
Abstract
Chronic cerebral ischaemia (CCI) is a high-incidence cardiovascular and cerebrovascular disease that is very common in clinical practice. Although many pathogenic mechanisms have been explored, there is still great controversy among neuroscientists regarding the pathogenesis of CCI. Therefore, it is important to elucidate the mechanisms of CCI occurrence and progression for the prevention and treatment of ischaemic cerebrovascular disorders. Autophagy and inflammation play vital roles in CCI, but the relationship between these two processes in this disease remains unknown. Here, we review the progression and discuss the functions, actions and pathways of autophagy and inflammation in CCI, including a comprehensive view of the transition from acute disease to CCI through ischaemic repair mechanisms. This review may provide a reference for future research and treatment of CCI.
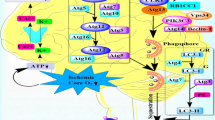
Graphical Abstract
Schematic diagram of the interplay between autophagy and inflammation in CCI. CCI lead to serious, life-threatening complications. This review summarizes two factors in CCI, including autophagy and inflammation, which have been focused for the mechanisms of CCI. In short, the possible points of intersection are shown in the illustration. CCI, Chronic cerebral ischaemia; ER stress, Endoplasmic reticulum stress; ROS, Reactive oxygen species.

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ischemic cerebrovascular disease is mainly caused by system hypoperfusion, cerebral vascular stenosis, thrombosis or embolism (Caplan 2006), including cerebral infarction (CI) and chronic cerebral ischaemia (CCI). CI is also called ischemic stroke. It occurs when a blood vessel in the brain becomes blocked (Sherman et al. 2021), which leads to hypoxic-ischemic necrosis of brain tissue, resulting in clinically corresponding neurological defects. CCI, which mostly occurs in elderly individuals, refers to a decrease in the overall blood supply to the brain (Alia et al. 2017). When this condition is not cured, a series of pathological changes can be observed in brain tissue over time, such as changes in glial activity, inflammation and the degeneration of neurons (Du et al. 2017). The pathological process of CCI damage is complicated and includes excitotoxicity, calcium excess, oxidative stress, cell death, necrosis, autophagy, and inflammation.
Inflammation is a protective reaction in the body and an immune response in tissues to various stimuli, including microbial pathogens and endogenous molecules. However, long-term chronic inflammation can cause damage due to high concentrations of inflammatory mediators in exposed tissues. As a vital mechanism related to the pathophysiological process, inflammation has an important function in the onset and progression of CCI (Deng et al. 2018; Yang et al. 2017b). An FDA-approved medicine alleviates spatial memory deficits in CCH rats, which may be linked to anti-inflammatory impacts (Yan et al. 2021). By taking part in the disposal of injured organelles and denatured proteins and degrading pathogens through the lysosomal pathway, autophagy functions to preserve homeostasis in cells. Increasing evidence has revealed that autophagy also plays a role in CCI (Wang et al. 2017).
Autophagy and inflammation are considered to be interdependent processes. Recently, autophagy-dependent mechanisms have been associated with the onset of a number of inflammatory disorders, including infectious disorders, Crohn’s disease, pulmonary hypertension, and malignancy (Ceccariglia et al. 2020; Cosin-Roger et al. 2017; Lee et al. 2018; Schlemmer et al. 2018; Sorbara et al. 2013; Tang et al. 2018). These studies indicate that there are some unclear crosstalk mechanisms between autophagy and the inflammatory signalling cascade. Although some progress has been made in elucidating the function of autophagy in inflammation, the molecular mechanism of the reaction between autophagy and inflammation in CCI are still relatively poorly understood. Inflammation and autophagy may be critical players in CCI, which causes neuronal loss and vascular cognitive impairment (Wang et al. 2020). In this review, we analysed the connection between autophagy and inflammation in the pathogenesis of CCI to provide novel insights into CCI.
Relationship Between Inflammation and CCI
Inflammation is Involved in the Repair Process of CCI
During cerebral ischaemia, the brain responds to the injury and triggers an inflammatory cascade response by releasing harmful materials, particularly necrotic cell debris, which lead to not only subsequent tissue injury but also the repair process (Li et al. 2022). In addition, subsequent regeneration can be triggered in tissues with secondary injury signals, such as free radicals and inflammatory cytokines (Carmichael 2016). This finding shows that damage and repair processes occur not only in the local stroke site but also in the neuronal connection network at the stroke site. This means that damage signals can be transmitted through the brain network. These transmitted injury signals can increase reactive astrocytes and inflammation in distant brain regions, leading to the formation of new connections. The inflammatory cytokine TNF is a valuable and useful neuroprotective factor in the acute stage, and it may serve as a regulator of neuroinflammation after cerebral ischaemia, which is beneficial in the recovery of cerebral ischaemia (Clausen et al. 2016), suggesting that inflammation is one of the repair mechanisms of CCI.
Inflammation Involved in CCI Injury
Recently, evidence has shown that stroke infarcts are zones of chronic inflammation (Zbesko et al. 2018). Important variables that influence the onset and development of inflammatory responses are inflammatory mediators. Multiple inflammatory mediators have been revealed to play significant roles in the inflammatory reaction to CCI. The chronic phase is the main stage of cognitive dysfunction caused by chronic cerebral hypoperfusion (CCH). CCH is associated with highly activated inflammatory pathways and inflammation-mediated effects, which induce neuroinflammation and lead to white matter lesions, neuronal loss, and learning and memory disorders. Therefore, we should pay more attention to chronic inflammation in CCH models (Sattayakhom et al. 2022).
Yoshizaki et al. (2008) found that interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumour necrosis factor-α (TNF-α) levels were substantially upregulated in the ischaemic hemisphere at 2 h after surgery in a CCH model, and monocyte chemotactic protein-1 (MCP-1), IL-6, and TNF-α were also detected at 24 h after surgery; the anti-inflammatory cytokines IL-4 and IL-10 were not altered until 3 days following occlusion (Kitamura et al. 2012). After CCI, the level of TNF-α in serum and brain tissue was increased, and the production of IL-6 was also elevated. Inhibiting the production of the proinflammatory cytokines TNF-α and IL-6 may protect against cerebral ischaemia (Pan et al. 2012). These findings reveal that IL-4 and IL-10 deserve attention as anti-inflammatory cytokines in CCI.
