
Abstract
Detecting population subdivision when apparent barriers to gene flow are lacking is important in evolutionary and conservation biology. Recent research indicates that intraspecific population complexity can be crucial for maintaining a species′ evolutionary potential, productivity, and ecological role. We monitored the genetic relationships at 14 allozyme loci among ~4,000 brown trout (Salmo trutta) collected during 19 years from two small interconnected mountain lakes (0.10 and 0.17 km2, respectively) in central Sweden. There were no allele frequency differences between the lakes. However, heterozygote deficiencies within lakes became obvious after a few years of monitoring. Detailed analyses were then carried out without a priori grouping of samples, revealing unexpected differentiation patterns: (i) the same two genetically distinct (F ST ≥ 0.10) populations occur sympatrically at about equal frequencies within both lakes, (ii) the genetic subdivision is not coupled with apparent phenotypical dichotomies, (iii) this cryptic structure remains stable over the two decades monitored, and (iv) the point estimates of effective population size are c. 120 and 190, respectively, indicating that genetic drift is important in this system. A subsample of 382 fish was also analyzed for seven microsatellites. The genetic pattern does not follow that of the allozymes, and in this subsample the presence of multiple populations would have gone undetected if only scoring microsatellites. Sympatric populations may be more common than anticipated, but difficult to detect when individuals cannot be grouped appropriately, or when markers or sample sizes are insufficient to provide adequate statistical power with approaches not requiring prior grouping.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Accurate identification of intra-specific population subdivision is of central importance in evolutionary and conservation biology and constitutes a prerequisite for sound management of species. Growing evidence indicate that a diverse “portfolio” of genetically separate populations is needed for maintaining viability of species (Hilborn et al. 2003; Lindley et al. 2009; Schindler et al. 2010) and to ensure ecosystem resilience (e.g. Luck et al. 2003; Frankham 2005; Reusch et al. 2005; Hajjar et al. 2008).
Assessment of intra-specific population structuring typically involves comparison of allele frequencies among individuals that have been grouped based on a priori information such as sampling site or morphotype. Alternatively, a model based a posteriori approach is used with no such prior grouping.
So far, the a priori approach has dominated empirical population genetics research and has provided information on substructuring in a wide range of organisms from insects and plants to fishes and large vertebrates (e.g. Clarke and O’Dwyer 2000; Huang and Zhang 2000; Wilson et al. 2003; Van Rossum and Prentice 2004; Knutsen et al. 2010, 2012). During recent years, however, model based approaches for assessing the number of subpopulations and for assignment of individuals to those populations have become available (e.g. Pritchard et al. 2000) and increasingly popular (e.g. Lee et al. 2010; Martien et al. 2012; Phinchongsakuldit et al. 2013).
With respect to the statistical properties of the two approaches for detecting genetic heterogeneity the a priori approach is often powerful when based on a reasonable number of genetic markers, also when examining a rather restricted number of individuals (Ryman et al. 2006; Waples and Gaggiotti 2006). There are several reasons for the generally high power of this approach. (1) The statistical null hypothesis (H0) can be specified explicitly with respect to the allele frequency differences that might exist among predefined groups (i.e. H0 specifies identical allele frequencies among groups). (2) The test is two-sided, the null hypothesis can thus be rejected regardless of the direction of the differences observed, an aspect particularly important when combining information from multiple loci. (3) Statistical tests for allele frequency heterogeneity are generally based on the number of sampled genes (2n) rather than the number of individuals (n), and the “statistical sample size” can therefore be quite large (Ryman et al. 2006).
In contrast, when testing for structuring in an a posteriori situation the null hypothesis must be less specific, for example with respect to the possible number of populations and their relative contribution to the sample, and therefore less powerful. Further, only some deviations from random association of alleles or genotypes are indicative of subdivision (e.g. deficiency of heterozygotes, but not excess) which may complicate the joint evaluation of information from multiple loci. Finally, the statistical sample size equals the number of individuals sampled, a number that may be insufficient for detecting anything but quite substantial heterogeneity when examining, say, 50–100 individuals at a particular location, which is generally adequate for allele frequency comparisons using the a priori approach (cf. Waples and Gaggiotti 2006; Waples and Do 2010).
Thus, the statistical power is typically lower when applying an a posteriori approach, but erroneous assumptions underlying the grouping of individuals for an a priori approach might prevent detection of true genetic structuring. Because of the lower statistical power, large differences or extensive sampling might be required to detect genetic structuring in a posteriori situations, and it is possible that “hidden” or “cryptic” population structuring over short geographical distances is more common than generally acknowledged.
In this paper, we delineate a situation of intra-specific diversity in brown trout (Salmo trutta) in two small inter-connected mountain lakes that exemplifies how a prior grouping can prevent detection of true genetic structuring. We identify sympatric populations in these lakes. This is not uncommon in brown trout and other salmonids, but empirical identification of genetically distinct sympatric populations has typically been associated with a priori grouping of individuals due to (1) morphological differences (e.g. Hindar et al. 1986; Taylor and Bentzen 1993a,b; Power et al. 2009), (2) ecological separation including different times or locations for spawning (e.g. Child 1984; Spruell et al. 1999; Gerlach et al. 2001) and different life history strategies such as resident versus anadromous forms of salmonid fishes (e.g. Verspoor and Cole 1989; Taylor et al. 1997), or (3) geographical distance (e.g. Bernatchez and Martin 1996; Fraser and Bernatchez 2005; Dupont et al. 2007; see Online Resource 1 for a review of empirical evidence of sympatry in salmonids and other fishes detected using a priori versus a posteriori approaches).
The only empirical examples of a posteriori detection of distinct sympatrically occurring populations that we have found refer to the documentation of multiple populations of brown trout in Lakes Bunnersjöarna in Sweden (Allendorf et al. 1976; Ryman et al. 1979) and of Arctic char (Salvelinus alpinus) in the Scottish Lochs Maree and Stack (Wilson et al. 2004; Adams et al. 2008; Online Resource 1). In both these cases large genetic differences were found among previously unrecognized co-existing populations.
We monitored our system of brown trout through annual sampling over 19 consecutive years. We first applied the a priori approach but found no differentiation between the two tiny mountain lakes. However, heterozygote deficiencies within lakes became obvious after a few years of monitoring, and using the a posteriori approach we report (1) the existence of the same two sympatrically occurring populations within each lake, (2) the amount of divergence between populations, (3) their effective size, and (4) the temporal stability of the detected genetic patterns.
