
Abstract
Identifying the spatial distribution of genetic variation across the landscape is an essential step in informing species conservation. Comparison of closely related and geographically overlapping species can be particularly useful in cases where landscape may similarly influence genetic structure. Congruent patterns among species highlight the importance that landscape heterogeneity plays in determining genetic structure whereas contrasting patterns emphasize differences in species-specific ecology and life-history or the importance of species-specific adaptation to local environments. We examined the interacting roles of demography and adaptation in determining spatial genetic structure in two closely related and geographically overlapping species in a pristine environment. Using single nucleotide polymorphism (SNP) loci exhibiting both neutral and putative adaptive variation, we evaluated the genetic structure of sockeye salmon in the Copper River, Alaska; these data were compared to existing data for Chinook salmon from the same region. Overall, both species exhibited patterns of isolation by distance; the spatial distribution of populations largely determined the distribution of genetic variation across the landscape. Further, both species exhibited largely congruent patterns of within- and among-population genetic diversity, highlighting the role that landscape heterogeneity and historical processes play in determining spatial genetic structure. Potential adaptive differences among geographically proximate sockeye salmon populations were observed when high FST outlier SNPs were evaluated in a landscape genetics context. Results were evaluated in the context of conservation efforts with an emphasis on reproductive isolation, historical processes, and local adaptation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Identifying the distribution of genetic variation across the landscape is an essential step in informing species conservation. Landscape genetics methods facilitate our understanding of the role that landscape heterogeneity plays in shaping genetic structure in natural populations (Manel et al. 2003; Storfer et al. 2007). Comparison of closely related and geographically overlapping species can be particularly useful in cases where the landscape may similarly (or differentially) influence the genetic structure of multiple species (Marten et al. 2006; Olsen et al. 2011). Congruent patterns among species may highlight the importance that landscape, including historical processes, plays in determining spatial genetic structure through demographic processes such as gene flow and genetic drift (Waples 1987; Petren et al. 2005; Gagnon and Angers 2006). Conversely, contrasting patterns may emphasize differences in species-specific ecology and life history (i.e. differential dispersal abilities; Gomez-Uchida et al. 2009) or the importance of species-specific adaptation to local environments (Short and Caterino 2009). In either regard, an understanding of the interacting roles that gene flow, genetic drift, and local adaptation play in shaping genetic structure can greatly improve our knowledge of species and improve conservation efforts (Schwartz et al. 2009; Olsen et al. 2011).
Traditional landscape genetics analyses have focused on divergence at loci assumed to be neutral to infer demographic history and population structure (Manel et al. 2003). However, overemphasis on the role of reproductive isolation and under-emphasis on adaptive differences among populations has received scrutiny in conservation applications (Hard 1995; Pearman 2001; Schwartz et al. 2009). Here, we examined the interacting roles of demography and putative local adaptation in determining the spatial distribution of genetic variation in two closely related and geographically overlapping species in a largely pristine environment. We used single nucleotide polymorphism (SNP) loci that exhibit both neutral and putative adaptive variation to evaluate the spatial structure of sockeye salmon (Oncorhynchus nerka) in the Copper River, Alaska; these data were then compared to existing data available for Chinook salmon (O. tshawytscha), a congener from the same drainage (Seeb et al. 2009).
Pacific salmon are distributed throughout most river systems along the west coast of North America from central California north to Alaskan and Canadian rivers flowing into the Arctic Ocean (Behnke and Tomelleri 2002). Five species of Pacific salmon are anadromous and semelparous. The two species in this study, sockeye salmon and Chinook salmon, support economically and culturally valuable fisheries in the Copper River, Alaska, and elsewhere. The high fidelity of Pacific salmon to their natal spawning and rearing environments results in genetic variation among discrete populations (Taylor 1991; Dittman and Quinn 1996; Quinn 2005). Juvenile sockeye salmon primarily spend a year or more rearing in freshwater before emigrating to the marine environment; although a ‘sea-type’ occurs in which juveniles may migrate to the marine environment in their first year (Quinn 2005; Wood et al. 2008). Adult sockeye salmon usually mature and return to spawn in their natal freshwater environment at 4–5 years of age. Chinook salmon exhibit a similar life-history with one notable exception; juvenile sockeye salmon generally rear in lakes whereas juvenile Chinook salmon generally rear in fluvial environments (Behnke and Tomelleri 2002; Quinn 2005).
The Copper River, with a drainage area of approximately 64,000 km2, provides a heterogeneous landscape to evaluate interactions between genetic and spatial variation in Pacific salmon. From its origins in the Wrangell, Chugach, and Alaska mountain ranges, the Copper River flows approximately 450 km south before emptying into the Gulf of Alaska (Fig. 1). During its course, the river drains two unique environments. The upper Copper River and its main tributaries, the Slana, Tanada, Chistochina and Gulkana rivers, are relatively low gradient and drain large portions of tundra. The middle and lower Copper River and its main tributaries, the Tazlina, Klutina, Tonsina, and Chitina rivers, drain a more forested, mountainous landscape, are largely fed by glacial meltwater, and are characterized by higher gradient, glacially turbid reaches. Based on environmental differences between the upper and middle/lower Copper River and similar research conducted on Pacific salmon in subarctic North America (Olsen et al. 2011), we hypothesize that sockeye salmon will exhibit an inland-coastal transition in the distribution of genetic variation, and that abrupt differences in allele frequencies (i.e. barriers) may occur between the two environments.

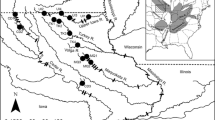
Gradient of allelic richness, population groups, and inferred barriers for sockeye salmon from the Copper River, Alaska. Population symbols indicate the groups defined by SAMOVA. Barriers indicate abrupt changes in allele frequencies transferred from the Delaunay triangulation in BARRIER to approximate locations on the map. Numbers inside the parentheses indicate the robustness of each barrier based on 100 bootstrap samples. The “Adaptive Barrier” observed above population #17 indicates the additional barrier that was observed when including outlier SNPs. The population symbols in parentheses for populations #17 and #19 indicate the groupings that were determined when including the outlier SNPs. Population numbers indicate Population ID in Table 1
In this study we used a combination of population genetics and landscape genetics methods to assess the spatial genetic diversity of Pacific salmon in the Copper River. Our analyses and discussion focus primarily on sockeye salmon and evaluating the roles of spatial variation and putative local adaptation in shaping their hierarchical genetic structure in the Copper River drainage. The sockeye salmon data were drawn from a genetic baseline previously developed to document population structure and perform mixed stock analyses (Ackerman et al. 2011). In addition, a comparable dataset was available for Chinook salmon in the Copper River (Seeb et al. 2009). We used a subset of this dataset to evaluate how the landscape (including historical processes) may similarly or differentially shape the genetic structure of the two species. Results from each species were used to evaluate the interacting roles of landscape heterogeneity and putative local adaptation in determining genetic variation.
