyped at a standard set of 14 polymorphic allozyme loci [all of them bi-allelic except for one locus (AAT6) where a third allele has been found in six individuals]; these loci have been used within the monitoring project (for details of procedures and terminology see Jorde and Ryman 1996; Palm et al. 2003; for locus nomenclature see Palmé et al. 2013). The screenings have been carried out in our own lab which has scored allozymes since the 1970s (Allendorf et al. 1976; Ryman et al. 1979; Ryman 1983). All markers have been carefully evaluated with respect to genotypic interpretation and inheritance (cf. Allendorf et al. 1977; Ståhl and Ryman 1982; Ryman and Utter 1987; Jorde et al. 1991).
In addition to the allozymes, we obtained SNP data (3093 polymorphic loci) for 60 individuals collected in Lakes Östra and Västra Trollsvattnet in 2004–2005, 30 fish from each of the two sympatric clusters originally identified with allozyme loci and denoted A and B (Palmé et al. 2013). The individuals chosen for SNP genotyping were selected on the basis of membership coefficient to respective population and we only included fish with a membership coefficient of 0.90 or higher (based on allozyme genotyping and the structure software, see below, using data from Palmé et al. 2013). In total, each population (A and B) and lake (ÖT and VT) was represented by 15 individuals (i.e. 30 per lake and population).
DNA extraction and SNP genotyping
Muscle samples were frozen at −70 °C within 6 h of fish collection. Genomic DNA was extracted from 50 mg tissue samples (n = 60) using DNeasy Blood & Tissue Kit from Qiagen (Hilden, Germany) according to manufacturer’s instructions and eluted in 100 μl elution buffer. DNA quality was assessed by electrophoresing an aliquot through a 1% agarose gel and subjectively assessing the proportion of high-molecular weight DNA relative to degraded DNA. Samples showing relatively inferior quality DNA were not genotyped but replaced with DNA from another individual representing the same population and lake. Double-stranded DNA was quantified using a Qubit fluorometer (ThermoScientific, MA, USA) and normalised to 30–50 ng/μl.
SNP genotyping of 60 samples was performed according to manufacturer’s instructions using an Illumina iSelect SNP-array containing 5509 SNP assays (Illumina, CA, USA). Briefly, this array included SNPs detected in whole genome sequencing (WGS) data obtained from 16 individuals representing both domestic families and wild populations, acquired from a Danish breeding program and Norwegian rivers. The final set of SNPs included on the custom array was arrived at after extensive filtering of WGS SNPs, and selection of SNP subsets based on their inter-SNP physical distance (60%), similarity to Salmo salar cDNA sequences (20%), and high sequence homology to larger S. salar scaffolds (20%; Sodeland et al. in prep). Genotypes were assigned using an existing custom cluster file in genomestudio (version 2011.1), and minor manual cluster correction was performed to improve calling and produce the best data. The cluster file we used had been constructed through earlier genotyping of several thousand samples from wild and farmed populations, and had also allowed us to subjectively classify markers into performance categories. This included “SNP”; defined as presenting three allele clusters (AA, AB, BB) with theta positions at 0.0, 0.5 and 1.0, “MSV3”; multisite-variant showing three clusters but skewed so that theta positions are 0.0, 0.25, 0.5 or 0.5, 0.75, 1.0 or “other”; including markers with low polymorphism rates and failed genotyping assays. Average call rate for the 60 samples was 0.997 and only polymorphic markers classified and behaving as SNPs (n = 2853) and MSV3s (n = 240) were included for subsequent analysis resulting in a total of 3093 SNP loci.
Statistical analysis
Allele frequencies and F ST (Weir and Cockerham 1984), quantifying spatial genetic heterogeneity, were estimated using genepop (version 4.3; Raymond and Rousset 1995; Rousset 2008). To obtain F ST significance levels genepop conducts an exact contingency test at each locus and combines the information from multiple loci using Fisher’s method. This approach frequently results in elevated Type I statistical errors (too many false significances; Ryman and Jorde 2001; Ryman et al. 2006), and particularly so for loci with few alleles; to avoid this problem we used chifish (Ryman 2006) for F ST significance testing. This program applies a contingency Chi square test at each locus separately and sums the corresponding Chi square values and their degrees of freedom when combining results from multiple loci. This approach has been shown to have a high power for detecting allele frequency differences while being less associated with excessive Type I errors (Ryman and Jorde 2001; Ryman et al. 2006; updated version of chifish allowing the joint evaluation of up to 10000 loci now available at http://www.zoologi.su.se/~ryman/). Deviations from Hardy–Weinberg proportions measured as F IS and their associated significance levels were obtained separately for each lake/cluster using genepop. Holm’s (1979) sequential Bonferroni approach was applied to adjust significance levels when evaluating the results from multiple testing as indicated in table legends.
We assessed the most likely number of populations (K) that would correspond to the genotypic distribution in the samples by individually based likelihood analyses using the structure software (Pritchard et al. 2000; Falush et al. 2003). The default model allowing population admixture and correlated allele frequencies was used. The burn-in length and the number of Markov chains (MCMC) were set to 250000 steps and 500000 replicates, respectively, when estimating Q (assignment probability; the estimated membership coefficient for each individual in each cluster) and likelihoods for different K-values (K = 1–10 for allozymes and K = 1–5 for SNPs). Estimation of the most likely value of K was repeated over ten runs and the output from structure was analysed using the structure harvester software (Earl and vonHoldt 2012). We based our estimation of the most likely number of K on ΔK (an ad hoc quantity related to the second order rate of change of the log probability of data; Evanno et al. 2005) given by structure harvester as well as the log probability of data (lnPrX│K), another ad hoc quantity given by structure. Mean individual membership coefficients (Q) to each population over the ten runs were derived from the clumpp software (Jakobsson and Rosenberg 2007).
We used structure on the following allozyme data sets: (1) each of the 9 lakes separately (Fig. 1; Table 1), (2) Lakes Östra and Västra Trollsvattnet pooled (n = 6159), (3) Lakes Östra and Västra Trollsvattnet pooled with fish collected in creeks draining to and from these lakes (n = 7047), (4) the 60 fish that were typed for SNPs. structure was also run for these 60 fish using SNP genotypes only.