Materials and methods
Material
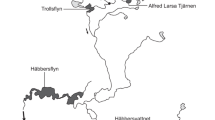
The trout examined were collected within an ongoing genetic monitoring project of brown trout populations in the County of Jämtland, central Sweden (Jorde and Ryman 1996; Laikre et al. 1998; Palm and Ryman 1999; Palm et al. 2003, Charlier et al. 2011, 2012). The major part of the material is from two closely connected lakes, Lake Östra Trollsvattnet (ÖT) and Lake Västra Trollsvattnet (VT; Fig. 1, Table 1), collectively referred to as Lakes Trollsvattnen. A total of n = 4,140 fish from these two lakes, representing 19 sampling years (1987–2005) and 26 cohorts (1977–2002; Table 1) are included in the study.

Lakes Östra (ÖT) and Västra (VT) Trollsvattnet that have been monitored over 19 years, and neighboring lakes including N1-N3 which were sampled during one of these years. The site is located in the Hotagen nature reserve, County of Jämtland, central Sweden. Sampled lakes are in black, open white arrows indicate approximate sampling localities within ÖT and VT, and small arrows indicate direction of waterflow
Lakes Trollsvattnet are oligotrophic mountain lakes at an elevation of 698 m, and have an area of about 0.10 (ÖT) and 0.17 (VT) km2, respectively. The two lakes belong to the uppermost part of the River Indalsälven drainage system flowing into the Baltic Sea. Brown trout and Arctic char are the only fish species occurring in the lakes. The last period of glaciation sets the upper limit for the age of Lakes Trollsvattnen at c. 7,000 years.
About 100 individuals have been collected annually in August from each of the two lakes (sampling locations are marked in Fig. 1), and fish were caught using gillnets of various mesh sizes. For each fish weight, total length, sex, and stage of maturation (breeding the year of collection or not) was recorded. Otoliths for age determination were collected (Jorde and Ryman 1996), as well as tissue samples (muscle, eye, liver; i.e. destructive sampling) for genotype analysis.
During one sampling year (2004) we also collected brown trout from three small neighboring lakes (Table 1) draining into Lakes Trollsvattnen (n = 448). These neighboring lakes are nameless and are here referred to as N1, N2, and N3 (Fig. 1). Spawning typically peaks in late September, and the outlet from Lake Västra Trollsvattnet and all the creeks connecting the five lakes that have been sampled appear to provide suitable spawning areas. We do not know whether or not spawning occurs in the lakes.
Genetic analyses
The monitoring study of Lakes Trollsvattnen started in the early 1980s when allozymes were the only genetic marker available for large scale screening of natural populations, and we have continued to score the same markers to provide consistency throughout the project. Screening was performed by horizontal starch gel electrophoresis (Allendorf et al. 1977). Using the nomenclature of Shaklee et al. (1990) the following 14 polymorphic loci with co-dominant gene expression were scored (older locus designations used in previous publications from our group are given in brackets): sAAT-4 [AAT-6], CK-A1 [CPK-1], DIA-1 [DIA], bGALA-2, bGLUA [BGA], G3PDH-2 [AGP-2], sIDDH-1 [SDH-1], sIDHP-1 [IDH-2], LDH-C1 [LDH-5], aMAN, sMDH-2 [MDH-2], ME [MEL], MPI-2 [PMI], PEPLT (Table 2).
In addition to the allozymes that were scored in all fish (n = 4,140 + 448 = 4,588) we also generated microsatellite data for a limited number of individuals from ÖT and VT (total n = 382; Table 1), representing cohorts 1984–1986 and 1996–1998, and sampling years 1988–1992 and 2000–2003. Individuals were genotyped for the following seven loci: One9 (Scribner et al. 1996), Ssa85, Ssa197 (O’Reilly et al. 1996), SsoSL417 (Slettan et al. 1995), STR15, STR60, and STR73 (Estoup et al. 1993; Table 2), according to procedures described by Dannewitz et al. (2003). To verify the microsatellite genotyping results, 96 of the 382 individuals were scored independently by a different laboratory. There was a good (98 %) consistency of the genotype results generated from the different labs.
Statistical treatment
Allele frequency differences between groups were tested by Chi square using chifish version 1.3 (Ryman 2006). F-statistics (Weir and Cockerham 1984) quantifying spatial and temporal genetic heterogeneity and deviation from Hardy–Weinberg proportions, with associated levels of significances, were appraised using genepop version 3.4 (Raymond and Rousset 1995), as were tests for gametic phase (linkage) disequilibrium. Hierarchical gene diversity analyses were performed using the program negst (Chakraborty et al. 1982), and tests for selective neutrality were conducted using the lositan software (Beaumont and Nichols 1996; Antao et al. 2008).
We assessed statistical power for detecting differentiation for the present set of marker loci using powsim (Ryman and Palm 2006; Ryman et al. 2006). With sample sizes of those corresponding to Lakes Östra and Västra Trollsvattnet, a true F ST between them of 0.001 would be detected (P < 0.05) with a probability of over 99 % for the present 14 allozyme loci. Similar power was provided by the seven microsatellite loci scored in 382 fish.
The most likely number of populations (clusters; K) compatible with the observed genotypic distribution was assessed by individually based likelihood analyses using structure (Pritchard et al. 2000; Falush et al. 2003). We used the four default models (1–4) referring to the four possible default combinations including/excluding the assumptions “admixture genomes within individuals” and “correlated allele frequencies among populations” (Online Resource 2; see Pritchard et al. 2007 for details). Estimation of the most likely value of K under different models was replicated over five runs (Online Resource 2a–e). To get accurate parameter estimates of P (estimated allele frequencies), Q (estimated membership coefficient for each individual in each cluster, i.e. assignment probability), and likelihood values for different numbers of K, the burn-in length and the number of Markov chains (MCMC) used in the simulations was 500,000 steps and 200,000 replicates, respectively.
We identified two populations, hereafter referred to as A and B. There was no contrasting homozygosity between clusters and each fish was assigned to the cluster to which its assignment probability (Q) was larger than 0.5 (Fig. 2). This is equivalent to applying a “cut-off” probability of 0.5 (resulting in all fish being assigned to a cluster), but we also applied 0.7 and 0.9 as cut-offs in some cases. Using these cut-offs of 0.5, 0.7, and 0.9 a total of 4,140, 3,189, and 1,843 fish, respectively, were assigned to a cluster. When not stated otherwise results presented refer to the 0.5 cut-off level.