On a large scale, we first predict that isolation by distance will be significant for sockeye salmon because the exchange of genetic material between geographically proximate populations will be more likely to occur than among more distant populations. Second, we predict that an inland-to-coastal transition of within-population diversity (allelic richness) will occur that coincides with the transition in environments between the upper and middle/lower Copper River. Olsen et al. (2011) hypothesized, tested, and confirmed a similar pattern for Pacific salmon in neighboring regions. Third, we predict that sockeye and Chinook salmon will exhibit largely congruent patterns of hierarchical genetic structure, highlighting the importance that landscape heterogeneity and historical geologic processes play in determining the genetic structure in multiple species. And fourth, we predict that important putative adaptive differences between geographically proximate populations will be observed when incorporating high FST outlier SNPs into the hierarchical structure analyses.
Spatial and genetic data were analyzed to test these four predictions. We found that the spatial distribution of sockeye salmon populations in the Copper River drainage primarily determined their genetic structure. Sockeye salmon populations throughout the upper Copper River exhibited less within-population diversity and greater population structure than populations in the lower Copper River, likely resulting from greater reproductive isolation and historical founder effects. Further, high FST outlier SNPs revealed potential adaptive differences among geographically proximate sockeye salmon populations. Chinook salmon populations exhibited largely congruent patterns of hierarchical genetic structure demonstrating that landscape heterogeneity and historical processes similarly influence genetic variation in both species in the Copper River.
Methods
Genetic data for sockeye salmon within the Copper River drainage were drawn from Ackerman et al. (2011). The data for 45 SNP loci included three mitochondrial and 42 nuclear loci described in Elfstrom et al. (2006) and Habicht et al. (2010). Chinook salmon data were drawn from Seeb et al. (2009); data for 52 SNPs included one mitochondrial and 51 nuclear loci (Smith et al. 2005a, b, 2007; Narum et al. 2008). All collections were obtained between 2004 and 2009 (Table 1). During field collections, a GPS waypoint or a physical description of each location was taken to later generate a GIS shapefile of the populations in ArcGIS 9.3 (ESRI, Redlands, California).
Sockeye salmon SNPs were evaluated for within-population diversity and among-population divergence in an effort to differentiate SNPs exhibiting neutral variation versus SNPs exhibiting greater divergence than expected under neutral processes (high FST outlier SNPs). Ackerman et al. (2011) performed an FST outlier analysis involving populations from the Copper River and adjacent drainages in southern Alaska (Alsek, Bering, and Eyak rivers and Prince William Sound), but as this study is only interested in adaptation occurring in geographically proximate populations within the Copper River, the analysis was repeated using the smaller dataset. The expected heterozygosity (HE) of each nuclear locus was estimated using GENALEX 6.3 (Peakall and Smouse 2006) and the (Weir and Cockerham 1984) FST for each locus was estimated using FSTAT v2.9.3.2. Outlier SNPs were identified using a genome scan approach implemented in ARLEQUIN v3.5 (Excoffier and Lischer 2010). ARLEQUIN v3.5 uses coalescent simulations to obtain a null distribution of FST as a function of expected heterozygosities. We performed 20,000 simulations using 100 demes per group under the assumptions of a finite island model. Any SNPs that fell above the 99 % quantile were determined to be high FST outliers. We were only interested in high FST outliers; low FST outliers suggestive of balancing selection (Beaumont and Balding 2004) were not considered in our analyses. High FST outlier SNPs are similarly referred to as outlier SNPs hereafter. For Chinook salmon, outlier SNPs were identified from analyses conducted by Seeb et al. (2009).
Fluvial distance and FST/(1 − FST) are expected to follow a linear model when gene flow is geographically limited (Rousset 1997; Guillot et al. 2009). Seeb et al. (2009) showed that this relationship holds true for Chinook salmon in the Copper River drainage. We wanted to determine whether this relationship was also significant for sockeye salmon in the region using a Mantel test implemented in GENALEX 6.3. A significant isolation-by-distance relationship would indicate that the spatial distribution of populations largely determines the genetic population structure, or in other terms, that straying individuals are more likely to reproduce with individuals from neighboring populations than with individuals from more distant populations. One hundred permutations were performed to test the statistical significance of the pairwise matrix correlations (Sokal and Rohlf 1995). As the goal was to examine the dispersal ability or patterns of straying, outlier SNPs were eliminated from the isolation-by-distance analysis.
Decreases in allelic richness may be indicative of demographic isolation among populations or population bottlenecks (including founder effects). To examine for potential gradients of within-population diversity across the landscape for each species, we spatially interpolated the observed allelic richness of populations across the drainage using the ordinary kriging (Isaaks and Srivastava 1989) function in the geostatistical analyst toolbox in ArcGIS 9.3. Further, allelic richness was plotted as a function of upstream migration distance for each species. Upstream migration distance was measured as the fluvial distance from the mouth of the Copper River to each population’s spawning habitat and was measured using ArcGIS 9.3. Allelic richness for each population was calculated in FSTAT v2.9.3.2 (Goudet 1995, 2001). Each population’s allelic richness was calculated by averaging the allelic richness across loci. Mitochondrial and outlier SNPs were eliminated from allelic richness analyses.
SAMOVA v1.0 (Spatial Analysis of Molecular Variance; Dupanloup et al. 2002) was used to evaluate patterns of spatial genetic structure for each species. SAMOVA performs a series of AMOVA analyses to define population groups that are geographically proximate and genetically similar and that are maximally differentiated from each other using a simulated annealing process (Kirkpatrick et al. 1983). The goal is to minimize the amount of variation that occurs within groups (FSC) and maximize the amount of variation that occurs among groups (FCT). The number of groups to be defined (k) is a user-defined variable; we conducted analyses of k equals 2 through 10 for each species and examined patterns of FSC and FCT for each k to determine the appropriate number of population groups. The analysis was initially conducted for each species using exclusively neutral SNPs. Following, analyses were re-run incorporating outlier SNPs to determine if putative adaptive variation among populations changed population groups suggesting signals of adaptive divergence among geographically proximate populations. Because latitude and longitude data do not reflect the true spatial relationships in a fluvial system (Olsen et al. 2011), we calculated proxy x and y coordinates using a pairwise matrix of waterway distances between collections to project the populations on a multidimensional scale (MDS) using the program NTSYS (Rohlf 1994). The populations were spatially referenced using the first and second MDS values (Manni et al. 2004).