We also performed a Discriminant Analysis of Principal Components (DAPC; Jombart et al. 2010) implemented in the adegenet package (version 1.4–2; Jombart 2008; Jombart and Ahmed 2011) in R (version 3.1.2; R Core Team 2014) for the SNP data. DAPC is a multivariate method for exploring the distribution of genetic diversity between groups that can be predefined. The method minimises the within-group variation and maximises among-group variation by first transforming the genotype data into principal components (PCs), followed by discriminant analysis (DA) to define the groups. We assessed the number of clusters using the find.clusters function, which runs successive K-means clustering with a maximum K set to 12. The best supported model for our data was K = 2 as it showed the lowest BIC (Bayesian Information Criterion) value. Forty axes of the PCA explained ~80% of the variation and were retained for the analysis.
We visualised the allozyme genetic relationship between the localities and populations detected within the main lakes (Östra and Västra Trollsvattnet) by constructing a neighbour-joining phylogenetic tree using the poptree2 software (Takezaki et al. 2010). The tree was based on sample size bias corrected F ST distance, because our samples varied substantially in size, and the default number of bootstrap replications (1000) was used.
To see if any of the allozymes and/or SNPs seem to deviate from neutrality we employed the software bayescan (v. 2.1; Foll and Gaggiotti 2008; Foll 2012). Default settings—samples size 5000, thinning interval 10, pilot runs 20, pilot run length 5000 and additional burn-in 50000—were used. Two separate bayescan analyses were conducted on fish assigned to the two cryptic populations (A and B). First, we analysed the SNP data set using the 60 fish scored at 3093 loci (30 fish per cluster). Next, we focused on the 14 allozyme loci using the pooled material from Lakes Östra and Västra Trollsvattnet (6159 fish assigned to cluster A and B, respectively, using assignment probability Q = 0.5 as a cut-off). Detection of selection with bayescan is based on comparisons of subpopulation-specific allele frequencies against the global level of differentiation (based on F ST coefficients; Fischer et al. 2011). Outliers were identified using a false discovery rate (q-value) threshold of 0.05.
Population-based outlier analysis (such as bayescan) can lead to biased results when there are admixed individuals present (Luu et al. 2016). Because of this, we also tested an alternative method implemented in the R-based pcadapt software (v.3.0.3; Luu et al. 2016) for the dataset of 3093 SNPs. Underlying population structure was first ascertained with principal component analysis by testing K values up to 25. Based on the obtained ’scree plot’ (Fig. S1), the optimal value of PCs to retain was between 2 and 4. Thus, the subsequent pcadapt analysis was conducted for K = 2, 3 and 4. A false discovery rate was set to 5%, and loci with minor allele frequency (MAF) < 0.05 (N = 361) were removed from the analysis leaving 2732 loci.
We had varying sample sizes from the studied localities (Table 1) and wanted to address the issue of statistical power for detecting a heterozygote deficiency at different sampling sizes. A modified version of the powsim software (Ryman and Palm 2006; cf. Palmé et al. 2013) was used to simulate the drawing of samples of various size from a mixture of two genetically diverged populations. We assumed equal proportions of the two populations and a divergence of F ST = 0.1 and allele frequencies as observed empirically (see “Results” section), and repeated the sampling 1000 times for each sample size.
Randomisation tests were used for exploring potential differences in the amount of heterozygote deficiency (relative to Hardy–Weinberg expectations) among young-of-the-year fish (parr) as compared to older age groups. The tests were performed through comparisons of F IS values using the poptools add-in for Excel (Hood 2010). The randomisation (reshuffling without replacement) was done within loci as well as across all loci.
Results
Genetic structure with SNP versus allozyme markers in Lakes Trollsvattnen
At the 14 allozyme loci we observed a significant heterozygote deficiency within each of the Lakes Östra and Västra Trollsvattnet (F IS = 0.035 and F IS = 0.039, respectively, with P ≪ 0.001 in both cases), and structure suggested K = 2 as the most likely number of populations within each of these lakes (Online Resource 1, Tables S2 and S3). When pooling the material from both lakes we obtained F IS = 0.037 (P ≪ 0.001) and a structure suggestion of K = 2. Using Q = 0.5 as the threshold for assignment to these clusters (A and B) yields 3263 and 2896 fish assigned to clusters A and B, respectively. These results are consistent with those reported by Palmé et al. (2013), and the addition of nine sampling years and 2019 individuals has thus not changed the previous observations.
The SNP data largely provide consistency with allozyme patterns (F IS = 0.022, P ≪ 0.001); the structure analysis combined with the Evanno method identified K = 2 as the highest hierarchical structure (Online Resource 1, Table S4) when using the 3093 SNP markers. However, the log likelihood approach for this data proposed K = 4 as the most likely number of genetic clusters (mean log likelihood = −7500.6; Table S4) indicating that additional substructuring might occur in Lakes Trollsvattnen. This four population solution suggests that additional clusters are nested within the two main clusters, although only two individuals are assigned to a third cluster (both within the original cluster A), and four to a fourth cluster (all four within the original cluster B; Online Resource 1, Fig. S2). Evanno et al. (2005) found the ΔK approach more reliable than that of log likelihoods for determining the true number of populations. Thus, for the purpose of the present paper, we conservatively focus on the highest hierarchical level identified using the Evanno approach. The substructuring was further supported by the pcadapt analysis of the SNP data where the first principal component separated the individuals into two distinct groups (Online Resource 1, Fig. S3).
With respect to assignment probabilities (Q) of individual fish to the two main clusters A and B, the SNP data also provide relatively high consistency with the allozyme data for the same 60 fish (Fig. 2). When K was set to 2 and using an assignment threshold of Q = 0.5, structure classified 55 of the 60 fish with SNP genotype to the same cluster as that based on allozyme data (Fig. 2a, b). A similar result was obtained using the DAPC approach (53 of 60 coinciding classifications; Fig. 2c). Mean assignment probability to each of the two clusters with SNPs was 0.95 for A and 0.81 for B, respectively (the 90% credible regions of individual assignment probabilities are presented in Online Resource 2).