The matrix metalloproteinase (MMP) family, which includes important inflammatory mediators, is the most important family in chronic hypoperfusion white matter lesions. MMP-2 and MMP-9 belong to the MMP family, are present in blood vessels, participate in vascular remodelling and have a vital function in the pathophysiology of CCH. Upregulation of MMP-2 and MMP-9 can promote cerebral ischaemia post-functional recovery (Wang et al. 2012). This effect may occur because MMP-2 promotes angiogenesis and compensation during CCH (Corbin et al. 2014).
Inflammation, as measured by VCAM-1 levels, may have an impact on poststroke cognitive problems (El Husseini et al. 2020). In addition, intercellular cell adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) are substantially increased in cerebral vascular endothelial cells in a permanent bilateral vessel occlusion (2-VOS) animal model, and the upregulation of VCAM-1 expression is the initial link between the inflammatory response and adhesion, suggesting that CCI can induce a microvascular inflammatory response and participate in CCI-induced cognitive impairment (Huang et al. 2010).
Activation of Related Inflammatory Pathways
TLRs and the NF-κB Signalling Pathway
Toll-like receptors (TLRs), a class of pattern-recognizing receptors, trigger inflammatory reactions and are expressed by astrocytes, microglia, and neurons. In the central nervous system (CNS), TLRs can recognize multiple types of pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) (Kawabori and Yenari 2015). PAMPs can bind to TLRs, activate receptors and initiate an immune response via the signalling cascade, further leading to the expression of effector molecules. The cascade mobilization of Toll/IL-1R (TIR) adapter proteins is primarily comprised MyD88, NF-κB-inducing kinase (NIK), and IκB kinase (IKK), which stimulate the downstream molecule NF-κB to join the nucleus and initiate the generation of proinflammatory cytokines, such as TNF-α, IL-1, and IL-12 (Zhao et al. 2020). TLRs rely on microglia in the ischaemic core area rather than astrocytes to produce ATP-dependent IL-1β in the CNS during cerebral ischaemia (Facci et al. 2014; Hao et al. 2016).
Recent studies have revealed that TLR4 triggers an inflammatory reaction in CCI and has a vital role in initiating glial activation in cerebral ischaemia‒reperfusion damage (Wang et al. 2016a). The inhibition of TLR4-NOX4 signalling significantly reduces the infarct volume after cerebral infarction and reperfusion and improves neurological functional scores, demonstrating that TLR4 represents a possible therapeutic target for CCI (Suzuki et al. 2012). Downregulation of TLR4 and its downstream signalling factor MyD88 has a neuroprotective impact against cerebral ischaemia (Sun et al. 2015). These findings suggest that the TLR4/MyD88/NF-κB may be the main signalling pathway that regulates the glial inflammatory response.
MAPK Signalling Pathway
Mitogen-activated protein kinase (MAPK) signalling, consisting of p38, ERK (extracellular signal-regulated kinase) and JNK (N-terminal kinase), plays a crucial role in glial cell activation. In a model of CCH, AMP-activated protein kinase (AMPK) and the Ca(2+)/calmodulin-dependent protein kinase β (CaMKKβ)-dependent signalling pathway, upstream of MAPK, were also implicated in the activation of microglia (Green et al. 2011; Yuan et al. 2017). Hypoxia increased MCP-1 expression, which activated microglia and the p38MAPK/PKC pathways, which are involved in the process of cognitive deficits produced by CCH (Li et al. 2016). Inhibiting p38 MAPK and MAPK2 (MK2) with pharmacological methods could alleviate the activation of microglia and the production of inflammatory cytokines (TNF-α, IL-6 and IL-1β), suggesting that this signalling pathway may be a therapeutic target for neuroinflammation-related diseases (Bhatia et al. 2017). Studies have revealed that attenuating the activation of hippocampal microglia and astrocytes and inhibiting inflammatory mediators and cytokines and their corresponding intracellular signalling pathways (TLR4/MyD88 and p38 MAPK) can significantly alleviate CCI-induced behavioural impairments (Cheng et al. 2021; Lee et al. 2015).
Autophagy Plays a Role in CCI
Autophagy has a vital function in preserving homeostasis in the cell and may be necessary for neuronal homeostasis, neuronal plasticity, and protein quality management (Smith et al. 2014; Wang et al. 2006). Cell autophagy can be induced by changes in the intracellular environment, such as organelle and cytoplasmic accumulation or damage, and extracellular stimulation, such as starvation, high temperature, hypoxia, and hormone stimulus (Yang and Klionsky 2010). Autophagy is primarily related to the circulation and reprocessing of macromolecular elements in cells, and the removal of foreign substances from the cytoplasm and damaged organelles by the autophagy lysosome system maintains cell function (Burman and Ktistakis 2010). By playing dual roles in cell survival during this process (Ravanan et al. 2017), autophagy is not entirely beneficial to the body (Liu and Levine 2015). Studies have revealed that autophagy plays a significant role in ischaemic brain injury because CCH can lead to hippocampal atrophy, lowering the Bcl-2/Bax ratio, neuronal apoptosis, and enhancement and redistribution of autophagy in rats (Liu et al. 2015).
Autophagy is activated and may play dual roles in cell death or survival during cerebral ischemia. Autophagy not only mediates direct microbial degradation but also exerts other protective mechanisms, such as mediating inflammation by affecting the progression, homeostasis and survival of inflammatory cells (such as macrophages, neutrophils and lymphocytes), which have a key function in inducing and eliminating the source of inflammatory stimuli together with inflammatory signals, or by affecting the transcription, processing, and secretion of various cytokines, thereby reducing the adverse effects of inflammatory responses (Heckmann et al. 2017; Lambelet et al. 2018; Netea-Maier et al. 2016; Pun and Park 2017). On the other hand, numerous pathological pathways, including mitochondrial dysfunction, acidosis, oxidative stress, calcium overload, excitotoxicity, and the inflammatory reaction, are implicated in the onset and progression of cerebral ischaemia reperfusion, resulting in inhibited glutamate accumulation in brain tissue and an increase in impaired cells, which evoke autophagy to varying degrees (Khoshnam et al. 2017). During this process, autophagy may be triggered through multiple-related signalling pathways, such as the AMPK/TSC/mTOR, Beclin 1/BNIP3/SPK2, PI3K/Akt-mTOR, and FoxO/NF-κB pathways (Qi et al. 2014; Sheng and Qin 2015; Yang et al. 2017d). P301L is the tau mutation most frequently observed in patients with frontotemporal dementia, and an interesting study showed that enhancing autophagy could alleviate stroke in elderly rats by modulating P301L-Tau, which reveals that autophagy could be a potential target for improving cognitive impairments in CCI (Yang et al. 2017a). In animal studies (Ma and Ji 2022; Wu et al. 2021; Zhang et al. 2021), targeting autophagy signalling could be a novel treatment approach to prevent and treat CCI. However, determining whether this treatment will be beneficial for humans requires further investigation.