Distribution of assignment probabilities (Q) to cluster A for brown trout from Lakes Östra and Västra Trollsvattnet (obtained from structure using allozymes, model 4, cf. Online Resource 2a–c). Fish assigned to cluster A with a probability of, say, 0.30 are assigned to cluster B with the probability 0.70. a Both lakes (4,140 fish), b Östra Trollsvattnet (2,058 fish), c Västra Trollsvattnet (2,082 fish)
The potential existence of multiple populations was also addressed by means of principal component analysis (PCA) based on allozyme data using statistica v.7 (StatSoft, Inc. 2005), coding the genotype of a particular fish and locus as 0, 0.5, or 1. To avoid cluttering when illustrating graphically the relation between the results obtained by structure and PCA we only used the smaller dataset of 382 fish that was scored for both sets of markers.
Estimation of effective size
Variance effective population size (N e) of the clusters A and B was estimated from allozyme data using the ‘temporal method’ as modified for overlapping generations (temporal shifts in allele frequencies between consecutive cohorts; Jorde and Ryman 1995). We used the same approach as Jorde and Ryman (1996), but with the unbiased estimator for genetic drift (F s) of Jorde and Ryman (2007) as applied in their software tempofs.
When estimating N e for organisms with overlapping generations, life table data are required for calculating a correction factor (C) and for assessing the generation interval (G; Jorde and Ryman 1995), and we followed the same procedures as Jorde and Ryman (1996) to generate this information (Online Resource 3).
Results
We focus first on the genetic structuring in the two primary lakes Östra and Västra Trollsvattnet (ÖT and VT) as revealed by the allozymes, which constitute the major part of the data. Allozymes provide strong evidence for subdivision within these lakes whereas the microsatellites are more ambiguous in this respect, and those observations are presented in the end of the Result section.
There is little or no allozyme allele frequency differentiation between lakes ÖT and VT (F ST = 0.001; Table 2, “lakes” columns). There is, however, a conspicuous heterozygote deficiency in the pooled material from the two lakes (F IT = 0.042) indicating a general deviation from random mating. This deficiency is also found within each of the two lakes, where F IS is 0.038 and 0.044 in ÖT and VT, respectively (average 0.041; Table 2, “lakes” columns). Allele frequency differences among sampling years or cohorts represent sources of variation that might result in heterozygote deficiencies, but in the present case those differences are too small to explain the heterozygote deficiencies observed (F ST among sampling years and cohorts is 0.001 and 0.002, respectively).
Two populations
The structure analysis strongly rejects the hypothesis of one single population (Pr(K = 1) = 0.00). Rather, the program consistently suggests two populations (clusters) as the most likely number (Pr(K = 2) = 1.00) in the total material as well as within each of the lakes ÖT and VT. The results were consistent between individual runs and regardless of the combination of ancestry and allele frequency model applied (Online Resource 2a–c).
A series of observations support the contention of the existence of two populations in lakes ÖT and VT. First, the distribution of individual Q-values, for both as well as for the separate lakes, reveals a clear U-shape (Fig. 2). Second, the principal component analysis reveals a clear distinction between individuals assigned to each of the two clusters (Fig. 3, depicting the outcome when using the allozyme data for the 382 fish that were scored for both sets of markers. The result from this restricted dataset is consistent with that for the total material from ÖT and VT (n = 4,140), but a plot of the larger dataset is too cluttered to be informative). Third, no heterozygote deficiency can be detected within any of the two clusters (rather, there is a weak but significant heterozygote excess within each cluster; Table 3). Finally, the amount of gametic (linkage) disequilibrium is conspicuously smaller within each of the two clusters than in the total material (ÖT + VT; n = 4,140), where 48 of the 91 allozyme locus pairs (53 %) displayed significant linkage disequilibrium, 28 remaining after Bonferroni adjustment. In contrast only 21 (6 after adjustment) and 15 (4) were observed within clusters A and B, respectively. The observation of a few disequilibria within each of the clusters remaining after correction for multiple comparisons is not unexpected considering that assignment to cluster is imperfect and that disequilibria are also generated in populations of restricted effective size (below).

Principal component plot for allozymes for 382 fish from ÖT and VT (Table 1). A and B denote cluster assignment when using structure. a Cut-off = 0.5 (382 fish), b cut-off = 0.7 (293 fish), c cut-off = 0.9 (183 fish). (Note the different Factor 2 scale in plate c)
Further, there was a high degree of consistency between assignment probabilities obtained with different structure models and over replicate runs within models (Online Resource 2a). The four series of 4,140 individual assignment probabilities (one from each structure model) resulted in six pair wise correlations which were all in the range r = 0.99–1.00. We also compared assignment probabilities for individual fish obtained in the five replicate runs generated under each of the four different models. Within each model the five replicate series of individual assignment probabilities resulted in ten pair wise correlations. All the 40 correlations were r = 1.00 (using two decimal points) indicating a high degree of consistency among replicate runs of the same model.
Similarly, assignment probabilities obtained when only including ÖT or VT in the structure analysis were almost identical to those obtained when combining ÖT and VT (r = 0.99 in both cases). Likewise, probabilities obtained for a particular sampling year generally correlated well with those from the entire data set (r = 0.86–0.99, except for sampling year 2005 with r = 0.10).
Amount of divergence and temporal stability of genetic patterns
The amount of genetic differentiation between the two co-existing populations (clusters A and B) is high (Table 2, “allozyme clusters” columns). Depending on the level of cut-off for assignment (0.5, 0.7, or 0.9) overall allozyme F ST between clusters is estimated as 0.10, 0.15, and 0.23 (Table 3), and these estimates stay reasonably stable over the entire study period of 19 years (Fig. 4).

Genetic differentiation (F ST ) between cluster A and B in Lakes Östra and Västra Trollsvattnet estimated from 14 allozyme loci and 19 sampling years (1987–2005). The lines represent the cut-off probability levels 0.5, 0.7, and 0.9 when assigning individuals to cluster
Quantifying different sources of allozyme variation by means of a hierarchical approach for gene diversity analysis indicates that “cluster” represents the by far most important source when considering the overall variation caused by “lake”, “cluster” (within lakes), and “cohort” (within clusters). Including all sources of explained variation, and regardless of the hierarchical order in the analysis, “lake” accounts for less than 1 %, and “cluster” and “cohort” account for 83 % and 16 %, respectively (with the residual “unexplained” variation representing 94 % of the total variation).
In spite of the considerable allozyme allele frequency differences between clusters, the levels of genetic variation within them are quite similar. All loci segregate for the same two alleles in both populations, except at sAAT-4 where cluster A has a third allele at a low frequency (0.001) that is missing in cluster B. Average heterozygosity (H e) over cohorts ranged in the interval 0.27–0.29 and 0.25–0.27 within A and B, respectively, and there was no apparent temporal trend in this variation in either cluster.