Membership (Q) coefficient plots showing individuals assigned to cluster A and B, respectively, for allozyme (a) and SNP data (b, c) of the 60 brown trout that were scored for both sets of markers. Membership coefficients were obtained from structure (a) and (b), and from DAPC (c) for K = 2. Each fish is represented by a vertical bar that denotes membership coefficient (Q) to the two clusters; the fish are in the same order in all three subfigures. The fish were collected from Lakes Östra (n = 30) and Västra Trollsvattnet (n = 30; cf. Fig. 1)
The amount of divergence between the two clusters differs for allozymes versus SNPs. Overall allozyme F ST between clusters using all the 6159 fish collected in Lakes Trollsvattnen (ÖT + VT; Table 1) is ~0.1, whereas F ST is ~0.3 for the 60 fish used for SNP analysis. In contrast, using SNP data for the same 60 fish (30 per allozyme cluster) we have F ST ~ 0.03 (P < < 0.001). The allozyme divergence remains around F ST = 0.1 over 28 sampling years and 30 cohorts (Online Resource Fig. S6), however there is a slightly decreasing trend that is statistically significant for sampling years (R = −0.66; P < 0.001) but not for cohorts (R = −0.34; P = 0.07). Individual based dendrograms for the 60 fish for the two separate sets of markers (Online Resource 1, Fig. S4) also revealed a stronger divergence between the two clusters for the allozymes than for the SNPs.
Investigating cryptic structures in lakes and creeks neighbouring Lakes Trollvattnen
Our statistical power simulations suggest that with a F ST = 0.1 and the allele frequencies observed in the two clusters we should be able to detect substructuring (i.e., heterozygote deficiencies) with a power of about 0.7 at sample sizes close to 200 individuals (Online Resource Fig. S5). At sample sizes of 300–400, the power increases to 0.8–0.9 (Fig. S5). Thus, the power for detecting heterozygote deficiency is reasonable for most of the lakes, but weak for most of the separate creeks.
Heterozygote deficiency was not detected in any of the seven neighbouring lakes when combining the information from all allozyme loci (Table S2). Similarly, structure only suggested K = 2 as the most likely number of populations within Lakes Trollsvattnen; in no other lake did we find support for the existence of more than a single population (Online Resource 1, Table S3).
When the young-of-the-year parr from all the creeks were pooled (n = 888) we found a significant heterozygote deficiency for allozymes (F IS = 0.02; P < 0.05; Online Resource 1, Table S5), but the significance was not retained after Bonferroni correction. In contrast, no heterozygote deficiency was observed when examining each creek separately. structure suggests one population as most likely for three of the four default models for the pooled creek material; for the most restrictive model (no admixture and uncorrelated allele frequencies) it suggests two populations.
Genetic relationship among populations, lakes and creeks
The phylogenetic tree visualising the genetic relationships among clusters A and B (of Lakes Trollsvattnen) and all the other sampling localities had two major branches. Cluster B is found together with two of the electrofished localities on one of the branches, and cluster A with all neighbouring lakes and the remaining spawning localities on the other (Fig. 3). Cluster B showed the greatest genetic uniqueness in relation to the neighbouring lakes, while cluster A groups with Lakes Alfred Larsa Tjärnen, Häbbersflyn, Häbbersvattnet and Trollsflyn. Lake Hästskotjärnen, located east of ÖT (Fig. 1), comes out as intermediate between clusters A and B (Fig. 3). Cluster A had the strongest genetic resemblance to creek localities A, D, and E, C, while cluster B was most similar to creeks G and F.

Phylogenetic tree illustrating genetic relationships among clusters A and B in Lakes Östra and Västra Trollsvattnet (Lakes Trollsvattnen), the seven neighbouring lakes, and the potential spawning creeks (A–G; cf. Fig. 1, note that creek sites B, H, I, and K are excluded due to small sample size). The tree is neighbour-joining and based on sample size corrected F ST. Numbers along the branches indicate bootstrap values in percentages
Heterozygote deficiency and assignment probabilities among parr versus adults in Lakes Trollsvattnen
To examine whether the heterozygote deficiency among adults in Lakes Trollsvattnen is greater than that observed in the parr (which could indicate selection during development) we performed randomisation tests on observed F IS values. We found no significant difference when comparing the heterozygote deficiency (F IS) between young-of-the-year parr (age class 0+) and older age classes (separately and combined) in those tests. However, F IS was consistently higher in each of the older age classes than in the 0+ age class (P < 0.05).
structure suggested K = 2 as the most likely number of populations when the young-of-the-year parr (n = 888) were pooled with adults from Lakes Östra and Västra Trollsvattnet (n = 7047; using both the ΔK and log likelihood approaches; Online Resource 1, Table S6). The mean of assignment probabilities (Q) to the two clusters were relatively high for both adults and parr (Fig. 4). For adults we find mean Q = 0.78 to cluster A, and mean Q = 0.79 to cluster B; 29% of the adults (n = 1778) had a Q of less than 0.7 to the most likely cluster, and 30% (n = 1867) had a Q of 0.9 or higher. For parr, mean Q were 0.78 and 0.74 to clusters A and B, respectively. In total, 35% of the parr (n = 307) had an assignment probability of less than 0.7 to the most likely cluster and 21% (n = 188) had a Q of 0.9 or higher. The 90% credible regions of individual assignment probabilities for both adults and parr are presented in Online Resource 2.

Distribution of assignment probabilities (Q; allozymes) to cluster A for a adults from Lakes Östra and Västra Trollsvattnet and b young-of-the-year parr from nearby creeks. Membership coefficients (Q) were obtained from structure, using the model allowing population admixture and correlated allele frequencies. Among adults (total n = 6159), 3242 and 2917 fish were classified as cluster A and B, respectively, using Q = 0.5 as cut-off. Among the parr (total n = 888), 561 individuals were classified as population A and 327 as population B
The creek localities A, B, C, D, E and K were dominated by parr with higher assignment probabilities to cluster A, whereas parr with higher membership coefficients to cluster B dominated the localities F, G, H and I (Fig. 5). Within each of the separate creeks, however, the assignment probability distributions suggested some degree of admixture between clusters A and B at all sampling sites (Fig. 5).