The program BARRIER v2.2 (Manni et al. 2004) was used to identify any barriers to gene flow on the landscape and to complement the SAMOVA analysis (Olsen et al. 2011). BARRIER uses Monmonier’s maximum difference algorithm (Monmonier 1973) to detect areas of abrupt change in population divergence along edges of a Delaunay triangulation (Delaunay 1934) connecting population pairs. Simulations suggest the Monmonier’s approach is better than the simulated annealing approach (SAMOVA) for identifying barriers, especially in cases where population divergence follows an isolation-by-distance model (Dupanloup et al. 2002; Manni et al. 2004, Olsen et al. 2011). The first and second MDS values used in the SAMOVA analysis were also used to create the Delaunay triangulation used in BARRIER. We used a matrix of residuals (based on the observed isolation-by-distance relationship) rather than the observed FSTs as suggested by Manni et al. (2004) to account for patterns of isolation-by-distance (Olsen et al. 2011). By using the residuals, the influence of isolation-by-distance is removed during the identification of barriers. To determine the robustness of identified barriers, 100 matrices of residuals were generated by bootstrapping across loci. Bootstrap sampling across loci and calculation of pairwise observed, expected, and residual FSTs were performed in R v2.9.2. R-scripts for bootstrapping residuals were written and provided by J. Olsen and J. Bromaghin (USFWS, Anchorage, Alaska). Similar to the SAMOVA analysis, the BARRIER analysis was first conducted for each species using only neutral SNPs to identify any ‘demographic barriers’. The analysis was then re-run incorporating outlier SNPs to determine if any additional ‘adaptive barriers’ were identified, suggestive of abrupt differences in allele frequencies at outlier SNPs. We recognize barriers if they were consistently identified with greater than 50 % bootstrap support.
Results
One sockeye salmon SNP (Ackerman et al. 2011) and 14 Chinook salmon SNPs (Seeb et al. 2009) were removed from analyses due to lack of variation in Copper River populations or because SNP pairs exhibited linkage disequilibrium. Multi-locus genotype data were obtained from Ackerman et al. (2011) for 28 sockeye salmon (n = 4,100) populations from throughout the Copper River drainage for 44 SNPs (Tables 1, 2; Fig. 1). Additionally, Seeb et al. (2009) provided data from 13 Chinook salmon (n = 1,184) populations for 38 SNPs (Tables 1, 2; Fig. 2). No data were available for Chinook salmon from the lower Copper River (below the confluence with the Chitina River) as there is little evidence of Chinook salmon spawning in the region (Savereide 2005).

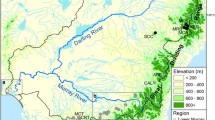
Gradient of allelic richness, population groups, and inferred barriers for Chinook salmon from the Copper River, Alaska. Population symbols indicate the groups defined by SAMOVA. Barriers indicate abrupt changes in allele frequencies transferred from the Delaunay triangulation in BARRIER to approximate locations on the map. Numbers inside the parentheses indicate the robustness of each barrier based on 100 bootstrap samples. Population numbers indicate Population ID in Table 1
Among the sockeye salmon SNPs, One_MHC2_190 and One_MHC2_251 exhibit linkage disequilibrium (Ackerman et al. 2011), but both were retained for analyses as each were high FST outliers. A composite phenotype for the two loci was used for both the SAMOVA and BARRIER analyses. The phenotype was generated by combining the two genotypes, ordering them alphabetically, and then assigning a numeric code to the resulting composite phenotype (Habicht et al. 2010).
Based on the FST outlier analysis, three sockeye salmon SNPs (One_GHII-2165, One_MHC2_190, and One_MHC2_251) exhibited greater among-population diversity (above 99 % quantile) than expected under neutral processes; we classified these as high FST outliers. For Chinook salmon, Seeb et al. (2009) identified one SNP (Ots_MHC-2) as a high FST outlier at the α = 0.05 level using the infinite-alleles model implemented in FDIST2 (M.A. Beaumont, University of Reading, UK; http://www.rubic.rdg.ac.uk/~mab/software.html). Figure 3a, b highlight outlier SNPs for both sockeye salmon and Chinook salmon, respectively.

FST as a function of heterozygosity (ARLEQUIN v3.5; Excoffier and Lischer 2010) for a 41 sockeye salmon SNPs and b 37 Chinook salmon SNPs. The dashed line represents the median and the solid lines represent the 99 % confidence interval boundaries based on coalescent simulations using a finite island model. Any SNPs that fell above the 99 % quantile were identified as high FST outliers
Both sockeye salmon (r2 = 0.241; p value <0.001) and Chinook salmon (r2 = 0.362; p value <0.001; Seeb et al. 2009) exhibited significant isolation by distance (Fig. 4). That is, according to the neutral variation observed among populations, gene flow was more likely to occur between geographically proximate populations than between more distant populations. For sockeye salmon, the linear relationship was FST/(1 − FST) = (0.00009 × rkm) + 0.0206 where rkm is the number of fluvial kilometers between a pair of populations. For Chinook salmon, the linear relationship was FST/(1 − FST) = (0.00008 × rkm) + 0.01. Pairwise residual matrices used in the BARRIER analyses were calculated based on the observed relationships.

Pairwise genetic divergence among populations as a function of fluvial distance for a sockeye salmon and b Chinook salmon from the Copper River, Alaska. Chinook salmon data adapted from Seeb et al. (2009)
Sockeye and Chinook salmon populations that spawn farther inland exhibited lesser allelic richness than populations that spawn nearer to the coast. Among sockeye salmon populations, the minimum allelic richness (1.61) was observed in the Fish Creek (8) population of the Gulkana River and the maximum (1.92) was observed in the Clear Creek at 40-mile (24) population in the lower Copper River (Table 1). Allelic richness decreased as upstream migration distance increased for populations in both species, although the relationships did not appear to be linear. In particular, a marked decrease in allelic richness was observed in populations >375 km upriver for both species (Fig. 5). Similarly, Seeb et al. (2009) showed that significant differences in allelic richness occurred among three geographic regions in Chinook salmon (upper Copper River < Gulkana River < lower Copper River) and that regions farther inland exhibited lesser allelic richness. Spatial patterns of allelic richness interpolated across the Copper River landscape (Figs. 1, 2) further supported these findings.