The Link Between Autophagy and Inflammation in CCI
The above evidence shows that CCI is associated with inflammation and autophagy. DAMPs released in local tissue due to neuronal and axonal injury further recruit and activate leukocytes to induce secondary brain injury (Huber-Lang et al. 2018; Takeuchi and Akira 2010). Intriguingly, autophagy can modulate the production of DAMPs and participate in the inflammatory regulation mediated by PAMPs or DAMPs by some unclear mechanism. On the other hand, PAMPs and DAMPs can activate autophagy through particular mechanisms, whereas autophagy can suppress an excessive inflammatory response via PAMPs and DAMPs and regulate TLR or NLR signalling to restrict tissue damage (Into et al. 2012). This evidence shows that there is an interaction between autophagy and inflammation, which may present a potential link between CCI, autophagy, and inflammation.
Autophagy Regulates the Inflammatory Response in CCI
Autophagy plays dual regulatory roles in the inflammatory response to CCI. In a moderate manner, autophagy is effective at anti-inflammation by eliminating inflammatory proteins and proinflammatory cytokines (Bussi et al. 2017). In contrast, excessive autophagy promotes inflammatory responses. As shown in a related study, activation of excessive autophagy induced by CCH simultaneously triggers inflammatory responses that further aggravate cognitive impairment (Yang et al. 2014).
Glial cells are widely distributed in the CNS and mainly include astrocytes, microglia and peritubular glia. Astrocyte differentiation is crucial for advanced embryonic brain development processes, along with activating autophagy. Atg5, an autophagy-related gene, regulates the differentiation of astrocytes in the developing mouse cortex. Deficiency in this gene inhibits the production of astrocytes in vitro and in vivo. In contrast, Atg5 overexpression considerably increases the number of astrocytes (Wang et al. 2014). Altogether, these results demonstrate that autophagy may exert positive effects on glial differentiation.
As a type of immune cell that resides in the CNS, triggered microglia are a sign of neuroinflammation. Autophagy could be triggered by both trehalose and rapamycin in BV2 microglia, therefore decreasing proinflammatory cytokine and nitric oxide (NO) induced by LPS and alpha-synuclein. In addition, autophagy alters the phosphorylation of p38 and ERK1/2 MAPKs in BV2 cells, which is necessary for NO generation (Bussi et al. 2017). Hypoxia inducible factor 1α (HIF-1α), an overexpressed factor in CCH, reduces the density of astrocytes and microglia in the cortex and hippocampus, inhibits oxidative stress and inflammation in the brain, and provokes mitochondrial autophagy, which is frequently accompanied by suppression of the mTOR signalling, thereby enhancing neuronal survival in CCH (Gong et al. 2014). This research implies that HIF-1 may govern the status of glial activation and inflammation in CCH via autophagy. It has been found that a lack of autophagy under nutritional shortage may enhance microglial activation and the severity of inflammation (Bray et al. 2022; Li et al. 2019; Plaza-Zabala et al. 2017). Another study revealed that autophagy is a regulator of endothelial cell-leukocyte trafficking machinery that aims to stop physiological inflammation (Reglero-Real et al. 2021). Therefore, autophagy may regulate the neuroinflammatory response by regulating the activation of glia after CCH.
Inflammation Regulates Autophagy Through Cytokines and ROS
Cytokines and reactive oxygen species (ROS) are inflammatory mediators that regulate autophagy by altering the intracellular environment. Th1 family cytokines (IL-2, TNF-α, etc.) induce autophagy production, while Th2 family cytokines (IL-4, IL-5, IL-6, IL-10, and IL-13) and anti-inflammatory cytokines have inhibitory effects on autophagy (Harris 2011). In addition, IL-1, TGF-β, and IFN-γ are cytokines that can induce autophagy.
PRRs, such as Toll-like receptors (TLRs), NOD-like receptors (NLRs), RIG-I-like receptors, and C-type lectin receptors, can activate primary inflammatory signals and promote autophagy activation by recognizing PAMPs or DAMPs (Liu et al. 2017). With lung ischaemia–reperfusion damage, DAMPs induce autophagy and increase inflammatory reactions by promoting K63-linked TRAF6 ubiquitination and stimulating MAPK and NF-kappa B signalling (Liu et al. 2017).
Xu et al. (2007) demonstrated that TLR ligands not only contribute to the inflammatory process but also trigger autophagy. Autophagy is also induced by the NLR family members nucleotide-binding oligomerization domain 1 (NOD1) and NOD2. Through adaptor kinase receptor interacting protein 2 (RIP2), these molecular signals drive NF-κB and MAPK signalling. Upregulation of NOD2 signalling can provide feedback to inhibit the activation of NOD2-RIP2 signalling and inflammatory bodies by inducing autophagy in alveolar macrophages and negatively regulating lung pulmonary inflammation (Wen et al. 2014). A recent study showed the role of microglia in prolonged cerebral hypoperfusion. Voltage-gated proton channel (Hv1) is expressed in microglia and is involved in the generation of ROS and proinflammatory cytokines in microglia (Yu et al. 2020). The orphan nuclear receptor TLX may exert protective effects on cognitive disorders caused by CCH by regulating the SIRT1/NF-κB pathway (Qu et al. 2021).
This evidence shows that cytokines, ROS, and inflammation-related signalling molecules can activate autophagy through multiple pathways. Therefore, controlling inflammation following CCI is essential for reversing brain injury.
TLR Signalling Mediates the Interaction Between Autophagy and Inflammation
As the main innate immunity sensors that detect various pathogen-specific molecular patterns, TLRs activate downstream signals mainly through ubiquitin-dependent mechanisms. TLR2 or TLR4 agonists may induce autophagy by supporting the reaction between Beclin1 and MyD88 or TRIF and reducing Beclin1 binding to Bcl-2 (Chuang et al. 2013). Inhibition of the TLR4/MyD88/TRAF6 signalling pathways decreases the inflammatory reaction and autophagy and has a neuroprotective effect on cerebral ischaemia‒reperfusion damage (Chan et al. 2016; Cheng et al. 2019; Nazio et al. 2013; Schneider et al. 2012; Verstak et al. 2014; Wang et al. 2016b).