Assignment probability distributions to cluster A for young-of-the-year parr from the potential spawning localities (creeks; cf. Fig 1). Assignment probabilities were obtained using the structure software (Pritchard et al. 2000) for K = 2 with parr pooled with adults from Lakes Östra and Västra Trollsvattnet. Note that the scaling on the y-axis at localities B, G, I and K differ from the remaining localities
Potential outlier loci
No allozyme loci were suggested as being under selection (F ST = 0.16–0.17, q = 0.89–0.91, log10(PO) = −1.13–0.93) whereas four SNP loci were suggested to be under diversifying selection, i.e. q-values under 0.05 from the bayescan analysis; Gdist:S177676_2877, F ST = 0.126, q = 0.001, log10(PO) = 2.92; Gdist:S424263_9861, F ST = 0.107, q = 0.014, log10(PO) = 1.557; SalHit:S355030_2257, F ST = 0.134, q = 0.019, log10(PO) = 1.534; and Gdist:S455821_3746_1, F ST = 0.115, q = 0.043, log10(PO) = 0.889. To assess whether there could be a physical relationship between the SNP outliers and allozyme loci we compared their location in the genome (chromosome and position). In the absence of an annotated brown trout genome this was done using the closely related Atlantic salmon (Salmo salar) genome. Allozyme gene ID matches were found distributed across 15 S. salar chromosomes for all but 2 of the 14 analysed allozymes (Online Resource 3). The four outlier SNPs were most likely located on four different chromosomes, and three separate allozymes appeared to be located on three of these chromosomes. However, the positions did not overlap and the smallest physical distance between a SNP and an allozyme on same chromosome was 30 Mb; thus, we do not anticipate that the outlier SNPs were associated with allozyme genes.
The pcadapt analysis of SNP data suggested other outliers than bayescan. Depending on the K value, the number of supported outliers varied from eight (with K = 3) to fourteen (with K = 2 or 4). Five loci—cDNA:S242162_9689, cDNA:S36850_3607, Gdist:S153878_9142, SalHit:S440995_2172, and SalHit:S453385_2176 were consistently identified as outliers. These loci were mapped to five salmon chromosomes, three of which coincide with likely chromosomes for allozymes. However, in no case do the positions overlap and the smallest physical distance for SNP and allozyme estimated to be located on the same chromosome is c. 6 Mb.
Discussion
We explored genetic patterns of cryptic, sympatric brown trout genetic clusters identified with allozyme data in tiny mountain lakes in a nature reserve in central Sweden. We found that (a) these clusters have remained in relatively stable sympatry over at least 28 sampling years (c. 4 generations), (b) the structures can also be detected with the 3093 SNPs applied here, but in line with previous results from microsatellites (Palmé et al. 2013), the amount of divergence is much smaller, (c) the spatial distribution of the clusters is very different—one of them has a very restricted distribution when looking at adults across a wide geographic range, (4) the structuring is also observed in juveniles, and (5) parr from particular creeks tend to group preferentially with either of the two clusters, suggesting a possible role of different spawning sites in maintaining this structuring.
We did not find support for more than a single population occurring in any of the surrounding lakes indicating that the cryptic, sympatric clusters, A and B, only occur in sympatry in Lakes Trollsvattnen. However, for two of the seven lakes sample sizes were relatively small and the statistical power for detecting heterozygote deficiencies was only around 0.5–0.6 (cf. Table 1 vs. Fig S4). Cluster B appears relatively isolated; it is the most genetically unique group and only shows relatively close genetic relationship (F ST ≈ 0.027) with Lake Hästskotjärnen—a tiny lake south-east of Lake Östra Trollsvattnet (Fig. 1); with all other sampled populations, including population A, pairwise F ST range from 0.043 to 0.092 (Table 2). In contrast, cluster A shows high genetic similarity with the downstream lakes Alfred Larsa Tjärnen, Häbbersvattnet, Trollsflyn and Häbbersflyn; F ST between these lakes and population A is very low (0.001–0.005) suggesting that there is a high degree of connectivity among those lakes and this population. The magnitude of this differentiation among population A and the neighbouring lakes is similar to or smaller than what is typically observed between brown trout populations within the same water system when using allozyme markers (Ryman 1983).
Cryptic, sympatric salmonid populations
To our knowledge, this is the first time that brown trout cryptic genetic structures are mapped over several interconnected lakes permitting detection of disparate connectivity patterns of cryptic populations. There are many reports of non-cryptic sympatric populations of brown trout and other salmonids coexisting over relatively short distances, such as within the same lake (e.g. Ferguson and Taggart 1991; Østbye et al. 2006; Power et al. 2009; May-McNally et al. 2015). However, such lakes are often substantially larger and deeper than those in the present study, potentially permitting ecological divergence between the populations. In our case, we find no indications of ecological niche separation between populations A and B (Andersson et al. 2016). To our knowledge, there is only one additional case where cryptic, sympatric populations of brown trout have been detected within a very restricted geographical area, i.e. that of Lakes Bunnersjöarna in central Sweden (Allendorf et al. 1976; Ryman et al. 1979). Neither of the populations within Lakes Trollsvattnen and within Lakes Bunnersjöarna can be discriminated by the naked eye (Ryman et al. 1979; Andersson et al. 2016), a feature that separates these two cases from other reports on sympatric brown trout populations.
Cryptic population structure is difficult to uncover because sample sizes must be of a magnitude that allow statistical detection of heterozygote deficiencies and/or detection of more than one genetic group when using Bayesian clustering techniques such as the one applied by the software structure (Pritchard et al. 2000). In a large genetic structure study of another salmonid species—the Arctic char (Salvelinus alpinus)—involving 43 lakes in Iceland, Scandinavia and Great Britain, significant heterozygote deficiencies suggesting multiple populations were detected in 10 of the lakes (Wilson et al. 2004). These findings, together with the present example and that of Lakes Bunnesjöarna might indicate that cryptic structuring is more common in salmonids than previously anticipated.