Test for selection
The lositan software suggested one locus (sIDHP-1) as a candidate for directional selection (0.05 > P > 0.01) when analyzing the distribution of allozyme F ST values between clusters. This locus also shows the highest degree of differentiation between clusters (F ST = 0.26), followed by G3PDH-2 (F ST = 0.20) which is not classified as a candidate for selection. Exclusion of sIDHP-1 has no effect on the conclusions of this paper except that the overall F-statistics calculated across the 14 allozyme loci tend to get somewhat smaller. For the major comparison of clusters in the two lakes, for example, allozyme F ST is reduced from 0.100 (Table 2, “allozyme clusters” columns) to 0.080, and F IT is reduced from 0.089 (Table 2, “allozyme clusters”) to 0.073 when using the 0.5 cut-off level for assignment. Because of the minor change of the F-statistics when excluding sIDHP-1, the allozyme results have been consistently presented with all loci included, except when estimating effective population size where estimates were calculated both with and without sIDHP-1 (below).
Spatial distribution of clusters A and B
Both clusters occur in both lakes and in similar frequencies (Fig. 2), and over sampling years the overall proportion of fish assigned to cluster A varied in the range 0.45–0.63, with an average of 0.53 (using a cut-off level of 0.5 for assignment). Similarly, within each of the lakes, the proportion assigned to cluster A ranged in the intervals 0.47–0.71 and 0.41–0.68 for ÖT and VT, respectively, with averages of 0.57 and 0.50.
The notion that the same two clusters occur in both lakes is supported by an apparent lack of divergence between fish assigned to the same cluster but caught in different lakes. For example, using a cut-off level of 0.5 for assignment, the estimated allozyme divergence between lakes within clusters is F ST = 0 and F ST = 0.001 for clusters A and B, respectively, and both values are associated with F IS- and F IT-estimates close to zero (for other cut-off levels see Table 3). Similar results were obtained when making comparisons within cohorts. F ST-estimates between cluster A in ÖT and cluster A in VT vary in the range −0.006–0.014 for the 19 cohorts comprising at least 15 fish in each lake, and for cluster B the corresponding values are −0.026–0.010 (18 cohorts), all of them non-significant.
No statistically significant allozyme heterozygote deficiency was found within any of the three small neighboring lakes (N1-N3), although F IS point estimates were positive ranging from 0.01 to 0.03. Nevertheless, structure analyses indicate that neither of these lakes constitutes a single panmictic population. The program suggests two populations as the most likely number in N1 and N3, and four populations in N2 (Pr(K = 2) = 0.99, Pr(K = 2) = 1.00, and Pr(K = 4) = 0.70, respectively).
When pooling allozyme data from all the five sampling localities (n = 4,588) structure still suggests two clusters as the most likely number (Pr(K = 2) = 1.00), and the two populations co-exist in the entire area. In the neighboring lakes the proportion of the two clusters appears to be largely of the same magnitude as in ÖT and VT, and the proportion of cluster A is estimated as 0.54, 0.69, and 0.53 for N1, N2, and N3, respectively. Within both clusters F ST is close to zero over all the five localities as well as among the three neighboring lakes, whereas considerable divergence is observed between clusters (Table 3).
Phenotypic differences
We can see no apparent phenotypic differences hinting about the existence of two separate populations in Lakes Trollsvattnen. Fish in cluster A are consistently somewhat larger than those in B (Online Resource 4), and the difference is statistically significant (P < 0.05) in 13 of the 14 comparisons between clusters within lakes and age classes. This difference, however, is insufficient for assigning a particular fish to cluster with any degree of acceptable accuracy.
In all age classes the proportion of breeders (fish mature to breed the year of collection) was consistently lower in cluster A, with overall proportions of 35 and 54 % for A and B, respectively. This tendency of a higher proportion of breeders in cluster B was found in both lakes, among all age classes, and in both sexes (males maturating a bit earlier than females within both clusters). Generation length is estimated as G = 7.0 and G = 6.8 for cluster A and B, respectively (calculated from Online Resource 3).
Effective size and migration
Estimates of effective population size are similar for the two clusters. Based on allozyme data for cohorts with 30 or more individuals, effective sizes (with 95 % confidence limits) are estimated as \( \hat{N}_{e} \)=121 (77–282) and \( \hat{N}_{e} \)=193 (101–2,225) for A and B, respectively, when using 0.5 as cut-off for assignment. The point estimates of N e are of similar magnitudes when increasing the cut-off level, resulting in estimates of 113 (69–304) and 127 (65–3,037) for A and 212 (96-∞) and 329 (63-∞) for B when applying the 0.7 and 0.9 cut-offs, respectively. The standard errors are larger for these estimates, however, as expected with the smaller sample sizes resulting from higher cut-offs. Further, the effect of excluding the locus considered a potential candidate for selection was negligible, resulting in estimates of \( \hat{N}_{e} \)=123 (77–308) and \( \hat{N}_{e} \)=173 (92–1,494) for the A and B clusters, respectively (cut-off = 0.5).
The exchange of individuals between the two clusters appears fairly small. We crudely assessed the amount of migration between them assuming mutation-migration-drift equilibrium and using recursion equations for gene diversity (Nei 1975; Li 1976) applied as exemplified in Ryman and Leimar (2008; eq. A15). For an island model of migration, two subpopulations, a mutation rate of 10−6 (for allozymes), and an N e = 157 in both clusters (representing the average of N e = 121 and N e = 193 for the two clusters, respectively), the observed F ST (0.10–0.23) would correspond roughly to each cluster receiving a proportion of 0.001–0.004 migrants per generation from the other one (depending on the cut-off and F ST-value applied). Clearly, this assessment is based on several assumptions, the validity of which cannot be evaluated; nevertheless, it provides a hint of the amount of isolation required to result in the divergence observed.
Microsatellites
The information with respect to population structure obtained from the microsatellites is more ambiguous and difficult to interpret than that from the allozymes. There is no overall heterozygote deficiency indicating the potential existence of more than a single population in the total material for the two lakes (subset of n = 382 fish scored for both types of markers), and F IT suggests a heterozygote excess rather than a deficit (F IT = −0.013, non-significant; Table 2, “lakes” columns). Three of the four structure models suggest a single population (Pr(K = 1) = 1.00), whereas the most likely one suggests two populations (Pr(K = 2) = 1.00; Supplementary Table S1d) with a significant degree of differentiation (F ST = 0.060, Table 2, “microsatellite clusters” columns).