Selective autophagy degrades TRIF and negatively regulates TLR3/4-mediated natural immune reactions (Shi and Kehrl 2008; Yang et al. 2017c). Another study revealed that basal autophagy reduced the expression of monomeric MyD88, thus preventing the self-activation of inflammatory signals (Into et al. 2017). This evidence partly explains the bidirectional communication between TLR and autophagy. Because TLR activation is present in CCI (Khan et al. 2022; Wang et al. 2016a), further research must be conducted to determine whether it is also related to the excessive stimulation of autophagy in CCI.
ROS Produced by Mitochondria May be a Link Between Autophagy and Inflammation
A recent study demonstrated that ROS contribute to the progression of CCI. In CCI pathophysiological mechanisms, inflammatory cascades exacerbate mitochondrial damage, causing increased ROS production and the blocking of autophagosome breakdown, thus initiating a vicious cycle (Zeng et al. 2022). As the most abundant organelle, mitochondria generate large amounts of ROS in cells, which regulate the intracellular redox state and energy metabolism (Busija et al. 2016; Ham and Raju 2017). Studies have revealed that the loss of intracellular balance in redox states and the development of free radicals can lead to neuronal damage (Sutkowy et al. 2021), while dysfunctional mitochondria produce ROS (Busija et al. 2016). Under physiological conditions, ROS mediate beneficial responses, while excess ROS are harmful to the body. Especially with I/R, ROS trigger tissue inflammation and activate NLRP3 inflammasomes, thereby inducing neurological dysfunction and inflammatory responses in brain tissue (Minutoli et al. 2016; Zhang et al. 2017).
By limiting the production of ROS, autophagy maintains mitochondrial integrity and inhibits the activation of NALP3-dependent inflammation (Meng et al. 2019; Nakahira et al. 2011). Inhibiting mitochondrial autophagy results in the accumulation of ROS in damaged mitochondria, which in turn triggers the NLRP3 inflammasome (Zhao et al. 2020; Zhou et al. 2011). Therefore, coordinated interactions between autophagy activation and ROS-dependent signalling are necessary for the regulation of innate defence and homeostatic inflammation.
In the cytoplasm of CA1 neurons in CCH rat models, abnormal mitochondrial aggregation and morphological changes have been observed (Hai et al. 2013). ROS-mediated oxidative stress and neuroinflammation are closely correlated with chronic ischaemia‒reperfusion injury in mice (Han et al. 2022), suggesting that mitochondria participate in CCI. Can mitochondria balance cell autophagy and inflammation by increasing or decreasing ROS in CCI? This question has not yet been answered, so further studies are warranted.
Concluding Remarks
CCI is a major cause of global disability and can lead to permanent tissue damage. The study of CCI in regarding to inflammation and autophagy continues to evolve in both fundament and translation. However, few neuroprotective approaches have been successfully translated into the clinic due to its complex pathogenesis. Here, we reviewed the possible mechanisms of autophagy and inflammation, and we also discussed whether and how these factors affect CCI, which will help us understand the occurrence of CCI and find new ways to target autophagy to control brain tissue injury induced by excessive inflammation. The current study demonstrates that autophagy affects brain that drive or suppress inflammatory responses during CCI, which causes internal changes in CCI. This study may also potentially serve as a guide for future research and therapeutics of CCI, and it should be noted that monitoring autophagy may represent a key aspect of protecting the brain from CCI injury.
Data Availability
Not applicable.
References
Alia C, Spalletti C, Lai S, Panarese A, Lamola G, Bertolucci F et al (2017) Neuroplastic Changes Following Brain Ischemia and their Contribution to Stroke Recovery: Novel Approaches in Neurorehabilitation. Front Cell Neurosci 11. https://doi.org/10.3389/fncel.2017.00076
Bhatia HS, Roelofs N, Munoz E, Fiebich BL (2017) Alleviation of Microglial Activation Induced by p38 MAPK/MK2/PGE(2) Axis by Capsaicin: Potential Involvement of other than TRPV1 Mechanism/s. Sci Rep 7:116. https://doi.org/10.1038/s41598-017-00225-5
Bray CE, Witcher KG, Adekunle-Adegbite D, Ouvina M, Witzel M, Hans E et al (2022) Chronic cortical inflammation, cognitive impairment, and immune reactivity associated with diffuse brain injury are ameliorated by forced turnover of microglia. J Neurosci 42(20):4215–4228. https://doi.org/10.1523/JNEUROSCI.1910-21.2022
Burman C, Ktistakis NT (2010) Autophagosome formation in mammalian cells. Semin Immunopathol 32(4):397–413. https://doi.org/10.1007/s00281-010-0222-z
Busija DW, Rutkai I, Dutta S, Katakam PV (2016) Role of mitochondria in cerebral vascular function: energy production, cellular protection, and regulation of vascular tone. Compr Physiol 6(3):1529–1548. https://doi.org/10.1002/cphy.c150051
Bussi C, Peralta Ramos JM, Arroyo DS, Gaviglio EA, Gallea JI, Wang JM et al (2017) Autophagy down regulates pro-inflammatory mediators in BV2 microglial cells and rescues both LPS and alpha-synuclein induced neuronal cell death. Sci Rep 7:43153. https://doi.org/10.1038/srep43153
Caplan LR (2006) The last 50 years of cerebrovascular disease: Part 1. Int J Stroke 1(2):104–108. https://doi.org/10.1111/j.1747-4949.2006.00028.x
Carmichael ST (2016) The 3 Rs of stroke biology: radial, relayed, and regenerative. Neurotherapeutics 13(2):348–359. https://doi.org/10.1007/s13311-015-0408-0