Why different degrees of genetic divergence between the clusters using allozymes versus SNPs?
Both allozymes and SNPs detect largely the same genetic clusters, but the degree of genetic divergence between the structures (F ST) differs by at least one order of magnitude between allozymes and SNPs. These results are compatible with a model of two partly reproductively isolated populations where diversifying selection has acted on a restricted part of the genome (including allozyme loci in this case) whereas the dynamics of the remaining part is primarily determined by genetic drift and migration (reflected for most of the SNPs as well as at eight microsatellites previously reported by Palmé et al. 2013). Palmé et al. (2013) estimated effective population size (N e) for each of the two clusters to be in the range 100–200. Assuming a finite island model with two subpopulations of N e = 150 exchanging two migrants per generation we would expect, using equation A15 of Ryman and Leimar (2008), an F ST ≈ 0.03 at migration-drift equilibrium. This value corresponds to the results obtained for both SNPs and microsatellites, whereas the allozymes, which may reflect selection, show a markedly higher F ST. A similar type of observation has been reported for herring (Clupea harengus) in the Baltic Sea (Lamichhaney et al. 2012).
Many of the allozymes used in the present study are involved in cell respiration, and these metabolic processes incorporate both nuclear-coded proteins and proteins encoded by the mitochondrial DNA which must be compatible to work efficiently (Wolff et al. 2014). Compatibility between the nuclear and mitochondrial genes involved is imperative, and individuals with mitonuclear mismatches would have a non-optimal metabolism and experience a selective disadvantage, which, in turn would lead to heterozygote deficiency and genetic differentiation (Wolff et al. 2014). We currently lack mitochondrial DNA data, and thus cannot resolve whether this type of mechanism is operating in our system. Observations in line with such a mechanism of ontogenetic selection include that F IS is consistently lower in parr than in all adult age classes (although randomisation tests do not yield statistical significance when comparing F IS among 0+ to each older age class), and that structure suggests a single population when analysing parr only.
Could the allozymes be erroneously scored and heterozygote deficiencies and structuring only reflect artifacts and false genotyping? We do not consider this a likely explanation because the allozymes in this study have been evaluated thoroughly and used for decades within the present and other projects since the 1970s in our lab (e.g. Allendorf et al. 1977; Ryman et al. 1979; Ryman 1983; Jorde and Ryman 1996; Palm et al. 2003), as well as in many other studies of brown trout (e.g. Ferguson and Mason 1981; Ferguson and Taggart 1991; Duguid et al. 2006). We do not find heterozygote deficiencies in other lakes in the area, neither the ones included in the present study nor in other lakes that have been monitored as long as Lakes Trollsvattnen with the same markers and for which the same huge amount of data is available (e.g., Jorde and Ryman 1996; Charlier et al. 2012). Thus, we are confident that the patterns reported here for allozymes reflect true genetic patterns of these markers in these waters.
Evolutionary origin of the sympatric populations
One possible scenario for two sympatric populations in Lakes Trollsvattnen is that they represent two independent colonisation events. It is presently unclear if brown trout in the Fennoscandic mountain regions have a common, recent evolutionary origin, or if present populations represent biodiversity shaped over longer evolutionary time scales than since the last glaciation c. 7000 years ago. If such lineages are coupled to mitonuclear mismatches the structures might remain even with extensive gene flow between populations.
Parapatric origin with multiple colonisation events is suggested as a likely explanation for co-existence in several studies of sympatric populations with genetic differentiation of the same magnitude as in ours, (e.g. Fraser and Bernatchez 2005a; May-McNally et al. 2015). It is possible that such an explanation applies to the two clusters in Lakes Trollsvattnen as well, and the differentiation pattern observed appears most consistent with such a model. We find that clusters A and B are more diverged from each other (largest pairwise F ST, Table 2) than from neighbouring lakes, and this might suggest that they represent two distinct lineages that colonised this area when ice of the last glaciation disappeared around 7000 years ago. If they had arisen in sympatry we would have expected them to be more similar to each other than to the neighbouring lakes, unless selection of some sort is involved. Further, we have observed a minor decline in F ST between the clusters over time (Online Resource 1, Fig. S6), and this may suggest that the populations are experiencing increasing interbreeding upon a recent secondary contact, supporting a parapatric origin hypothesis. Alternatively, the decrease of F ST might reflect changes in divergent selection pressures at allozyme loci over time. We note, however, that the absolute change of F ST is minor, and in the last year of sampling (2014) there is an increase that makes F ST approach its starting value in 1987.
Spawning sites
Different spawning sites for the two clusters A and B are suggested by the phylogenetic tree (Fig. 3); young-of-the-year collected at creek localities A, D, E and C are most genetically similar to cluster A, whereas parr at G and F cluster more closely to B. One possible explanation for this pattern is that populations A and B differ with respect to outflow versus inflow spawning. The creeks dominated by population A are downstream (with the exception of creeks D and E), while predominant B creeks are upstream from the main lakes. Such upstream–downstream spawning segregation has been documented in brook char (Fraser and Bernatchez 2005b), as well as in brown trout (Ferguson and Taggart 1991).
However, when examining the assignment probability distributions in the young-of-the-year samples, we find parr with relatively high assignment to both clusters at all sites, possibly suggesting that spawners from both populations use all the creeks to some extent. Typically, young brown trout stay in their natal stream during the first year of their life, starting to disperse around the age of 1–3 years (Elliott 1994; Elfman et al. 2000). Thus, it does not appear likely that occurrence of parr from both clusters in the creeks is caused by movement of young fish from their natal site to the place of collection. Records of migrating parr under one year of age do exist (Thorpe 1974; Bagliniére et al. 1994; Limburg et al. 2001), but such parr are reported to migrate in autumn–winter, after at least one growing season (Trophe 1974; Bagliniére et al. 1994). We collected the young fish in late summer-early autumn; thus, the bulk of our samples should have been collected before such dispersal started. We also acknowledge that sampling of full sibs in the parr material may pose potential problems in the deduction of the correct number of genetic clusters, which could be a limitation of the present study.