Allelic richness as a function of upstream migration distance for sockeye and Chinook salmon populations from the Copper River, Alaska
Nine sockeye salmon and six Chinook salmon population groups were suggested as appropriate by SAMOVA results when attempting to maximize among-group variation (FCT) and minimize within-group variation (FSC; Figs. 1, 2, respectively) using exclusively neutral SNPs. Although the FCT continued to increase slightly and the FSC continued to decrease slightly as the number of groups was increased for each species, the FCT largely plateaued and the FSC markedly decreased at these respective groupings (Table 3). Findings from the SAMOVA analysis reflected what would be expected based on the within-population diversity (allelic richness) analyses. Two large groups of sockeye salmon populations dominated the middle/lower Copper River region where greater within-population diversity was observed among populations. The two groups include; (1) seven populations that all spawn less than 150 rkm from the mouth of the Copper River (below the confluence of the Chitina and Copper Rivers) and (2) nine populations throughout the Klutina, Tonsina, and Chitina river drainages within the middle Copper River. Among the sockeye salmon populations that spawn >375 km upstream of the mouth of the Copper River that exhibit decreased allelic richness, we identified six population groups, none of which were larger than three populations. The final population group for sockeye salmon was a single population, Long Lake (20), that spawns in the Chitina River drainage. Among Chinook salmon populations, which all spawn above the confluence of the Chitina and Copper Rivers, we identified six population groups of which none were larger than four populations.
Results suggested that nine sockeye salmon and six Chinook salmon population groups were still appropriate after incorporating outlier SNPs into the SAMOVA analysis. For Chinook salmon, the population groups remained the same after Ots_MHC-2 was included with the neutral SNPs. However, two sockeye salmon populations changed groups, suggesting putative adaptive differences between geographically proximate populations. First, the Klutina Lake outlet (17) population that previously clustered with the large group encompassing the Klutina River, Tonsina River, and two populations from the Chitina River transferred to a group including the Keg Creek (6), East Fork Gulkana River—early run (9), and East Fork Gulkana River—late run (10) populations in the upper Copper River when incorporating outlier SNPs. Similarly, the Bear Island (19) population originally in the same large group as the Klutina Lake outlet population moved to a group with the Mendeltna Creek (11) population in the Tazlina River.
BARRIER results for both species largely corroborated findings from SAMOVA. For sockeye salmon, five barriers consistently occurred with greater than or equal to 50 % bootstrap support, and all barriers occurred between SAMOVA population groups (Fig. 1). Of the five barriers, four occurred in the upper Copper River above the 375 km transition area. The remaining barrier occurred below the Long Lake population identified as its own distinct population group in the Chitina River according to the SAMOVA analysis. Further, similar to the SAMOVA analysis, an additional ‘adaptive barrier’ was observed above the population that spawns in the mainstem Klutina River below Klutina Lake (17) when incorporating the outlier SNPs, separating itself from the five other populations that spawn in tributaries above Klutina Lake. For Chinook salmon, four barriers consistently occurred with greater than 50 % bootstrap support, and each of the barriers occurred between SAMOVA population groups (Fig. 2) one of which occurred near the 375 km region in the upper and the middle/lower Copper River ‘transition zone’ (see “Discussion” for explanation). After incorporating Ots_MHC-2 into the Chinook salmon barrier analysis, no additional ‘adaptive barriers’ were identified.
Discussion
Genetic differences among populations are inversely related to the exchange of genetic information, and under an isolation by distance model, the exchange of genetic information depends largely on the geographic proximity between populations. Decreases in gene flow owing to isolation by distance can result from two factors: (1) individuals are more likely to stray into the most geographically proximate populations (Olsen et al. 2008), and (2) barriers may limit gene flow in particular locations (Dupanloup et al. 2002; Guillot et al. 2009). In Pacific salmon, approximately 95–99 % of individuals home to their natal spawning habitat as adults; the remainder are termed ‘strays’ and may reproduce with non-natal populations (Quinn 2005). Generally, strays are more likely to reproduce with proximate populations than with more distant populations (Quinn 1993; Unwin and Quinn 1993). Further, the presence of in-stream velocity or ecological barriers has been shown to hinder the exchange of genetic material between proximate populations in Pacific salmon (Ramstad et al. 2004; Lin et al. 2008). For sockeye and Chinook salmon in the Copper River, the observed isolation by distance pattern appears to result from a combination of spatial patterns of straying and the presence of barriers to gene flow. Along a continuum, populations tend to be more similar to proximate populations than they are to more distant populations, suggesting that the exchange of genetic information is more common in geographically proximate populations. Also, in each species, abrupt changes in allele frequencies were also observed in geographically proximate populations, suggestive of barriers. In either regard, it appears that the spatial distribution of populations in the Copper River largely determines the distribution of hierarchical genetic variation in both species.
Three barriers identified in the sockeye and Chinook salmon BARRIER analyses show areas of abrupt changes in allele frequencies in proximate populations. For example, a barrier was detected (100 % bootstrap support) downstream of the sockeye salmon population in Fish Creek (8) in the Gulkana River (Fig. 1). The barrier was also supported by the SAMOVA analysis; the Fish Creek population formed its own distinct population group (Table 3). The Fish Creek population occurs in close proximity (~13 km) to two downstream populations, the East Fork Gulkana River—early (9) population and East Fork Gulkana River—late (10) population that are genetically similar to each other. However, the Fish Creek population appeared to be genetically distinct suggesting that little gene flow occurs between the two groups despite their close proximity.
Similarly, a barrier (94 % bootstrap support) was identified below the Long Lake (20) sockeye salmon population. The Long Lake population formed its own distinct population group (Table 3), despite the fact that the population spawns between two other populations in the Chitina River (Fig. 1), each of which were identified as part of the large middle Copper River population group. The Long Lake population is unique in that individuals reach the spawning grounds in August and September, similar to many other populations in the region, but have the longest known spawning duration (August through April) of any sockeye salmon population in North America, spawning through the winter and into spring during large returns (http://www.nps.gov/wrst/parkmgmt/long-lake-fish-weir.htm). The extended spawning duration in Long Lake is possible because the northwest corner of the lake rarely freezes; presumably due to a spring outlet in the area. In Pacific salmon, temporal differences in spawning between populations can be a major contributing factor in determining genetic population structure (O’Malley et al. 2007; Creelman et al. 2011). It must be noted that the Long Lake population was sampled in 2005, 3–4 years previous (approximately one generation) to a majority of sockeye salmon collections in the study (including the other two Chitina River collections). However, Ackerman et al. (2011) documented temporal genetic stability across approximately 4 generations in sockeye salmon populations from the lower Copper River; thus year of sampling likely does not explain the observed genetic structure in the Chitina River. Moreover, Gomez-Uchida et al. (2012) documented that temporal changes (across 25–42 years) were radically smaller than spatial changes in nuclear SNP allele frequencies in sockeye salmon populations from southwest Alaska. The genetic distinction of the Long Lake population is likely a result of its interannual temporal separation (spawn timing differences) from surrounding populations. This is in contrast to the general spatial pattern observed throughout the Copper River and highlights the role that differences in spawning timing can play in shaping genetic structure on a local scale.