Ceccariglia S, Cargnoni A, Silini AR, Parolini O (2020) Autophagy: a potential key contributor to the therapeutic action of mesenchymal stem cells. Autophagy 16(1):28–37. https://doi.org/10.1080/15548627.2019.1630223
Chan ST, Lee J, Narula M, Ou JHJ (2016) Suppression of host innate immune response by hepatitis C virus via induction of autophagic degradation of TRAF6. J Virol 90(23):10928–10935. https://doi.org/10.1128/Jvi.01365-16
Cheng Z, Zhang M, Ling C, Zhu Y, Ren H, Hong C et al (2019) Neuroprotective effects of ginsenosides against cerebral ischemia. Molecules. https://doi.org/10.3390/molecules24061102
Cheng CY, Huang HC, Kao ST, Lee YC (2021) Angelica sinensis extract promotes neuronal survival by enhancing p38 MAPK-mediated hippocampal neurogenesis and dendritic growth in the chronic phase of transient global cerebral ischemia in rats. J Ethnopharmacol 278:114301. https://doi.org/10.1016/j.jep.2021.114301
Chuang SY, Yang CH, Chou CC, Chiang YP, Chuang TH, Hsu LC (2013) TLR-induced PAI-2 expression suppresses IL-1beta processing via increasing autophagy and NLRP3 degradation. Proc Natl Acad Sci USA 110(40):16079–16084. https://doi.org/10.1073/pnas.1306556110
Clausen BH, Degn M, Sivasaravanaparan M, Fogtmann T, Andersen MG, Trojanowsky MD et al (2016) Conditional ablation of myeloid TNF increases lesion volume after experimental stroke in mice, possibly via altered ERK1/2 signaling. Sci Rep 6:29291. https://doi.org/10.1038/srep29291
Corbin ZA, Rost NS, Lorenzano S, Kernan WN, Parides MK, Blumberg JB et al (2014) White matter hyperintensity volume correlates with matrix metalloproteinase-2 in acute ischemic stroke. J Stroke Cerebrovasc Dis 23(6):1300–1306. https://doi.org/10.1016/j.jstrokecerebrovasdis.2013.11.002
Cosin-Roger J, Simmen S, Melhem H, Atrott K, Frey-Wagner I, Hausmann M et al (2017) Hypoxia ameliorates intestinal inflammation through NLRP3/mTOR downregulation and autophagy activation. Nat Commun 8:98. https://doi.org/10.1038/s41467-017-00213-3
Deng YL, Ma YL, Zhang ZL, Zhang LX, Guo H, Qin P et al (2018) Astrocytic N-Myc downstream-regulated gene-2 is involved in nuclear transcription factor kappaB-mediated inflammation induced by global cerebral ischemia. Anesthesiology 128(3):574–586. https://doi.org/10.1097/ALN.0000000000002044
Du SQ, Wang XR, Xiao LY, Tu JF, Zhu W, He T, Liu CZ (2017) Molecular mechanisms of vascular dementia: what can be learned from animal models of chronic cerebral hypoperfusion? Mol Neurobiol 54(5):3670–3682. https://doi.org/10.1007/s12035-016-9915-1
El Husseini N, Bushnell C, Brown CM, Attix D, Rost NS, Samsa GP et al (2020) Vascular cellular adhesion molecule-1 (VCAM-1) and memory impairment in African-Americans after small vessel-type stroke. J Stroke Cerebrovasc Dis 29(4):104646. https://doi.org/10.1016/j.jstrokecerebrovasdis.2020.104646
Facci L, Barbierato M, Marinelli C, Argentini C, Skaper SD, Giusti P (2014) Toll-like receptors 2,-3 and-4 prime microglia but not astrocytes across central nervous system regions for ATP-dependent interleukin-1 beta release. Sci Rep 4:6824. https://doi.org/10.1038/srep06824
Gong G, Xiang L, Yuan L, Hu L, Wu W, Cai, L et al (2014) Protective effect of glycyrrhizin, a direct HMGB1 inhibitor, on focal cerebral ischemia/reperfusion-induced inflammation, oxidative stress, and apoptosis in rats. PLoS One 9(3):e89450. https://doi.org/10.1371/journal.pone.0089450
Green MF, Scott JW, Steel R, Oakhill JS, Kemp BE, Means AR (2011) Ca2+/calmodulin-dependent protein kinase kinase beta is regulated by multisite phosphorylation. J Biol Chem 286(32):28066–28079. https://doi.org/10.1074/jbc.M111.251504
Hai J, Wu YF, Lin Q, Huang XS, Zhang GY (2013) Cerebral blood flow and metabolic changes in hippocampal regions of a modified rat model with chronic cerebral hypoperfusion. Acta Neurol Belg 113(3):313–317. https://doi.org/10.1007/s13760-012-0154-6
Ham PB, Raju R (2017) Mitochondrial function in hypoxic ischemic injury and influence of aging. Prog Neurobiol 157:92–116. https://doi.org/10.1016/j.pneurobio.2016.06.006
Han Y, Li X, Yang L, Zhang D, Li L, Dong X et al (2022) Ginsenoside Rg1 attenuates cerebral ischemia-reperfusion injury due to inhibition of NOX2-mediated calcium homeostasis dysregulation in mice. J Ginseng Res 46(4):515–525. https://doi.org/10.1016/j.jgr.2021.08.001
Hao XZ, Yin LK, Zhang XX, Tian JQ, Li CC, Feng XY et al (2016) Combining systemic and stereotactic MEMRI to detect the correlation between gliosis and neuronal connective pathway at the chronic stage after stroke. J Neuroinflamm 13:156. https://doi.org/10.1186/s12974-016-0622-7
Harris J (2011) Autophagy and cytokines. Cytokine 56(2):140–144. https://doi.org/10.1016/j.cyto.2011.08.022
Heckmann BL, Boada-Romero E, Cunha LD, Magne J, Green DR (2017) LC3-associated phagocytosis and inflammation. J Mol Biol 429(23):3561–3576. https://doi.org/10.1016/j.jmb.2017.08.012
Huang YH, Zhang WW, Lin L, Feng J, Chen F, Wei W et al (2010) Is endothelial dysfunction of cerebral small vessel responsible for white matter lesions after chronic cerebral hypoperfusion in rats? J Neurol Sci 299(1–2):72–80. https://doi.org/10.1016/j.jns.2010.08.035
Huber-Lang M, Lambris JD, Ward PA (2018) Innate immune responses to trauma. Nat Immunol 19(4):327–341. https://doi.org/10.1038/s41590-018-0064-8
Into T, Inomata M, Takayama E, Takigawa T (2012) Autophagy in regulation of Toll-like receptor signaling. Cell Signal 24(6):1150–1162. https://doi.org/10.1016/j.cellsig.2012.01.020