If both populations are spawning in all creeks, one potential explanation of the observed genetic structure could be that they differ with respect to some microgeographical habitat factors such as water velocity and/or gravel size. For example, Quinn et al. (1995) found a positive association between egg size and the preferred size of incubation gravels in sockeye salmon, and suggest that while larger eggs result in better survival of the young, they also require more oxygen, which is disadvantageous at sites with a denser substrate resulting from smaller gravel size.
In all, our current data suggest two sympatric genetic clusters occurring only in Lakes Trollsvattnen. Separate spawning locations of these clusters are indicated, but the results are not fully conclusive. Further, divergence patterns differ markedly between allozymes and SNPs; this issue also needs further exploration, and such work is currently underway.
Conservation implications
Our results add information on the complexity of brown trout population genetic structuring over restricted geographic areas in Scandinavian mountain lakes. Clearly, the biodiversity of this species can be strongly and quite counterintuitively structured. In the present system the largest genetic differences occur within lakes, whereas relatively minor heterogeneities appear to exist over large parts of the study area. This contrasts to what would be expected from the strong homing behaviour of the species. Our data suggest that the largest loss of diversity in this system would occur if the cryptic population B were reduced or removed. Thus, loss of separate brown trout populations, even within a single small lake, can apparently be associated with considerable reduction of genetic variation. At the same time, genetic exchange can occur relatively extensively in a brown trout metapopulation system, as suggested by the connectivity pattern of the cryptic population A in the present study.
Recognising the complexity that can occur over even microgeographical scales is important in conservation management of brown trout. Without genetic data it is difficult to predict the effects on biodiversity that will occur from removing or reducing parts of a brown trout metapopulation. A general recommendation in such situations is to apply the precautionary principle, and make sure that the full water system is recognised and taken into account in the management planning.
References
Allendorf FW, Ryman N, Stennek A, Ståhl G (1976) Genetic variation in Scandinavian brown trout (Salmo trutta L.): evidence of distinct sympatric populations. Hereditas 83:73–82
Allendorf FW, Mitchell N, Ryman N, Ståhl G (1977) Isozyme loci in brown trout (Salmo trutta L.): detection and interpretation from population data. Hereditas 86:179–190
Allendorf FW, Luikart GH, Aitken SN (2013) Conservation and the genetics of populations, 2nd edn. Wiley-Blackwell, Hoboken
Andersson A, Johansson F, Sundbom M, Ryman N, Laikre L (2016) Lack of trophic polymorphism despite substantial genetic differentiation in sympatric brown trout (Salmo trutta) populations. Ecol Freshw Fish. doi:10.1111/eff.12308
Baglinière J-L, Prévost E, Maisse G (1994) Comparison of population dynamics of Atlantic salmon (Salmo salar) and brown trout (Salmo trutta) in a small tributary of the River Scorff (Brittany, France). Ecol Freshw Fish 3:25–34
Barshis DJ, Ladner JT, Oliver TA, Seneca FO, Taylor-Knowles N, Palumbi SR (2013) Genomic basis for coral resilience to climate change. Proc Natl Acad Sci Biol 110:1387–1392
Bergek S, Björklund M (2007) Cryptic barriers to dispersal within a lake allow genetic differentiation of Eurasian perch. Evol Int J Org Evol 61:2035–2041
Charlier J, Palmé A, Laikre L, Andersson J, Ryman N (2011) Census (Nc) and genetically effective (Ne) population size in a lake resident population of brown trout Salmo trutta. J Fish Biol 79:2074–2082
Charlier J, Laikre L, Ryman N (2012) Genetic monitoring reveals temporal stability over 30 years in a small, lake resident brown trout population. Heredity 109:246–253
Cook-Patton SC, McArt SH, Parachnowitsch AL, Thaler JS, Agrawal AA (2011) A direct comparison of the consequences of plant genotypic and species diversity on communities and ecosystem function. Ecology 92:915–923
Duguid RA, Ferguson A, Prodöhl P (2006) Reproductive isolation and genetic differentiation of ferox trout from sympatric brown trout in Loch Awe and Loch Laggen, Scotland. J Fish Biol 69:89–114
Dupont P-P, Bourret V, Bernatchez L (2007) Interplay between ecological, behavioural and historical factors in shaping the genetic structure of sympatric walleye populations (Sander vitreus). Mol Ecol 16:937–951
Earl DA, von Holdt BM (2012) Structure harvester: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conservation Genet Resour 4:359–361
Elfman M, Limburg KE, Kristiansson P, Svedäng H, Westin L, Wickström H, Malmquist K, Pallon J (2000) Complex life histories of fishes revealed through natural information storage devices: case studies of diadromous events recorded by otoliths. Nucl Instrum Meth B 161–163:877–881
Elliott JM (1994) Quantitative ecology of the brown trout. Oxford University Press, Oxford
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587
Ferguson A, Mason FM (1981) Allozyme evidence for reproductively isolated sympatric populations of brown trout Salmo trutta L. in Lough Melvin, Ireland. J Fish Biol 18:629–642
Ferguson A, Taggart JB (1991) Genetic differentiation among the sympatric brown trout (Salmo trutta) populations of Lough Melvin, Ireland. Biol J Linn Soc 43:221–237
Ficke AD, Myrick CA, Hansen LJ (2007) Potential impacts of global climate change on freshwater fisheries. Rev Fish Biol Fisher 17:581–613
Fischer MC, Foll M, Excoffier L, Heckel G (2011) Enhanced AFLP genome scans detect local adaptation in high-altitude populations of a small rodent (Microtus arvalis). Mol Ecol 20:1450–1462