Finally, we detected a barrier (97 % bootstrap support) downstream of the Mendeltna Creek Chinook salmon population (9) located in close proximity (~12 km) to the Kaina Creek population (10). The Kaina Creek population was in a group that included two populations from the adjacent Klutina and Tonsina rivers according to the SAMOVA analysis. The three above mentioned barriers highlight the contribution of in-stream barriers, including ‘temporal barriers’, to the observed isolation by distance pattern in each species.
Both sockeye and Chinook salmon exhibited a similar inland-to-coastal gradient of within-population diversity; populations in the upper Copper River had less within-population diversity than populations from the middle and lower Copper River. Similar patterns have been observed in Pacific salmon species elsewhere in subarctic North America (Habicht et al. 2004; Olsen et al. 2011). In our study, populations >375 km upriver showed a marked decrease in allelic richness (Fig. 5) and generally exhibited lower expected heterozygosity (Table 1). The transition in allelic richness observed in both species coincides with the confluence of the Tazlina and Copper rivers ‘transition zone’ where the Copper River drainage transitions from a primarily tundra environment in the upper drainage to a more forested, mountainous environment in the middle/lower Copper River. An example of the influence of this transition zone are the sockeye (11) and Chinook (9) salmon populations that spawn in Mendeltna Creek within the Tazlina River drainage; each of these populations formed its own discrete population group in the SAMOVA analyses. Results from the among-population analyses conducted in SAMOVA and BARRIER support the observed transition zone, particularly in sockeye salmon. Sockeye salmon populations above the transition zone exhibited greater hierarchical population structure and four of the five barriers identified using neutral SNPs were located in the upper Copper River. Populations of both species exhibited decreased within-population diversity above the transition zone (Fig. 5).
The lower within-population diversity of salmon populations in Mendeltna Creek and the upper Copper River may be attributable to two primary factors: (1) influence from genetic drift due to smaller, geographically isolated populations (Olsen et al. 2011) and (2) founder effects (Ramstad et al. 2004; Olsen et al. 2011). Above the transition area, populations tend to be geographically isolated relative to populations below (Wade et al. 2008) promoting decreased gene flow and increased genetic drift. However, historic processes contributing to founder effects also likely contribute to the decreased within-population diversity. Sockeye salmon populations in the upper Copper River may have been founded by individuals that persisted in a northern glacial refuge (Lindsey and McPhail 1986). During the height of the most recent glaciation, known as the McConnell/McCauley Glaciation, ice-free areas persisted north of the ice in regions near the present day upper Alsek, Copper, Mackenzie and Yukon river drainages (Smith et al. 2001). Although it is unclear whether sockeye and Chinook salmon occupied the region during the glaciations, Smith et al. (2001) provide evidence that coho salmon (Oncorhynchus kisutch) populations may have persisted in the region in small numbers. Olsen et al. (2011) suggest that individuals from the same refugia may have once founded Pacific salmon populations in the upper Yukon River owing to decreased within-population diversity within that region. If sockeye and Chinook salmon individuals were able to persist in the refugia, populations in the upper Copper River may have been founded by individuals from the northern glacial refuge as ice receded. Similarly, as ice receded north, populations in the upper Copper River may have been founded by strays from the middle/lower Copper River or coastal populations. In either regard, the observed decrease in within-population diversity in salmon populations in the upper Copper River is likely a result of a combination of contemporary (gene flow and genetic drift) and historical (founder effects) demographic processes.
Analyses conducted by Seeb et al. (2009) for Chinook salmon in the Copper River using STREAMTREE (Kalinowski et al. 2008) supported the BARRIER results reported in this study. Seeb et al. (2009) used STREAMTREE to map (Cavalli-Sforza and Edwards 1967) chord distances onto sections of the Copper River drainage connecting populations within the drainage. The two stream reaches identified by Seeb et al. (2009) as having the greatest genetic discontinuities were identified by us (barriers 1 and 4) with greater than 50 % bootstrap support. Further, the remaining two barriers we identified (barriers 2 and 3) were also located in areas identified by Seeb et al. (2009) as having increased chord distances relative to surrounding stream reaches. Finally, Seeb et al. (2009) noted increased chord distances in the transition area above the confluence of the Tazlina and Copper rivers; reaches of the Copper River above the Tazlina and the lower Gulkana River each showed genetic discontinuities. Both STREAMTREE and BARRIER proved to be useful for identifying putative barriers to gene flow.
We identified interesting putative adaptive differences in sockeye salmon in the Klutina River drainage when incorporating outlier SNPs into SAMOVA and BARRIER analyses. The sockeye salmon population that spawns in the mainstem Klutina River below the outlet of Klutina Lake (17) grouped with populations throughout the middle Copper River (including five populations that spawn in tributaries above Klutina Lake) when using exclusively neutral SNPs. However, the mainstem Klutina River population changed groups and joined three populations from the Gulkana River drainage (Fig. 1) when outlier SNPs were incorporated. An additional ‘adaptive barrier’ was identified above this population using BARRIER. The potential adaptive differences observed between the population below Klutina Lake and the five populations that spawn in tributary habitats above Klutina Lake are primarily due to allele frequency differences observed at One_MHC2_190 and One_MHC2_251. Interestingly, the Klutina Lake outlet population may be of the sea/river ecotype as defined by Wood et al. (2008) whereas the five populations spawning above Klutina Lake are presumably of the lake ecotype (Ackerman et al. 2011). The lake ecotype is the typical form of sockeye salmon that spends about half its life in a nursery lake before emigrating to the marine environment to mature, whereas the sea/river ecotype is a rarer form that rears in the freshwater environment for a much shorter and more variable period of time. Large differences in MHC allele frequencies between geographically proximate populations of the two ecotypes have been observed elsewhere throughout the range of sockeye salmon (Creelman et al. 2011; Gomez-Uchida et al. 2011; McGlauflin et al. 2011). The two sockeye salmon MHC SNPs are found in one exon (One_MHC2_190) and one intron (One_MHC2_251) within a Major Histocompatibility Complex (MHC) class II gene (Miller and Withler 1996; Miller et al. 2001; Limborg et al. 2011). Several studies support the adaptive nature of MHC polymorphisms related to pathogen-mediated selection (Dionne et al. 2009; Evans and Neff 2009; McGlauflin et al. 2011; Gomez-Uchida et al. 2011). Differences in the spawning and rearing habitats (i.e. temperature regimes, flow velocity) between the two ecotypes may provide a mechanism for the adaptive differences observed among populations occurring in the Klutina River drainage.