Into T, Horie T, Inomata M, Gohda J, Inoue J, Murakami Y, Niida S (2017) Basal autophagy prevents autoactivation or enhancement of inflammatory signals by targeting monomeric MyD88. Sci Rep 7:1009. https://doi.org/10.1038/s41598-017-01246-w
Kawabori M, Yenari MA (2015) Inflammatory responses in brain ischemia. Curr Med Chem 22(10):1258–1277. https://doi.org/10.2174/0929867322666150209154036
Khan ZA, Sumsuzzman DM, Choi J, Kamenos G, Hong Y (2022) Pre- and post-conditioning with poly I: C exerts neuroprotective effect against cerebral ischemia injury in animal models: a systematic review and meta-analysis. CNS Neurosci Ther 28(8):1168–1182. https://doi.org/10.1111/cns.13851
Khoshnam SE, Winlow W, Farzaneh M, Farbood Y, Moghaddam HF (2017) Pathogenic mechanisms following ischemic stroke. Neurol Sci 38(7):1167–1186. https://doi.org/10.1007/s10072-017-2938-1
Kitamura A, Fujita Y, Oishi N, Kalaria RN, Washida K, Maki T et al (2012) Selective white matter abnormalities in a novel rat model of vascular dementia. Neurobiol Aging 33(5):1012.e25. https://doi.org/10.1016/j.neurobiolaging.2011.10.033
Lambelet M, Terra LF, Fukaya M, Meyerovich K, Labriola L, Cardozo AK, Allagnat F (2018) Dysfunctional autophagy following exposure to pro-inflammatory cytokines contributes to pancreatic beta-cell apoptosis. Cell Death Dis 9:96. https://doi.org/10.1038/s41419-017-0121-5
Lee KM, Bang J, Kim BY, Lee IS, Han JS, Hwang BY, Jeon WK (2015) Fructus mume alleviates chronic cerebral hypoperfusion-induced white matter and hippocampal damage via inhibition of inflammation and downregulation of TLR4 and p38 MAPK signaling. BMC Complement Altern Med 15:125. https://doi.org/10.1186/s12906-015-0652-1
Lee YR, Kuo SH, Lin CY, Fu PJ, Lin YS, Yeh TM, Liu HS (2018) Dengue virus-induced ER stress is required for autophagy activation, viral replication, and pathogenesis both in vitro and in vivo. Sci Rep 8:489. https://doi.org/10.1038/s41598-017-18909-3
Li D, Huang S, Yin Z, Zhu J, Ge X, Han Z et al (2019) Increases in miR-124-3p in microglial exosomes confer neuroprotective effects by targeting FIP200-mediated neuronal autophagy following traumatic brain injury. Neurochem Res 44(8):1903–1923. https://doi.org/10.1007/s11064-019-02825-1
Li YW, Li QY, Wang JH, Xu XL (2016) Contribution of p38 MAPK to the ameliorating effect of enriched environment on the cognitive deficits induced by chronic cerebral hypoperfusion. Cell Physiol Biochem 40(3–4):549–557. https://doi.org/10.1159/000452568
Li L, Chen Y, Sluter MN, Hou R, Hao J, Wu Y et al (2022) Ablation of Siglec-E augments brain inflammation and ischemic injury. J Neuroinflamm 19(1):191. https://doi.org/10.1186/s12974-022-02556-1
Liu Y, Levine B (2015) Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ 22(3):367–376. https://doi.org/10.1038/cdd.2014.143
Liu L, Li CJ, Lu Y, Zong XG, Luo C, Sun J, Guo LJ (2015) Baclofen mediates neuroprotection on hippocampal CA1 pyramidal cells through the regulation of autophagy under chronic cerebral hypoperfusion. Sci Rep 5:14474. https://doi.org/10.1038/srep14474
Liu CH, Liu HY, Ge BX (2017) Innate immunity in tuberculosis: host defense vs pathogen evasion. Cell Mol Immunol 14(12):963–975. https://doi.org/10.1038/cmi.2017.88
Ma X, Ji C (2022) Remote ischemic conditioning: a potential treatment for chronic cerebral hypoperfusion. Eur Neurol 85(4):253–259. https://doi.org/10.1159/000521803
Meng Y, Pan MX, Zheng BJ, Chen Y, Li W, Yang QJ et al (2019) Autophagy attenuates angiotensin II-induced pulmonary fibrosis by inhibiting redox imbalance-mediated NOD-like receptor family pyrin domain containing 3 inflammasome activation. Antioxid Redox Signal 30(4):520–541. https://doi.org/10.1089/ars.2017.7261
Minutoli L, Puzzolo D, Rinaldi M, Irrera N, Marini H, Arcoraci V et al (2016) ROS-mediated NLRP3 inflammasome activation in brain, heart, kidney, and testis ischemia/reperfusion injury. Oxid Med Cell Longev. https://doi.org/10.1155/2016/2183026
Nakahira K, Haspel JA, Rathinam VAK, Lee SJ, Dolinay T, Lam HC et al (2011) Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12(3):222-U257. https://doi.org/10.1038/ni.1980
Nazio F, Strappazzon F, Antonioli M, Bielli P, Cianfanelli V, Bordi M et al (2013) mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat Cell Biol 15(4):406. https://doi.org/10.1038/ncb2708
Netea-Maier RT, Plantinga TS, van de Veerdonk FL, Smit JW, Netea MG (2016) Modulation of inflammation by autophagy: consequences for human disease. Autophagy 12(2):245–260. https://doi.org/10.1080/15548627.2015.1071759
Pan LN, Zhu W, Li C, Xu XL, Guo LJ, Lu Q (2012) Toll-like receptor 3 agonist Poly I: C protects against simulated cerebral ischemia in vitro and in vivo. Acta Pharmacol Sin 33(10):1246–1253. https://doi.org/10.1038/aps.2012.122
Plaza-Zabala A, Sierra-Torre V, Sierra A (2017) Autophagy and microglia: novel partners in neurodegeneration and aging. Int J Mol Sci 18(3):598. https://doi.org/10.3390/ijms18030598
Pun NT, Park PH (2017) Role of p62 in the suppression of inflammatory cytokine production by adiponectin in macrophages: involvement of autophagy and p21/Nrf2 axis. Sci Rep 7:393. https://doi.org/10.1038/s41598-017-00456-6
Qi ZF, Yan F, Shi WJ, Zhang CC, Dong W, Zhao YM et al (2014) AKT-related autophagy contributes to the neuroprotective efficacy of hydroxysafflor yellow A against ischemic stroke in rats. Transl Stroke Res 5(4):501–509. https://doi.org/10.1007/s12975-014-0346-x
Qu C, Qu C, Xu L, Shen J, Lv D, Li Y et al (2021) Nuclear receptor TLX may be through regulating the SIRT1/NF-kappaB pathway to ameliorate cognitive impairment in chronic cerebral hypoperfusion. Brain Res Bull 166:142–149. https://doi.org/10.1016/j.brainresbull.2020.11.006