Foll M (2012) BayeScan v2.1 User Manual. University of Bern. http://cmpg.unibe.ch/software/BayeScan/files/BayeScan2.1_manual.pdf. Accessed 14 March 2016
Foll M, Gaggiotti OE (2008) A genome scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180:977–993
Frankham R (2005) Ecosystem recovery enhanced by genotypic diversity. Heredity 95:183
Fraser DJ, Bernatchez L (2005a) Allopatric origins of sympatric brook charr populations: colonization history and admixture. Mol Ecol 14:1497–1509
Fraser DJ, Bernatchez L (2005b) Adaptive migratory divergence among sympatric brook charr populations. Evol 59:611–624
Gowell CP, Quinn TP, Taylor EB (2012) Coexistence and origin of trophic ecotypes of pygmy whitefish, Prosopium coulterii, in a south-western Alaskan lake. J Evol Biol 25:2432–2448
Hajjar R, Jarvis DI, Gemmill-Herren B (2008) The utility of crop genetic diversity in maintaining ecosystem services. Agric Ecosyst Environ 123:261–270
Hellmair M, Kinziger AP (2014) Increased extinction potential of insular fish populations with reduced life history variation and low genetic diversity. PLoS ONE 9:1–10
Holm S (1979) A simple sequentially rejective multiple test procedure. Scand J Stat 6:65–70
Hood GM (2010) PopTools version 3.2.5. Available from http://www.poptools.org
Hughes JB, Daily GC, Ehrlich PR (1997) Population diversity; its extent and extinction. Science 278:689–692
Jakobsson M, Rosenberg NA (2007) Clumpp: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806
Jetz W, Fine PVA (2012) Global gradients in vertebrate diversity predicted by historical area-productivity dynamics and contemporary environment. PLoS Biol 10:e1001292
Johannesson K, Smolarez K, Grahn M, André C (2011) The future of Baltic Sea populations: local extinction or evolutionary rescue? AMBIO 40:179–190
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405
Jombart T, Ahmed I (2011) Adegenet 1.3–1: new tools for the analysis of genome-wide SNP data. Bioinform 27:3070–3071
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet 11:94
Jorde PE, Ryman N (1996) Demographic genetics of brown trout (Salmo trutta) and estimation of effective population size from temporal change of allele frequencies. Genet 143:1369–1381
Jorde PE, Gitt A, Ryman N (1991) New biochemical genetic markers in the brown trout (Salmo trutta L.). J Fish Biol 39:451–454
Karlsson J, Byström P, Ask J, Ask P, Persson L, Jansson M (2009) Light limitation of nutrient-poor lake ecosystems. Nature 460:506–509
Laikre L (ed) (1999) Conservation genetic management of brown trout (Salmo trutta) in Europe. Report by the concerted action on identification, management and exploitation of genetic resources in the brown trout (Salmo trutta), TROUTCONCERT; EU FAIR CT97-3882
Laikre L, Jorde PE, Ryman N (1998) Temporal change of mitochondrial DNA haplotype frequencies and female effective size in a brown trout (Salmo trutta) population. Evol Int J Org Evol 52:910–915
Laikre L, Larsson LC, Palmé A, Charlier J, Josefsson M, Ryman N (2008) Potentials for monitoring gene level biodiversity: using Sweden as an example. Biodivers Conserv 17:893–910
Lamichhaney S, Barrio AM, Rafati N, Sundström G, Rubin C-J, Gilbert ER, Berglund J, Wetterbom A, Laikre L, Webster MT, Grabherr M, Ryman N, Andersson L (2012) Population-scale sequencing reveals genetic differentiation due to local adaptation in Atlantic herring. Proc Natl Acad Sci 109:19345–19350
Limburg KE, Landergren P, Westin L, Elfman M, Kristiansson P (2001) Flexible models of anadromy in Baltic sea trout: making the most of marginal spawning streams. J Fish Biol 59:682–695
Lindley ST, Grimes CB, Mohr MS, Peterson W, Stein J, Anderson JT, Botsford LW, Bottom DL, Busack CA, Collier TK, Ferguson J, Garza JC, Grover AM, Hankin DG, Kope RG, Lawson PW, Low A, MacFarlane RB, Moore K, Palmer-Zwahlen M, Schwing FB, Smith J, Tracy C, Webb R, Wells BK, Williams TH (2009) What caused the Sacramento River fall chinook stock collapse? Pre-publication report, Pacific Fishery Management Council. http://swfsc.noaa.gov/publications/TM/SWFSC/NOAA-TM-NMFS-SWFSC-447.PDF Accessed 16 March 2016
Luu K, Bazin E, Blum MGB (2016) pcadapt: an R package to perform genome scans for selection based on principal component analysis. Mol Ecol Resour. doi:10.1111/1755-0998.12592
Marco-Rius F, Sotelo G, Caballero P, Moran P (2013) Insights for planning an effective stocking program in anadromous brown trout (Salmo trutta). Can J Fish Aquat Sci 70:1092–1100
May-McNally SL, Quinn TP, Woods PJ, Taylor EB (2015) Evidence of genetic distinction among sympatric ecotypes of Arctic char (Salvelinus alpinus) in south-western Alaskan lakes. Ecol Freshw Fish 24:562–574
McGinnity P, Jennings E, deEyto E, Allott N, Samuelsson P, Rogan G, Whelan K, Cross T (2009) Impact of naturally spawning captive-bred Atlantic salmon on wild populations: depressed recruitment and increased risk of climate-mediated extinction. Proc R Soc B 276:3601–3610
Østbye K, Amundsen P-A, Bernatchez L, Klemetsen A, Knudsen R, Kristoffersen R, Næsje TF, Hindar K (2006) Parallel evolution of ecomorphological traits in the European whitefish Coregonus lavaretus (L.) species complex during postglacial times. Mol Ecol 15:3983–4001
Palm S, Ryman N (1999) Genetic basis of phenotypic differences between transplanted stocks of brown trout. Ecol Freshw Fish 8:169–180
Palm S, Laikre L, Jorde P-E, Ryman N (2003) Effective population size and temporal genetic change in stream resident brown trout (Salmo trutta, L.). Conserv Genet 4:249–264
Palmé A, Laikre L, Ryman N (2013) Monitoring reveals two genetically distinct brown trout populations remaining in stable sympatry over 20 years in tiny mountain lakes. Conserv Genet 14:795–808