The correspondence is poor between the patterns for subdivision suggested by structure for the two sets of markers. There is, however, a weak but significant correlation between the assignment probabilities obtained for the allozymes, and those obtained for the microsatellites in the only model suggesting two populations (n = 382, r = 0.276, P < 0.001). Further, there is a significant microsatellite allele frequency difference between the two clusters A and B defined on the basis of allozymes. This microsatellite differentiation is much smaller than that found for the allozymes, though (F ST, micros = 0.016 vs. F ST, allozymes = 0.100 for cut-off 0.5, Table 2, “allozyme clusters” columns). All in all, we conclude that the microsatellite results do neither support or strongly contradict the existence of the two populations identified by the allozymes. It is clear, however, that had we only had access to the microsatellite data we would most likely have concluded that there was little or no evidence for the occurrence of more than a single population.
There is no indication that the weak correspondence between the patterns provided by the two marker types is due to the subset of 382 fish not being representative of the total material. For example, all the four structure models suggest two populations for the 382 fish when considering the allozymes alone (Pr(K = 2) = 1.00; Online Resource 2e), and the F-statistics match closely those for clusters A and B in the total material (F ST = 0.107, F IT = 0.109, and F IS = 0.001). Similarly, there is a strong correlation between the allozyme assignment probabilities obtained for the total material and those for the subset (n = 382, r = 0.975, P < 0.001). Further, when testing for selective neutrality with lositan there is no suggestion of stabilizing selection operating at any of the microsatellites in the dataset of 382 individuals (clusters identified by allozyme data). When using microsatellite data for identifying clusters, lositan suggests one locus (SsoSL417) as a candidate for directional selection (0.05 > P > 0.01).
Discussion
We monitored the genetic pattern of brown trout in some small, inter-connected mountain lakes in northern Scandinavia and found two genetically distinct populations occurring in about equal frequency in all lakes. The populations are characterized by a high degree of genetic divergence for allozyme loci; the pattern is supported by significant differentiation between populations (clusters A and B) also for microsatellites and total body length. There is no anecdotal or other information suggesting the possible existence of multiple morphs or populations of brown trout in Lakes Trollsvattnen or neighboring lakes and the detection of the two populations was incidental.
Genetic monitoring confirms the temporal stability of these sympatrically occurring populations over 19 years representing about three brown trout generations. This is an important observation as most previous studies revealing cryptic populations have included samples separated by a much shorter time span, thus preventing taking the temporal aspects into account (e.g. Allendorf et al. 1976; Ryman et al. 1979; Wilson et al. 2004; Adams et al. 2008).
The amount of genetic differentiation between the two clusters falls within the range of what is typically found among apparently isolated local populations within this region (Ryman 1983; Jorde and Ryman 1996; Palm et al. 2003), and is somewhat larger than what was found between the three sympatrically occurring morphotypes in the classic study of brown trout in Lough Melvin, Ireland (Ferguson and Taggart 1991) where allozymes were also used. The statistically significant difference in total body length between clusters in Lakes Trollsvattnen is smaller than the body size difference observed between two sympatrically existing brown trout demes identified in Lakes Bunnersjöarna in central Sweden (Ryman et al. 1979). In both cases the size differences were detected after grouping individuals based on genetic data.
Detecting multiple, co-existing populations
The existence of multiple genetically divergent sympatric populations might be more common than anticipated, also in seemingly homogenous environments (Ryman et al. 1979; Bergek and Björklund 2007). They can be difficult to detect in the absence of obvious morphological or ecological differentiation, particularly when contrasting homozygosities are also lacking.
Our findings exemplify that the seemingly natural way of grouping individuals—by lake—can be misleading and actually hide the genetic signal of multiple populations. With no natural starting point for between group allele frequency comparisons, only a consistent heterozygote deficiency at several loci might indicate the existence of multiple populations, but the statistical power for detecting deviations from panmixia can be low with commonly applied sample sizes (Waples and Gaggiotti 2006). Rather, larger samples than typically collected might be needed for reliable estimation of the number of populations.
In our case, the existence of the two populations would most likely have gone undetected with a single sampling year effort; overall heterozygote deficiencies for allozyme data (F IT significantly larger than zero) was only observed in five of the 19 sampling years, and in 15 years no single locus exhibited a significant deficiency after Bonferroni correction (details not shown). To estimate the number of consecutive sampling years necessary for detecting the two populations (K = 2) of Lakes Trollsvattnen with a reasonable likelihood using allozyme data and our annual sampling size of 100 fish per lake, we applied structure analyses to each combination of 2, 3, 4, etc. consecutive sampling years. The observed likelihood for different K-values (K = 1 − 5) indicated that at least four years (corresponding to a total sample of c. 800 fish) were needed to yield a likelihood for K = 2 of 0.95 or more in all the suites of consecutive sampling years available during this 19 year study.
Inconsistency between allozymes and microsatellites
Our set of microsatellite loci does not show any significant heterozygote deficiencies and, consequently, does not indicate the existence of more than one population, whereas the allozymes show heterozygote deficiency also in the smaller data set (n = 382) that was analyzed for microsatellites (Table 2). Further, the genetic divergence between cluster A and B (identified using allozyme data) is significant also for microsatellites but lower as compared to allozymes (F ST, micros = 0.016 vs. F ST, allozymes = 0.100; Table 2).
There are several possible explanations for the observed inconsistency between allozymes and microsatellites: (i) the allozymes are subject to directional selection, (ii) the microsatellites are linked to loci subject to stabilizing selection, (iii) scoring errors, (iv) there are statistical power differences between markers in detecting heterozygote deficiencies, and (v) random chance.
We rule out scoring errors. This is because a large part (96 out of 382 fish) of the microsatellites have been run at two independent laboratories with a consistency of 98 percent. As for the allozymes we have decades of experience from these markers (e.g. Ryman et al. 1979; Jorde and Ryman 1996; Palm et al. 2003; Charlier et al. 2011, 2012) that have also been used by others in many other studies of brown trout (e.g. Ferguson and Mason 1981; Crozier and Ferguson 1986; Ferguson and Taggart 1991). Also, we do not think that selection is the cause of the inconsistency. When testing for selection, one or two allozyme loci (depending on including microsatellites in the analysis or not) was suggested as candidates for directional selection, but removing these markers provide consistent results with respect to the two clusters. We find no indications that all allozymes are subject to the same type of selection. Similarly, there are no indications that the microsatellites (or closely linked loci) are subject to stabilizing selection.