Ravanan P, Srikumar IF, Talwar P (2017) Autophagy: the spotlight for cellular stress responses. Life Sci 188:53–67. https://doi.org/10.1016/j.lfs.2017.08.029
Reglero-Real N, Perez-Gutierrez L, Yoshimura A, Rolas L, Garrido-Mesa J, Barkaway A et al (2021) Autophagy modulates endothelial junctions to restrain neutrophil diapedesis during inflammation. Immunity 54(9):1989-2004 e1989. https://doi.org/10.1016/j.immuni.2021.07.012
Sattayakhom A, Kalarat K, Rakmak T, Tapechum S, Monteil A, Punsawad C et al (2022) Effects of ceftriaxone on oxidative stress and inflammation in a rat model of chronic cerebral hypoperfusion. Behav Sci (Basel). https://doi.org/10.3390/bs12080287
Schlemmer F, Boyer L, Soumagne T, Ridoux A, Chouaid C, Maitre B et al (2018) Beclin1 circulating levels and accelerated aging markers in COPD. Cell Death Dis 9:156. https://doi.org/10.1038/s41419-017-0178-1
Schneider M, Zimmermann AG, Roberts RA, Zhang L, Swanson KV, Wen HT et al (2012) The innate immune sensor NLRC3 attenuates Toll-like receptor signaling via modification of the signaling adaptor TRAF6 and transcription factor NF-kappa B. Nat Immunol 13(9):823–831. https://doi.org/10.1038/ni.2378
Sheng R, Qin ZH (2015) The divergent roles of autophagy in ischemia and preconditioning. Acta Pharmacol Sin 36(4):411–420. https://doi.org/10.1038/aps.2014.151
Sherman V, Martino R, Bhathal I, DeVeber G, Dlamini N, MacGregor D et al (2021) Swallowing, oral motor, motor speech, and language impairments following acute pediatric ischemic stroke. Stroke 52(4):1309–1318. https://doi.org/10.1161/STROKEAHA.120.031893
Shi CS, Kehrl JH (2008) MyD88 and Trif target Beclin 1 to trigger autophagy in macrophages. J Biol Chem 283(48):33175–33182. https://doi.org/10.1074/jbc.M804478200
Smith ED, Prieto GA, Tong LQ, Sears-Kraxberger I, Rice JD, Steward O, Cotman CW (2014) Rapamycin and interleukin-1 beta impair brain-derived neurotrophic factor-dependent neuron survival by modulating autophagy. J Biol Chem 289(30):20615–20629. https://doi.org/10.1074/jbc.M114.568659
Sorbara MT, Ellison LK, Ramjeet M, Travassos LH, Jones NL, Girardin SE, Philpott DJ (2013) The protein ATG16L1 suppresses inflammatory cytokines induced by the intracellular sensors Nod1 and Nod2 in an autophagy-independent manner. Immunity 39(5):858–873. https://doi.org/10.1016/j.immuni.2013.10.013
Sun MY, Deng B, Zhao XY, Gao CJ, Yang L, Zhao H et al (2015) Isoflurane preconditioning provides neuroprotection against stroke by regulating the expression of the TLR4 signalling pathway to alleviate microglial activation. Sci Rep 5:11445. https://doi.org/10.1038/srep11445
Sutkowy P, Wozniak A, Mila-Kierzenkowska C, Szewczyk-Golec K, Wesolowski R, Pawlowska M, Nuszkiewicz J (2021) Physical activity vs. redox balance in the brain: brain health, aging and diseases. Antioxidants (Basel). https://doi.org/10.3390/antiox11010095
Suzuki Y, Hattori K, Hamanaka J, Murase T, Egashira Y, Mishiro K et al (2012) Pharmacological inhibition of TLR4-NOX4 signal protects against neuronal death in transient focal ischemia. Sci Rep 2:896. https://doi.org/10.1038/srep00896
Takeuchi O, Akira S (2010) Pattern recognition receptors and inflammation. Cell 140(6):805–820. https://doi.org/10.1016/j.cell.2010.01.022
Tang DF, Yao RY, Zhao DD, Zhou L, Wu Y, Yang Y et al (2018) Trichostatin A reverses the chemoresistance of lung cancer with high IGFBP2 expression through enhancing autophagy. Sci Rep 8:3917. https://doi.org/10.1038/s41598-018-22257-1
Verstak B, Stack J, Ve T, Mangan M, Hjerrild K, Jeon J et al (2014) The TLR signaling adaptor TRAM interacts with TRAF6 to mediate activation of the inflammatory response by TLR4. J Leukoc Biol 96(3):427–436. https://doi.org/10.1189/jlb.2A0913-487R
Wang QJ, Ding YM, Kohtz S, Mizushima N, Cristea IM, Rout MP et al (2006) Induction of autophagy in axonal dystrophy and degeneration. J Neurosci 26(31):8057–8068. https://doi.org/10.1523/Jneurosci.2261-06.2006
Wang S, Li B, Qiao H, Lv X, Liang Q, Shi Z et al (2014) Autophagy-related gene Atg5 is essential for astrocyte differentiation in the developing mouse cortex. Embo Reports 15(10):1053–1061. https://doi.org/10.15252/embr.201338343
Wang ZF, Tsai LK, Munasinghe J, Leng Y, Fessler EB, Chibane F et al (2012) Chronic valproate treatment enhances postischemic angiogenesis and promotes functional recovery in a rat model of ischemic stroke. Stroke 43(9):2430. https://doi.org/10.1161/Strokeaha.112.652545
Wang R, Wang ST, Wang YD, Wu G, Du Y, Qian MQ et al (2016a) Stress-responsive heme oxygenase-1 isoenzyme participates in Toll-like receptor 4-induced inflammation during brain ischemia. NeuroReport 27(6):445–454. https://doi.org/10.1097/Wnr.0000000000000561
Wang YJ, Chen G, Yu XD, Li YC, Zhang L, He ZZ et al (2016b) Salvianolic acid B ameliorates cerebral ischemia/reperfusion injury through inhibiting TLR4/MyD88 signaling pathway. Inflammation 39(4):1503–1513. https://doi.org/10.1007/s10753-016-0384-5
Wang L, Wang JY, Wang FQ, Liu CH, Yang XN, Yang JJ, Ming D (2017) VEGF-mediated cognitive and synaptic improvement in chronic cerebral hypoperfusion rats involves autophagy process. Neuromol Med 19(2–3):423–435. https://doi.org/10.1007/s12017-017-8458-6
Wang XX, Zhang B, Xia R, Jia QY (2020) Inflammation, apoptosis and autophagy as critical players in vascular dementia. Eur Rev Med Pharmacol Sci 24(18):9601–9614. https://doi.org/10.26355/eurrev_202009_23048
Wen ZM, Fan LY, Li YH, Zou Z, Scott MJ, Xiao GZ et al (2014) Neutrophils counteract autophagy-mediated anti-inflammatory mechanisms in alveolar macrophage: role in posthemorrhagic shock acute lung inflammation. J Immunol 193(9):4623–4633. https://doi.org/10.4049/jimmunol.1400899