Pastor J (1996) Unsolved problems of boreal regions. Clim Change 33:343–350
Pigeon D, Dodson JJ, Bernatchez L (1998) A mtDNA analysis of spatiotemporal distribution of two sympatric larval populations of rainbow smelt (Osmerus mordax) in the St. Lawrence River estuary, Quebec, Canada. Can J Fish Aquat Sci 55:1739–1747
Power M, Power G, Reist JD, Banjo R (2009) Ecological and genetic differentiation among the Arctic charr of Lake Aigneau, Northern Québec. Ecol Freshw Fish 18:445–460
Præbel K, Knudsen R, Siwertsson A, Karhunen M, Kahilainen KK, Ovaskainen O, Østbye K, Peruzzi S, Fevolden S-E, Amundsen P-A (2013) Ecological speciation in postglacial European whitefish: rapid adaptive radiations into the littoral, pelagic, and profoundal lake habitats. Ecol Evol 3:4970–4986
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Prodöhl PA, Taggart JB, Ferguson A (1992) Genetic variability within and among brown trout (Salmo trutta) populations: multi-locus DNA fingerprint analysis. Hereditas 117:45–50
Prowse TD, Wrona FJ, Reist JD, Gibson JJ, Hobbie JE, Lévesque LMJ, Vincent WF (2006) Climate change effects on hydroecology of arctic freshwater ecosystems. AMBIO 35:347–358
Quinn TP, Hendry AP, Wetzel LA (1995) The influence of life history trade-offs and the size of incubation gravels on egg size variation in sockeye salmon (Oncorhynchus nerka). OIKOS 74:425–438
R Core Team (2014) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. http://www.R-project.org/
Raymond M, Rousset F (1995) GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Reusch THB, Ehlers A, Hämmerli A, Worm B (2005) Ecosystem recovery after climatic extremes enhanced by genotypic diversity. Proc Natl Acad Sci 102:2826–2831
Rogers SM, Bernatchez L (2007) The genetic architecture of ecological speciation and the association with signatures of selection in natural lake whitefish (Coregonus sp. Salmonidae) species pairs. Mol Biol Evol 24:1423–1438
Rousset F (2008) GENEPOP’007: a complete reimplementation of the genepop software for Windows and Linux. Mol Ecol Resour 8:103–106
Ryman N (1983) Patterns of distribution of biochemical genetic variation in salmonids: differences between species. Aquaculture 33:1–21
Ryman N (2006) CHIFISH: a computer program testing for genetic heterogeneity at multiple loci using chi-square and Fisher’s exact test. Mol Ecol Notes 6:285–287
Ryman N, Jorde PE (2001) Statistical power when testing for genetic differentiation. Mol Ecol 10:2361–2373
Ryman N, Leimar O (2008) Effect of mutation on genetic differentiation among nonequilibrium populations. Evol 62:2250–2259
Ryman N, Palm S (2006) POWSIM: a computer program for assessing statistical power when testing for genetic differentiation. Mol Ecol Notes 6:600–602
Ryman N, Utter F (eds) (1987) Population genetics and fishery management. Washington Sea Grant Program, Seattle (reprinted 2009 by The Blackburn Press, Caldwell)
Ryman N, Allendorf FW, Ståhl G (1979) Reproductive isolation with little genetic divergence in sympatric populations of brown trout (Salmo trutta). Genetics 92:247–262
Ryman N, Palm S, André C, Carvalho GR, Dahlgren TG, Jorde PE, Laikre L, Larsson LC, Palmé A, Ruzzante DE (2006) Power for detecting genetic divergence: differences between statistical methods and marker loci. Mol Ecol 15:231–245
Schindler DE, Hilborn R, Chasco B, Boatright CP, Quinn TP, Rogers LA, Webster MS (2010) Population diversity and the portfolio effect in an exploited species. Nature 465:609–612
Ståhl G, Ryman N (1982) Simple Mendelian inheritance at a locus coding for alpha-glycerophosphate dehydrogenase in brown trout (Salmo trutta). Hereditas 96:313–315
Takezaki N, Nei M, Tamura K (2010) POPTREE2: Software for constructing population trees from allele frequency data and computing other population statistics with Windows interface. Mol Biol Evol 27:747–752
Thorpe JE (1974) The movement of brown trout, Salmo trutta (L.) in Loch Leven, Kinross, Scotland. J Fish Biol 6:153–180
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evol 38:1358–1370
Wilson AJ, Gíslason D, Skúlason S, Snorrason S, Adams CE, Alexander G, Danzmann RG, Ferguson MM (2004) Population genetic structure of Arctic charr, Salvelinus alpinus from northwest Europe on large and small spatial scales. Mol Ecol 13:1129–1142
Wolff JN, Ladoukakis ED, Enríquez JA, Dowling DK (2014) Mitonuclear interactions: evolutionary consequences over multiple biological scales. Phil Trans R Soc B 369:20130443
Yang L, Callaway RM, Atwater DZ (2015) Root contact responses and the positive relationship between intraspecific diversity and ecosystem productivity. AoB Plants 7:plv053. doi:10.1093/aobpla/plv053
Acknowledgements
This research was funded by grants from the Swedish Research Council Formas (LL) and the Swedish Research Council (NR). Financial support from the Swedish Agency for Marine and Water Management is also acknowledged. We are grateful to Sten Karlsson, Chris Wheat, and Sven Jakobsson for valuable comments and suggestions. We also thank Kurt Morin, Patrik Näs, Tobias Sundelin, Karin Tahvanainen, Kristoffer Andersson, Rolf Gustavsson, Thomas Giegold and Peter Guban for assistance in the field and Francois Besnier for help with data analysis and the Associate Editor and two anonymous reviewers for very valuable comments. The study area is located in the Hotagen Nature Reserve, County of Jämtland, Sweden, and we acknowledge the cooperation from the Jämtland County Administrative Board and the Jiingevaerie Sami Community.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Andersson, A., Jansson, E., Wennerström, L. et al. Complex genetic diversity patterns of cryptic, sympatric brown trout (Salmo trutta) populations in tiny mountain lakes. Conserv Genet 18, 1213–1227 (2017). https://doi.org/10.1007/s10592-017-0972-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-017-0972-4