Defining a locus as a high FST outlier generally requires obtaining good estimates of genome-wide null distributions of FST as a function of heterozygosity (Excoffier et al. 2009). FST outlier analyses performed in this study for sockeye salmon were based on 41 SNPs and for Chinook salmon were based on 37 SNPs. Although the authors acknowledge that the SNP sets evaluated here for both species provide less than ideal genome wide coverage and that results based on outlier SNPs should be interpreted with caution, we believe that evaluation of results is merited. FST outlier analyses using FDIST2 and ARLEQUIN v3.5 have been shown to be effective in detecting high FST outliers using simulated datasets with relatively low number of loci (i.e. 100 SNPs, Narum and Hess 2011). Further, the contradicting results between analyses based on exclusively neutral SNPs and analyses based on both neutral and outlier SNPs in this study are largely driven by the 3 MHC SNPs screened in this study (2 for sockeye salmon, 1 for Chinook salmon; Fig. 3). As noted above, the adaptive nature of MHC markers associated with pathogen-mediated selection in salmon has been well documented.
Identifying the correct number of groups (k) in the SAMOVA analysis was partially subjective as there was not a clear drop in FSC or a marked plateau in FCT, particularly for the sockeye salmon dataset. Unlike other commonly used clustering programs (i.e. BAPS Corander et al. 2008 or STRUCTURE Pritchard et al. 2000), SAMOVA does not assign a likelihood value to each model that could be used in model selection. Thus, there is no objective “optimal k”. For instance, examining Table 3 for the sockeye salmon dataset, an argument could be made for k = 7 and k = 5a as the optimal values. Nine population groups for the neutral dataset were chosen because the among-group variation (FCT) reached a maximum at k = 9; however, a greater decrease in within-group variation (FSC) occurred as k was increased from 6 to 7 than when k was increased from 8 to 9. For the dataset combining both neutral and outlier SNPs, a similar pattern occurred in which the decrease in FSC going from 4a to 5a was greater than occurred between 8a and 9a. However, when increasing from k = 4a to 5a, FCT actually decreased. Thus, 9a was chosen as the optimal k in lieu of 5a when incorporating outlier SNPs. The basis for the selection of 9 and 9a as the ‘optimal k’ for sockeye salmon was due to placing particular emphasis in identifying a maximum or plateau in FCT for identifying the correct number of groups (Dupanloup et al. 2002). Interestingly, the barriers identified in the complimentary BARRIER analysis emerged primarily at lower values of k (namely k = 7 and 7a), lending support to lower values of k; the appropriate groupings from SAMOVA were determined a priori to BARRIER analyses. For clustering analyses such as SAMOVA and other similar software packages, it can be informative to evaluate how population groups ‘decompose’ as k is increased, rather than simply determining an optimal k. For example, during the transition from k = 6–7 in the neutral dataset, the Slana River populations [1–3] emerged as a population group; when transitioning from k = 8–9, the Mendeltna Creek population [11] emerged as a population group. These results may suggest that Slana River populations are more genetically distinct than the Mendeltna Creek population. By mapping how population groups decompose as k is increased over time, patterns of differentiation may be inferred. Table 3 shows that populations from the upper Copper River (and Long Lake) tend to ‘separate’ or ‘decompose’ at lower values of k, corroborating the patterns of increased among-population differentiation and decreased within-population diversity observed.
The description of conservation units has previously been criticized for the overemphasis of reproductive isolation based on information from loci assumed to be neutral and the resulting under-emphasis of adaptive variation among populations (Allendorf and Luikart 2007). The developing field of landscape genomics aims to include large numbers of loci exhibiting both neutral and adaptive variation to not only make inferences regarding gene flow and drift, but also to tease apart information regarding evolutionary forces and local adaptation influencing natural populations (Schwartz et al. 2009; Limborg et al. 2011). Although we do not suggest that the methods presented here constitute a landscape genomics analysis, we do emphasize that the putative adaptive differences observed among the Klutina Lake sockeye salmon populations highlight the role of local adaptation in determining genetic variation. In the past, landscape geneticists have largely viewed loci under selection as nuisances that should be avoided (Schwartz et al. 2009), but we show that incorporating outlier loci into landscape genetics analyses can provide valuable insight into adaptive processes. Variation at outlier loci should be recognized when planning conservation efforts (Limborg et al. 2011).
We identified largely congruent patterns of spatial genetic structure between sockeye and Chinook salmon in the Copper River, emphasizing the role that landscape heterogeneity plays in determining spatial patterns of genetic variation among the closely related species. On a large scale, both species exhibited significant patterns of isolation by distance demonstrating that the spatial distribution of populations was the primary determinant of the genetic structure in each species. Further, inland populations of both species exhibited decreased within-population diversity resulting from a combination of reproductive isolation and historical processes including founder effects. On a smaller scale, temporal differences observed in the Chitina River sockeye salmon populations and putative adaptive differences observed in the Klutina River sockeye salmon populations emphasized the role that differences in spawning timing and local adaptation can play in determining genetic structure. Our results demonstrate that including genetic divergence at outlier loci together with divergence at neutral loci can enhance and help determine priorities when identifying units of conservation.