Rather, we suspect that our observation is largely coincidental or due to statistical power differences between markers with respect to detecting heterozygote deficiency (c.f. Ryman et al. 2006; Larsson et al. 2009; Karl et al. 2012). We conducted preliminary simulations using a modified version of powsim to estimate the power of detecting heterozygote deficiency in a situation with two populations with an expected F ST of 0.1. To mimic allozymes we used 14 loci with a total of 29 alleles and expected heterozygosity (H T) of 0.28. The “microsatellites” were 7 loci with 32 alleles and H T = 0.50 (cf. Table 2). From a mix of these populations in equal proportions we simulated sampling of n = 100 using 100 runs. We found that the allozymes show a larger probability of detecting the heterozygote deficiency as compared to the microsatellites (0.5 vs. 0.4). Although the difference is not statistically significant it is an indication that our observation in Lakes Trollsvattnen with respect to the two sets of markers might be consistent with expectations.
Evolutionary origin of the two co-existing populations
We cannot conclude whether the evolutionary origin of the two identified clusters is sympatric or allopatric. The fact that both populations segregate for essentially the same alleles in both allozyme and microsatellite loci indicates that the split is fairly recent, or that reproductive isolation is incomplete (cf. Fig. 2). Application of the above mentioned recursion equations for gene identity suggests that time since the most recent glaciation is enough to reach the observed degree of differentiation (F ST = 0.10–0.23 depending on cut-off), assuming that migration rates expected under steady state (m = 0.001–0.004) have persisted since the split (details not shown). Therefore, a sympatric origin, with still diverging populations, cannot be excluded.
Conservation implications
Our findings contribute to the understanding of the dynamics and complexity of within species biodiversity. The two brown trout populations are almost as genetically distinct as separate species (e.g. Rüber et al. 2001; Barluenga et al. 2006; Allendorf and Luikart 2007), although they occur sympatrically in a very small mountain lake system. This diversity was not possible to detect without repeated sampling, emphasizing the need both for increased efforts to assess intraspecific variation, and to monitor such diversity over time. Such assessments are largely lacking, in spite of conservation of genetic variation being an explicit international goal through the United Nation′s Convention on Biological Diversity of which almost all countries in the world are parties (Laikre 2010; Laikre et al. 2010).
Our results reinforce the general conception that brown trout, like many other salmonids, tend to develop distinct local populations of restricted effective size. N e has been estimated for seven natural brown trout populations in the geographic area monitored within the framework of the Lakes Bävervattnen project, and all of them are smaller than 200 with a range of 19–193 (Jorde and Ryman 1996; Laikre et al. 1998; Palm et al. 2003, Charlier et al. 2011, 2012). A meta population structure with small amounts of migration between populations seems necessary to maintain variation within these systems.
Multiple, co-existing populations can have important implications for the resilience of brown trout communities (Hilborn et al. 2003; Lindley et al. 2009; Schindler et al. 2010). Scandinavian mountain lakes are often species poor, and the two genetically distinct populations found in Lakes Trollsvattnen might contribute in different ways to the stability of the ecosystem as a whole.
The potential for sympatrically occurring populations should be considered in management strategies because simultaneous harvest of multiple populations can result in over-harvest of some populations (Allendorf et al. 2008). Relying exclusively on morphologic or ecological data as a basis for management decisions when genetic data is lacking, can lead to unsustainable use of genetic resources.
Future studies of the identified co-existing populations
Salmonids show strong homing behavior and this seems to constitute the basis for the marked population differentiation observed in many species including the brown trout. Thus, separation in space or time for spawning is likely to be involved in the origin or maintenance of the two clusters. Natural components of future studies of these particular populations therefore include identification of place and time for spawning as well as ecological and morphological characterization of the two populations. A more detailed assessment of the degree of allelic overlap using microsatellites or SNPs for a larger part of the dataset is also warranted. Similarly, we will further investigate the statistical power of detecting heterozygote deficiencies under various scenarios.
References
Adams CE, Wilson AJ, Ferguson MM (2008) Parallel divergence of sympatric genetic and body size forms of Arctic charr, Salvelinus alpinus, from two Scottish lakes. Biol J Linn Soc 95:748–757
Allendorf FW, Luikart G (2007) Conservation and the genetics of populations. Blackwell, Malden
Allendorf F, Ryman N, Stennek A, Ståhl G (1976) Genetic variation in Scandinavian brown trout (Salmo trutta L.): evidence of distinct sympatric populations. Hereditas 83:73–82
Allendorf FW, Mitchell N, Ryman N, Ståhl G (1977) Isozyme loci in brown trout (Salmo trutta L.): detection and interpretation from population data. Hereditas 86:179–190
Allendorf FW, England PR, Luikart G, Ritchie PA, Ryman N (2008) Genetic effects of harvest on wild animal populations. Trends Ecol Evol 23:327–337
Antao T, Lopes A, Lopes RJ, Beja-Pereira A, Luikart G (2008) lositan: a workbench to detect molecular adaptation based on a F st-outlier method. BMC Bioinformatics 9:323
Barluenga M, Stölting KN, Salzburger W, Muschick M, Meyer A (2006) Sympatric speciation in Nicaraguan crater lake cichlid fish. Nature 439:719–723
Beaumont MA, Nichols RA (1996) Evaluating loci for use in the genetic analysis of population structure. Proc R Soc Lond B 263:1619–1626
Bergek S, Björklund M (2007) Cryptic barriers to dispersal within a lake allow genetic differentiation of Eurasian perch. Evolution 61:2035–2041
Bernatchez L, Martin S (1996) Mitochondrial DNA diversity in anadromous rainbow smelt, Osmerus mordax Mitchill: a genetic assessment of the member-vagrant hypothesis. Can J Fish Aquat Sci 53:424–433