Wu X, Zheng Y, Liu M, Li Y, Ma S, Tang W et al (2021) BNIP3L/NIX degradation leads to mitophagy deficiency in ischemic brains. Autophagy 17(8):1934–1946. https://doi.org/10.1080/15548627.2020.1802089
Xu Y, Jagannath C, Liu, XD, Sharafkhaneh A, Kolodziejska KE, Eissa N T (2007) Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 27(1):135–144. https://doi.org/10.1016/j.immuni.2007.05.022
Yan N, Xu Z, Qu C, Zhang J (2021) Dimethyl fumarate improves cognitive deficits in chronic cerebral hypoperfusion rats by alleviating inflammation, oxidative stress, and ferroptosis via NRF2/ARE/NF-kappaB signal pathway. Int Immunopharmacol 98:107844. https://doi.org/10.1016/j.intimp.2021.107844
Yang ZF, Klionsky DJ (2010) Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 22(2):124–131. https://doi.org/10.1016/j.ceb.2009.11.014
Yang Z, Zhang N, Shen HC, Lin CG, Lin L, Yuan BQ (2014) Microglial activation with reduction in autophagy limits white matter lesions and improves cognitive defects during cerebral hypoperfusion. Curr Neurovasc Res 11(3):223–229. https://doi.org/10.2174/1567202611666140520124407
Yang C, Zhang X, Gao J, Wang M, Yang Z (2017a) Arginine vasopressin ameliorates spatial learning impairments in chronic cerebral hypoperfusion via V1a receptor and autophagy signaling partially. Transl Psychiatry 7:e1174. https://doi.org/10.1038/tp.2017.121
Yang CH, Yen TL, Hsu CY, Thomas PA, Sheu JR, Jayakumar T (2017b) Multi-targeting andrographolide, a novel NF-kappaB inhibitor, as a potential therapeutic agent for stroke. Int J Mol Sci. https://doi.org/10.3390/ijms18081638
Yang Q, Liu TT, Lin H, Zhang M, Wei J, Luo WW et al (2017c) TRIM32-TAX1BP1-dependent selective autophagic degradation of TRIF negatively regulates TLR3/4-mediated innate immune responses. PLoS Pathog 13(9):e1006600. https://doi.org/10.1371/journal.ppat.1006600
Yang Y, Chen SC, Zhang YQ, Lin XX, Song YY, Xue ZL et al (2017d) Induction of autophagy by spermidine is neuroprotective via inhibition of caspase 3-mediated Beclin 1 cleavage. Cell Death Dis. https://doi.org/10.1038/cddis.2017.161
Yoshizaki K, Adachi K, Kataoka S, Watanabe A, Tabira T, Takahashi K, Wakita H (2008) Chronic cerebral hypoperfusion induced by right unilateral common carotid artery occlusion causes delayed white matter lesions and cognitive impairment in adult mice. Exp Neurol 210(2):585–591. https://doi.org/10.1016/j.expneurol.2007.12.005
Yu Y, Luo X, Li C, Ding F, Wang M, Xie M et al (2020) Microglial Hv1 proton channels promote white matter injuries after chronic hypoperfusion in mice. J Neurochem 152(3):350–367. https://doi.org/10.1111/jnc.14925
Yuan BQ, Shi H, Zheng K, Su ZL, Su H, Zhong M et al (2017) MCP-1-mediated activation of microglia promotes white matter lesions and cognitive deficits by chronic cerebral hypoperfusion in mice. Mol Cell Neurosci 78:52–58. https://doi.org/10.1016/j.mcn.2016.08.003
Zbesko JC, Nguyen TV, Yang T, Frye JB, Hussain O, Hayes M et al (2018) Glial scars are permeable to the neurotoxic environment of chronic stroke infarcts. Neurobiol Dis 112:63–78. https://doi.org/10.1016/j.nbd.2018.01.007
Zhang BC, Gao MQ, Shen J, He DK (2017) Inhaled methane protects rats against neurological dysfunction induced by cerebral ischemia and reperfusion injury: PI3K/Akt/HO-1 pathway involved. Arch Med Res 48(6):520–525. https://doi.org/10.1016/j.arcmed.2018.01.001
Zhang ZB, Xiong LL, Xue LL, Deng YP, Du RL, Hu Q et al (2021) MiR-127-3p targeting CISD1 regulates autophagy in hypoxic-ischemic cortex. Cell Death Dis 12(3):279. https://doi.org/10.1038/s41419-021-03541-x
Zhao SC, Ma LS, Chu ZH, Xu H, Wu WQ, Liu F (2017) Regulation of microglial activation in stroke. Acta Pharmacologica Sinica 38(4):445–458. https://doi.org/10.1038/aps.2016.162
Zeng X, Zhang YD, Ma RY, Chen YJ, Xiang XM, Hou DY et al (2022) Activated Drp1 regulates p62-mediated autophagic flux and aggravates inflammation in cerebral ischemia-reperfusion via the ROS-RIP1/RIP3-exosome axis. Mil Med Res 9(1):25. https://doi.org/10.1186/s40779-022-00383-2
Zhao S, Chen F, Yin Q, Wang D, Han W, Zhang Y (2020) Reactive oxygen species interact with NLRP3 inflammasomes and are involved in the inflammation of sepsis: from mechanism to treatment of progression. Front Physiol 11:571810. https://doi.org/10.3389/fphys.2020.571810
Zhou R, Yazdi AS, Menu P, Tschopp J (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469(7329):221–225. https://doi.org/10.1038/nature09663
Acknowledgements
Not applicable.
Funding
This research was funded by National Natural Science Foundation of China (Nos. 31960190, 82160688), the Natural Science Foundation of Jiangxi (No. 20192BAB205117) and the Innovation Team Foundation of Gannan Medical University (Nos. TD201705, TD2021JC06).
Author information
Authors and Affiliations
Contributions
H-qZ conducted the search and drafted the manuscript. L-mZ collected important background information. Z-hH and XL participated in the design of the study and revised the manuscript. All authors have read and approved the final version.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no known competing financial interests.
Ethical Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
All the authors give consent.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhou, Hq., Zhang, Lm., Li, X. et al. Crosstalk Between Autophagy and Inflammation in Chronic Cerebral Ischaemia. Cell Mol Neurobiol 43, 2557–2566 (2023). https://doi.org/10.1007/s10571-023-01336-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-023-01336-6