References
Ackerman MW, Habicht C, Seeb LW (2011) SNPs under diversifying selection provide increased accuracy and precision in mixed stock analyses of sockeye salmon from Copper River, Alaska. Trans Am Fish Soc 140(3):865–881
Allendorf FW, Luikart G (2007) Conserving global biodiversity? Conservation and the genetics of populations. Blackwell Publishing, Oxford
Beaumont MA, Balding DJ (2004) Identifying adaptive genetic divergence among populations from genome scans. Mol Ecol 13(4):969–980
Behnke RJ, Tomelleri JR (2002) Trout and salmon of North America. Free Press, New York
Cavalli-Sforza LL, Edwards AWF (1967) Phylogenetic analysis: models and estimation procedures. Evolution 21(3):550–570
Corander JJ, Sirén J, Arjas E (2008) Bayesian spatial modeling of genetic population structure. Comp Stat 23:111–129
Creelman EL, Hauser L, Seeb LW, Simmons R, Templin WD (2011) Temporal and geographic genetic divergence: characterizing sockeye salmon populations in the Chignik watershed, Alaska, using single nucleotide polymorphisms. Trans Am Fish Soc 140(3):749–762
Delaunay B (1934) Sur la sphere vide. Bulletin Acad Sci USSR 7:793–800
Dionne MK, Miller KM, Dodson JJ, Bernatchez L (2009) MHC standing genetic variation and pathogen resistance in wild Atlantic salmon. Philos Trans R Soc B Biol Sci 364(1523):1555–1565
Dittman AH, Quinn TP (1996) Homing in Pacific salmon: mechanisms and ecological basis. J Exp Biol 199(1):83–91
Dupanloup I, Schneider S, Excoffier L (2002) A simulated annealing approach to define the genetic structure of populations. Mol Ecol 11(12):2571–2581
Elfstrom CM, Smith CT, Seeb JE (2006) Thirty-two single nucleotide polymorphism markers for high-throughput genotyping of sockeye salmon. Mol Ecol Notes 6(4):1255–1259
Evans ML, Neff BD (2009) Major histocompatibility complex heterozygote advantage and widespread bacterial infections in populations of Chinook salmon (Oncorhynchus tshawytscha). Mol Ecol 18:4716–4729
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Res 10(3):564–567
Excoffier L, Hofer T, Foll M (2009) Detecting loci under selection in a hierarchically structure population. Heredity 103:285–298
Gagnon MC, Angers B (2006) The determinant role of temporary proglacial drainages on the genetic structure of fishes. Mol Ecol 15(4):1051–1065
Gomez-Uchida D, Knight TW, Ruzzante DE (2009) Interaction of landscape and life history attributes on genetic diversity, neutral divergence and gene flow in a pristine community of salmonids. Mol Ecol 18(23):4854–4869
Gomez-Uchida D, Seeb JE, Smith MF, Habicht C, Quinn TP, Seeb LW (2011) Single nucleotide polymorphisms unravel hierarchical divergence and signatures of selection among Alaskan sockeye salmon (Oncorhynchus nerka) populations. BMC Evol Biol 11:48
Gomez-Uchida D, Seeb JE, Habicht C, Seeb LW (2012) Allele frequency stability in large, wild exploited populations over multiple generations: insights from Alaska sockeye salmon (Oncorhynchus nerka). Can J Fish Aquat Sci 69:916–929
Goudet J (1995) FSTAT (Version 1.2): a computer program to calculate F-statistics. J Hered 86(6):485–486
Goudet J (2001) FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3). Available at http://www.unil.ch/izea/softwares/fstat.html
Guillot G, Leblois R, Coulon A, Frantz AC (2009) Statistical methods in spatial genetics. Mol Ecol 18(23):4734–4756
Habicht C, Olsen JB, Fair L, Seeb JE (2004) Smaller effective population sizes evidenced by loss of microsatellite alleles in tributary-spawning populations of sockeye salmon from the Kvichak River, Alaska drainage. Environ Biol Fish 69(1–4):51–62
Habicht C, Seeb LW, Myers KW, Farley E, Seeb JE (2010) Summer-fall distribution of stocks of immature sockeye salmon in the Bering Sea as revealed by single nucleotide polymorphisms (SNPs). Trans Am Fish Soc 139(4):1171–1191
Hard JJ (1995) A quantitative genetic perspective on the conservation of intraspecific diversity. In: Nielsen JL (ed) Evolution and the aquatic ecosystem: defining unique units in population conservation. American Fisheries Society symposium 17, Bethesda, pp 304–326
Isaaks EH, Srivastava RM (1989) An introduction to applied geostatistics. Oxford University Press, New York
Kalinowski ST, Meeuwig MH, Narum SR, Taper ML (2008) Stream trees: a statistical method for mapping genetic differences between populations of freshwater organisms to the sections of streams that connect them. Can J Fish Aquat Sci 65(12):2752–2760
Kirkpatrick S, Gelatt CD, Vecchi MP (1983) Optimization by simulated annealing. Science 220(4598):671–680
Limborg MT, Blankenship SM, Young SF, Utter FM, Seeb LW, Hansen MHH, Seeb JE (2011) Signatures of natural selection among lineages and habitats in Oncorhynchus mykiss. Ecol Evol 2(1):1–18
Lin J, Quinn TP, Hilborn R, Hauser L (2008) Fine-scale differentiation between sockeye salmon ecotypes and the effect of phenotype on straying. Heredity 101(4):341–350
Lindsey CC, McPhail JD (1986) Zoogeography of fishes of the Yukon and Mackenzie basins. In: Hocutt CH, Wiley EO (eds) The zoogeography of North American freshwater fishes. Wiley, New York, pp 639–674
Manel S, Schwartz MK, Luikart G, Taberlet P (2003) Landscape genetics: combining landscape ecology and population genetics. Trends Ecol Evol 18(4):189–197
Manni F, Guerard E, Heyer E (2004) Geographic patterns of (genetic, morphologic, linguistic) variation: how barriers can be detected by using Monmonier’s algorithm. Hum Biol 76(2):173–190
Marten A, Brandle M, Brandl R (2006) Habitat type predicts genetic population differentiation in freshwater invertebrates. Mol Ecol 15(9):2643–2651
McGlauflin M, Schindler D, Seeb LW, Smith CT, Habicht C, Seeb JE (2011) Spawning habitat and geography influence population structure and juvenile migration timing of sockeye salmon in the Wood River Lakes, Alaska. Trans Am Fish Soc 140(3):763–782
Miller KM, Withler RE (1996) Sequence analysis of a polymorphic Mhc class II gene in Pacific salmon. Immunogenetics 43(6):337–351
Miller KM, Kaukinen KH, Beacham TD, Withler RE (2001) Geographic heterogeneity in natural selection on an MHC locus in sockeye salmon. Genetica 111(1–3):237–257
Monmonier MS (1973) Maximum-difference barriers: an alternative numerical regionalization method. Geogr Anal 5(3):245–261
Narum SR, Hess JE (2011) Comparison of FST outlier tests for SNP loci under selection. Mol Ecol Res 11(Suppl. 1):184–194
Narum SR, Banks M, Beacham TD, Bellinger MR, Campbell MR, Dekoning J, Elz A, Guthrie CM, Kozfkay C, Miller KM, Moran P, Phillips R, Seeb LW, Smith CT, Warheit K, Young SF, Garza JC (2008) Differentiating salmon populations at broad and fine geographical scales with microsatellites and single nucleotide polymorphisms. Mol Ecol 17(15):3464–3477