Chakraborty R, Haag M, Ryman N, Ståhl G (1982) Hierarchical gene diversity analysis and its application to brown trout population data. Hereditas 97:17–21
Charlier J, Palmé A, Laikre L, Andersson J, Ryman N (2011) Census (N C) and genetically effective (N e) population size in a lake-resident population of brown trout Salmo trutta. J Fish Biol 79:2074–2082
Charlier J, Laikre L, Ryman N (2012) Genetic monitoring reveals temporal stability over 30 years in a small, lake-resident brown trout population. Heredity 109:246–253
Child AR (1984) Biochemical polymorphism in charr (Salvelinus alpinus L.) from three Cumbrian lakes. Heredity 53:249–257
Clarke GM, O’Dwyer C (2000) Genetic variability and population structure of the endangered golden sun moth, Synemon plana. Biol Conserv 92:371–381
Crozier WW, Ferguson A (1986) Electrophoretic examination of the population structure of brown trout, Salmo trutta L., from the Lough Neagh catchment, Northern Ireland. J Fish Biol 28:459–477
Dannewitz J, Petersson E, Prestegaard T, Järvi T (2003) Effects of sea-ranching and family background on fitness traits in brown trout Salmo trutta reared under near-natural conditions. J Appl Ecol 40:241–250
Dupont P-P, Bourret V, Bernatchez L (2007) Interplay between ecological, behavioural and historical factors in shaping the genetic structure of sympatric walleye populations (Sander vitreus). Mol Ecol 16:937–951
Estoup A, Presa P, Krieg F, Vaiman D, Guyomard R (1993) (CT)n and (GT)n microsatellites: a new class of genetic markers for Salmo trutta L. (brown trout). Heredity 71:488–496
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587
Ferguson A, Mason FM (1981) Allozyme evidence for reproductively isolated sympatric populations of brown trout Salmo trutta L. in Lough Melvin, Ireland. J Fish Biol 18:629–642
Ferguson A, Taggart JB (1991) Genetic differentiation among the sympatric brown trout (Salmo trutta) populations of Lough Melvin, Ireland. Biol J Linn Soc 43:221–237
Frankham R (2005) Ecosystem recovery enhanced by genotypic diversity. Heredity 95:183
Fraser DJ, Bernatchez L (2005) Allopatric origins of sympatric brook charr populations: colonization history and admixture. Mol Ecol 14:1497–1509
Gerlach G, Schardt U, Eckmann R, Meyer A (2001) Kin-structured subpopulations in Eurasian perch (Perca fluviatilis L.). Heredity 86:213–221
Hajjar R, Jarvis DI, Gemmill-Herren B (2008) The utility of crop genetic diversity in maintaining ecosystem services. Agric Ecosyst Environ 123:261–270
Hilborn R, Quinn TP, Schindler DE, Rogers DE (2003) Biocomplexity and fisheries sustainability. Proc Natl Acad Sci USA 100:6564–6568
Hindar K, Ryman N, Ståhl G (1986) Genetic differentiation among local populations and morphotypes of artic charr, Salvelinus alpinus. Biol J Linn Soc 27:269–285
Huang QQ, Zhang YX (2000) Study on the genetic structure in Pinus massoniana (Masson Pine) populations. Silvae Genet 49:4–5
Jorde PE, Ryman N (1995) Temporal allele frequency change and estimation of effective size in populations with overlapping generations. Genetics 139:1077–1090
Jorde PE, Ryman N (1996) Demographic genetics of brown trout (Salmo trutta) and estimation of effective population size from temporal change of allele frequencies. Genetics 143:1369–1381
Jorde PE, Ryman N (2007) Unbiased estimator for genetic drift and effective population size. Genetics 177:927–935
Karl SA, Toonen RJ, Grant WS, Bowen BW (2012) Common misconceptions in molecular ecology: echoes of the modern synthesis. Mol Ecol 21:4171–4189
Knutsen H, Olsen EM, Jorde PE, Espeland SH, André C, Stenseth NC (2010) Are low but statistically significant levels of genetic differentiation in marine fishes ‘biologically meaningful’? A case study of coastal Atlantic cod. Mol Ecol 20:768–783
Knutsen H, Jorde PE, Bergstad OA, Skogen M (2012) Population genetic structure in a deepwater fish Coryphaenoides rupestris: patterns and processes. Mar Ecol-Prog Ser 460:233–246
Laikre L (2010) Genetic diversity is overlooked in international conservation policyimplementation. 2010. Cons Gen 11:349–354
Laikre L, Jorde PE, Ryman N (1998) Temporal change of mitochondrial DNA haplotype frequencies and female effective size in a brown trout (Salmo trutta) population. Evolution 52:910–915
Laikre L, Allendorf FW, Aroner LC, Baker CS, Gregovich DP, Hansen MM et al (2010) Neglect of genetic diversity in implementation of the Convention on Biological Diversity. Cons Biol 24:86–88
Larsson LC, Laikre L, André C, Dahlgren TG, Ryman N (2009) Temporally stable genetic structure of heavily exploited Atlantic herring (Clupea harengus) in Swedish waters. Heredity 104:40–51
Lee KE, Seddon JM, Corley SW, Ellis WAH, Johnston SD, de Villiers DL, Preece HJ, Carrick FN (2010) Genetic variation and structuring in the threatened koala populations of Southeast Queensland. Conserv Genet 11:2091–2103
Li W-H (1976) Effect of migration on genetic distance. Am Nat 110:841–847
Lindley ST, Grimes CB, Mohr MS, Peterson W, Stein J, Anderson JT et al (2009) What caused the Sacramento River fall chinook stock collapse? NOAA Tech Memo NMFS-SWFSC 447, 61 pp
Luck GW, Daily GC, Ehrlich PR (2003) Population diversity and ecosystem services. Trends Ecol Evol 18:331–336
Martien KK, Baird RW, Hedrick NM, Gorgone AM, Thieleking JL, McSweeney DJ, Robertson KM, Webster DL (2012) Population structure of island-associated dolphins: evidence from mitochondrial and microsatellite markers for common bottlenose dolphins (Tursiops truncatus) around the main Hawaiian Islands. Mar Mammal Sci 28:E208–E232
Nei M (1975) Molecular population genetics and evolution. North-Holland, Amsterdam
O’Reilly PT, Hamilton LC, McConnell SK, Wright JM (1996) Rapid analysis of genetic variation in Atlantic salmon (Salmo salar) by PCR multiplexing of dinucleotide and tetranucleotide microsatellites. Can J Fish Aquat Sci 53:2292–2298
Palm S, Ryman N (1999) Genetic basis of phenotypic differences between transplanted stocks of brown trout. Ecol Freshw Fish 8:169–180
Palm S, Laikre L, Jorde PE, Ryman N (2003) Effective population size and temporal genetic change in stream resident brown trout (Salmo trutta, L.). Cons Gen 4:249–264
Phinchongsakuldit J, Chaipakdee P, Collins JF, Jaroensutasinee M, Brookfield JFY (2013) Population genetics of cobia (Rachycentron canadum) in the Gulf of Thailand and Andaman Sea: fisheries management implications. Aquacult Int 21:197–217
Power M, Power G, Reist JD, Bajno R (2009) Ecological and genetic differentiation among the Arctic charr of Lake Aigneau, Northern Québec. Ecol Freshw Fish 18:445–460
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Pritchard JK, Wen X, Falush D (2007) Documentation for structure software: Version 2.2. Software available from http://pritch.bsd.uchicago.edu/software. Accessed May 2012
Raymond M, Rousset F (1995) genepop (version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Reusch TBH, Ehlers A, Hammerli A, Worm B (2005) Ecosystem recovery after climatic extremes enhanced by genotypic diversity. Proc Natl Acad Sci USA 102:2826–2831
Rüber L, Meyer A, Sturmbauer C, Verheyen E (2001) Population structure in two sympatric species of the Lake Tanganyika cichlid tribe Eretmodini: evidence for introgression. Mol Ecol 10:1207–1225
Ryman N (1983) Patterns of distribution of biochemical genetic variation in salmonids: differences between species. Aquaculture 33:1–21
Ryman N (2006) chifish: a computer program testing for genetic heterogeneity at multiple loci using Chi square and Fisher’s exact test. Mol Ecol Notes 6:285–287
Ryman N, Leimar O (2008) Effect of mutation on genetic differentiation among nonequilibrium populations. Evolution 62:2250–2259
Ryman N, Palm S (2006) powsim: a computer program for assessing statistical power when testing for genetic differentiation. Mol Ecol Notes 6:600–602
Ryman N, Allendorf FW, Ståhl G (1979) Reproductive isolation with little genetic divergence in sympatric populations of brown trout (Salmo trutta). Genetics 92:247–262
Ryman N, Palm S, André C, Carvalho GR, Dahlgren TG, Jorde PE et al (2006) Power for detecting genetic divergence: differences between statistical methods and marker loci. Mol Ecol 15:2031–2045
Schindler DE, Hilborn R, Chasco B, Boatright CP, Quinn TP, Rogers LA et al (2010) Population diversity and the portfolio effect in an exploited species. Nature 465:609–612
Scribner KT, Gust JR, Fields RL (1996) Isolation and characterization of novel salmon microsatellite loci: cross-species amplification and population genetic applications. Can J Fish Aquat Sci 53:833–841
Shaklee JB, Allendorf FW, Morizot DC, Whitt GS (1990) Gene nomenclature for protein-coding loci in fish. Trans Am Fish Soc 119:2–15
Slettan A, Olsaker I, Lie O (1995) Atlantic salmon, Salmo salar, microsatellites at the SSOSL25, SSOSL85, SSOSL311, SSOSL417 loci. Animal Genet 26:281–282
Spruell P, Rieman BE, Knudsen KL, Utter FM, Allendorf FW (1999) Genetic population structure within streams: microsatellite analysis of bull trout populations. Ecol Freshw Fish 8:114–121
StatSoft, Inc. (2005) statistica (data analysis software system), version 7.1. www.statsoft.com. Purchased in February 2006
Taylor EB, Bentzen P (1993a) Evidence for multiple origins and sympatric divergence of trophic ecotypes of smelt (Osmerus) in northeastern North America. Evolution 47:813–832
Taylor EB, Bentzen P (1993b) Molecular genetic evidence for reproductive isolation between sympatric populations of smelt Osmerus in Lake Utopia, south-western New Brunswick, Canada. Mol Ecol 2:345–357
Taylor EB, Harvey S, Pollard S, Volpe J (1997) Postglacial genetic differentiation of reproductive ecotypes of kokanee Oncorhynchus nerka in Okanagan Lake, British Columbia. Mol Ecol 6:503–517
Van Rossum F, Prentice HC (2004) Structure of allozyme variation in Nordic Silene nutans (Caryophyllaceae): population size, geographical position and immigration history. Biol J Linn Soc 81:357–371
Verspoor E, Cole LJ (1989) Genetically distinct sympatric populations of resident and anadromous Atlantic salmon, Salmo salar. Can J Zoo 67:1453–1461
Waples RS, Do C (2010) Linkage disequilibrium estimates of contemporary N e using highly variable genetic markers: a largely untapped resource for applied conservation and evolution. Evol Appl 3:244–262
Waples RS, Gaggiotti O (2006) What is a population? An empirical evaluation of some genetic methods for identifying the number of gene pools and their degree of connectivity. Mol Ecol 15:1419–1439
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Wilson PJ, Grewal S, Rodgers A, Rempel R, Saquet J, Hristienko H, Burrows F, Peterson R, White BN (2003) Genetic variation and population structure of moose (Alces alces) at neutral and functional DNA loci. Can J Zoolog 81:670–683
Wilson AJ, Gíslason D, Skúlason S, Snorrason SS, Adams CE, Alexander G et al (2004) Population genetic structure of Arctic charr, Salvelinus alpinus from northwest Europe on large and small spatial scales. Mol Ecol 13:1129–1142
Acknowledgments
We thank Per Erik Jorde, Robin Waples, Kerry Naish, and Stefan Palm for continuous and helpful discussions, and two anonymous reviewers for valuable comments on the manuscript. We are also grateful to Magne Friberg, Johan Charlier, Peter Guban, Morten Tange Olsen, and Dimitar Serbezov for assistance and suggestions. The study area is located in the Hotagen Nature Reserve, County of Jämtland, Sweden, and we acknowledge the cooperation from the Jämtland County Administrative Board and the Jiingevaerie Sami Community. Financial support was provided by Formas - the Swedish Research Council for Environment, Agricultural Sciences and Spatial Planning (LL, NR), the Swedish Research Council (LL, NR), and Carl Tryggers Stiftelse (LL). Parts of the analyses were conducted within the framework of the BaltGene project funded by BONUS Baltic Organisations’ Network for Funding Science EEIG. This study benefitted from discussions and collaborations within the Working Group on Genetic Monitoring supported by the National Evolutionary Synthesis Center (NSF #EF-0423641) and the National Center for Ecological Analysis and Synthesis (NSF #DEB-0553768), chaired by Drs. Fred W. Allendorf and Michael K. Schwartz.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
10592_2013_475_MOESM4_ESM.ppt
Online Resource 4 Mean body length (± SE) in different age classes in clusters A and B in Lakes Östra and Västra Trollsvattnet (ÖT and VT; cut-off = 0.5) (PPT 128 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Palmé, A., Laikre, L. & Ryman, N. Monitoring reveals two genetically distinct brown trout populations remaining in stable sympatry over 20 years in tiny mountain lakes. Conserv Genet 14, 795–808 (2013). https://doi.org/10.1007/s10592-013-0475-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-013-0475-x