Olsen JB, Flannery BG, Beacham TD, Bromaghin JF, Crane PA, Lean CF, Dunmall KM, Wenburg JK (2008) The influence of hydrographic structure and seasonal run-timing on genetic diversity and isolation-by-distance in chum salmon (Oncorhynchus keta). Can J Fish Aquat Sci 65(9):2026–2042
Olsen JB, Crane PA, Flannery BG, Dunmall K, Templin WD, Wenburg JK (2011) Comparative landscape genetic analysis of three Pacific salmon species from subarctic North America. Conserv Genet 12:223–241
O’Malley KG, Camara MD, Banks MA (2007) Candidate loci reveal genetic differentiation between temporally divergent migratory runs of Chinook salmon (Oncorhynchus tshawytscha). Mol Ecol 16(23):4930–4941
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6(1):288–295
Pearman PB (2001) Conservation value of independently evolving units: sacred cow or testable hypothesis? Conserv Biol 15(3):780–783
Petren K, Grant PR, Grant BR, Keller LF (2005) Comparative landscape genetics and the adaptive radiation of Darwin’s finches: the role of peripheral isolation. Mol Ecol 14(10):2943–2957
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Quinn TP (1993) A review of homing and straying of wild and hatchery-produced salmon. Fish Res 18(1–2):29–44
Quinn TP (2005) The behavior and ecology of Pacific salmon and trout. American Fisheries Society and University of Washington Press, Bethesda and Seattle
Ramstad KM, Woody CA, Sage GK, Allendorf FW (2004) Founding events influence genetic population structure of sockeye salmon (Oncorhynchus nerka) in Lake Clark, Alaska. Mol Ecol 13(2):277–290
Rohlf FJ (1994) NTSYS-pc. Numerical taxonomy system for cluster and ordination analyses. Version 2.2. Exeter Software, Setauket, New York
Rousset F (1997) Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145(4):1219–1228
Savereide JW (2005) Inriver abundance, spawning distribution, and run timing of Copper River Chinook salmon, 2002–2004 final report for study 02-015. U.S. Fish and Wildlife Service, Office of Subsistence Management, Fishery Information Service Division, Anchorage
Schwartz MK, Luikart G, McKelvey KS, Cushman SA (2009) Landscape genomics: a brief perspective. In: Cushman SA, Huettmann F (eds) Spatial complexity, informatics, and wildlife conservation. Springer, Berlin
Seeb LW, DeCovich NA, Barclay AW, Smith CT, Templin WD (2009) Timing and origin of Chinook salmon stocks in the Copper River and adjacent coastal fisheries using DNA markers. Annual report for study 04-207. UWFWS Office of Subsistence Management, Fisheries Resource Monitoring Program. Alaska Department of Fish and Game, Fishery Data Series No. 09-58, Anchorage
Short AEZ, Caterino MS (2009) On the validity of habitat as a predictor of genetic structure in aquatic systems: a comparative study using California water beetles. Mol Ecol 18(3):403–414
Smith CT, Nelson RJ, Wood CC, Koop BF (2001) Glacial biogeography of North American coho salmon (Oncorhynchus kisutch). Mol Ecol 10(12):2775–2785
Smith CT, Elfstron CM, Seeb LW, Seeb JE (2005a) Use of sequence data from rainbow trout and Atlantic salmon for SNP detection in Pacific salmon. Mol Ecol 14(13):4193–4203
Smith CT, Seeb JE, Schwenke P, Seeb LW (2005b) Use of the 5′-nuclease reaction for single nucleotide polymorphism genotyping in Chinook salmon. Trans Am Fish Soc 134(1):207–217
Smith CT, Antonovich A, Templin WD, Elfstron CM, Narum SR, Seeb LW (2007) Impacts of marker class bias relative to locus-specific variability on population inferences in Chinook salmon: a comparison of single-nucleotide polymorphisms with short tandem repeats and allozymes. Trans Am Fish Soc 136(6):1674–1687
Sokal RR, Rohlf FJ (1995) Biometry. Freeman, San Francisco
Storfer A, Murphy MA, Evans JS, Goldberg CS, Robinson S, Spear SF, Dezzani R, Delmelle E, Vierling L, Waits LP (2007) Putting the ‘landscape’ in landscape genetics. Heredity 98(3):128–142
Taylor EB (1991) A review of local adaptation in salmonidae, with particular reference to Pacific and Atlantic salmon. Aquaculture 98(1–3):185–207
Unwin MJ, Quinn TP (1993) Homing and straying patterns of Chinook salmon (Oncorhynchus tshawytscha) from a New Zealand hatchery: spatial distribution of strays and effects of release date. Can J Fish Aquat Sci 50(6):1168–1175
Wade GD, Smith JJ, van den Broek KM, Savereide JW (2008) Spawning distribution and run timing of Copper River sockeye salmon, 2007 final report for study 05-501. U.S. Fish and Wildlife Service, Office of Subsistence Management, Fisheries Resource Monitoring Program, Anchorage
Waples RS (1987) A multispecies approach to the analysis of gene flow in marine shore fishes. Evolution 41(2):385–400
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38(6):1358–1370
Wood CC, Bickham JW, Nelson RJ, Foote CJ, Patton JC (2008) Recurrent evolution of life history ecotypes in sockeye salmon: implications for conservation and future evolution. Evol Appl 1(2):207–221
Acknowledgments
This project was a collaborative effort between the University of Washington, School of Aquatic and Fishery Sciences (UW SAFS) and the Alaska Department of Fish and Game (ADFG), Gene Conservation Laboratory. Special thanks to Daniel Schindler, University of Washington, for insightful contributions that greatly improved the manuscript. We thank Christian Smith for his valuable contribution to the Chinook salmon dataset. Thanks to Jon Hess for ideas on evaluating genetic data using ArcGIS. We are grateful for laboratory contributions from staff at UW SAFS and ADFG. This report was partially funded by the Alaska Sustainable Salmon Fund under Study #45869 from the National Oceanic and Atmospheric Administration, U.S. Department of Commerce, administered by the Alaska Department of Fish and Game. The statements, findings, conclusions, and recommendations are those of the authors and do not necessarily reflect the views of the National Oceanic and Atmospheric Administration, the U.S. Department of Commerce, or the Alaska Department of Fish and Game. Additional project funding was provided by the Gordon and Betty Moore Foundation and the State of Alaska. Funding for MWA was provided by the H. Mason Keeler Endowment for Excellence (UW).
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Data accessibility: Sockeye salmon SNP genotype data are deposited at Dryad (doi:10.5061/dryad.ng93k). Chinook salmon SNP genotype data are deposited at Dryad (doi:10.5061/dryad.8063).
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Ackerman, M.W., Templin, W.D., Seeb, J.E. et al. Landscape heterogeneity and local adaptation define the spatial genetic structure of Pacific salmon in a pristine environment. Conserv Genet 14, 483–498 (2013). https://doi.org/10.1007/s10592-012-0401-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-012-0401-7