
Abstract
The molar masses and molar mass distributions of three commercial regenerated cellulose samples, viscose rayon, Tencel, and Bemliese (or cuprammonium nonwoven), have been determined by dissolution in 8% (w/w) lithium chloride/N,N-dimethylacetamide (LiCl/DMAc) and subsequent size-exclusion chromatography with multi-angle laser-light scattering detection (SEC/MALLS). Before dissolution in LiCl/DMAc, the regenerated cellulose samples were pretreated by the following three methods: (1) soaking in ethylene diamine (EDA) and subsequent solvent exchange to N,N-dimethylacetamide (DMAc) through methanol, (2) soaking in water and subsequent solvent exchange to DMAc through ethanol, and (3) soaking in water and subsequent solvent exchange to tert-butyl alcohol through ethanol and freeze dying. The pretreated samples were dissolved in 8% (w/w) LiCl/DMAc by stirring the cellulose/LiCl/DMAc mixtures for 1–3 weeks followed by dilution to 1% (w/v) LiCl/DMAc for SEC/MALLS analysis. The EDA- and water-pretreated samples gave almost the same SEC-elution pattens and molar mass plots, resulting in similar number- and mass-average molar masses. However, the freeze-dried samples gave 10%‒20% lower mass recovery ratios than those obtained for the EDA- or water-pretreated samples, probably because of incomplete dissolution of the freeze-dried samples in 8% (w/w) LiCl/DMAc. The average mass-average degree of polymerization values of viscose rayon, Tencel, and Bemliese were 340, 530, and 880, respectively. The slopes of the conformation plots were 0.58–0.62, showing that all of the molecules in the three regenerated cellulose samples were dissolved in 1% (w/v) LiCl/DMAc, forming linear random-coil conformations.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Regenerated cellulose fiber is important for textiles, engineering filament yarns, and various medical and healthcare applications. Viscose rayon, Tencel (or Lyocell), and Bemliese (or cuprammonium nonwoven) are typical regenerated cellulose materials produced at the industrial level, and they have contributed to our cultural lives and technologies for a long time (Fink et al. 2001; Sayyed et al. 2019; Veit 2022). Although viscose rayon production and wastewater treatment systems have some environmental issues caused by H2S emission into the atmosphere, viscose rayon is still the main regenerated cellulose fiber, and it is called artificial silk in man-made fibers (Li et al. 2018; Kuchtoá et al. 2023). Tencel has been developed to overcome some of the shortcomings of the viscose rayon production process and to improve the fiber quality (Fink et al. 2001; Kreze and Malei 2003; Abu-Rous et al. 2007; Borbély 2008; Sayyed et al. 2019; Veit 2022). Bemliese is another category of regenerated cellulose materials.
Viscose rayon and Tencel are produced by dissolution of wood dissolving pulps with high α-cellulose contents (> 93%) in aqueous CS2/NaOH at room temperature and thermally melted N-methylmorpholine N-oxide (NMMO) hydrate, respectively, and subsequent spinning, regeneration in aqueous media, washing with water, and drying. Bemliese is produced from cotton linters cellulose by dissolution in aqueous Cu(NH3)4(OH)2, spinning, fabrication, regeneration in an aqueous medium, washing with water, and drying to form cuprammonium nonwoven, which is mainly used for medical, healthcare, and agribusiness applications (Veit 2022).
The molar masses and molar mass distributions of these regenerated cellulose materials are fundamental and important factors that influence the mechanical and other key properties of the materials. Size-exclusion chromatography combined with multi-angle laser-light scattering and refractive index detection (SEC/MALLS/RI) gives molar-mass distributions and number- and mass-average molar mass values (Mn and Mw, respectively). In this case, the cellulose samples should be completely dissolved in a solvent at the individual molecular level, and the solution should be transparent (without fluorescence formed by laser-light irradiation). Furthermore, the cellulose molecules should be stable in the solvent without depolymerization or side reactions during the dissolution process and storage of the cellulose solution. Lithium chloride/N,N-dimethylacetamide (LiCl/DMAc) is the only solvent system that satisfies the above requirements (Bikova and Treimanis 2002; Potthast et al. 2003; Dupont 2003; Ono and Isogai 2020). Some activation methods or pretreatments of the cellulose sample are required before dissolution treatment in 8% (w/w) LiCl/DMAc for complete dissolution (Bikova and Treimanis 2002; Potthast et al. 2003; Dupont 2003). However, it has been reported that complete dissolution of regenerated cellulose materials is often difficult, resulting in inaccurate molar mass data by SEC/MALLS (Henninges et al. 2014; Siller et al. 2014; Silbermann et al. 2017).
The water-activation method followed by solvent exchange to DMAc through acetone has been used to pretreat some cellulose samples, such as hardwood bleached kraft pulps, softwood and hardwood bleached sulfite and prehydrolyzed kraft pulps, and cotton linters cellulose samples, for dissolution in 8% (w/w) LiCl/DMAc (Bikova and Treimanis 2002; Potthast et al. 2003; Dupont 2003; Henninges et al. 2014; Siller et al. 2014). However, softwood bleached kraft pulps (SBKPs), cotton lint, bacterial, tunicate, and algal cellulose samples, hemicellulose-rich plant holocellulose samples, and some regenerated cellulose samples cannot be completely dissolved in 8% (w/w) LiCl/DMAc by the water-activation method (Henninges et al. 2014; Siller et al. 2014; Silbermann et al. 2017).
In previous work, we succeeded in completely dissolving all of the above native cellulose and plant holocellulose samples in 8% (w/w) LiCl/DMAc by 100% ethylene diamine (EDA) pretreatment. The cellulose and holocellulose samples were first soaked in EDA, and EDA was then solvent exchanged to DMAc through methanol. Complete dissolution was achieved by stirring the EDA-pretreated cellulose and holocellulose samples in 8% (w/w) LiCl/DMAc at room temperature for a few days, weeks, or months depending on the sample. The key for complete dissolution of all of the native cellulose and holocellulose samples is conversion of the cellulose I crystal structure in the sample to cellulose III or disordered structures by EDA treatment followed by methanol washing. The cellulose and holocellulose solutions in 8% (w/w) LiCl/DMAc are then diluted to 1% (w/v) LiCl/DMAc and subjected to SEC/MALLS analysis. Consequently, some important information about the native cellulose samples and plant holocelluloses have been obtained by SEC/MALLS (Ono et al. 2016a, b, 2017, 2018, 2021, 2022a, b; c; Ono and Isogai 2020). It is possible that low-molar-mass hemicellulose molecules slightly dissolve in EDA during pretreatment and are excluded from the SEC/MALLS data, which should be taken into account (Yamamoto et al. 2011).
In this study, two regenerated cellulose fibers, viscose rayon and Tencel, and one regenerated nonwoven cellulose, Bemliese, were selected, and the following three pretreatments were applied to the regenerated cellulose samples: EDA soaking used in our previous studies, conventional water soaking, and water-soaking/freeze-drying. In the EDA- and water-soaking pretreatments, some mass losses may be unavoidable during repeated solvent exchange and centrifugation, resulting in inaccurate cellulose concentrations in the obtained LiCl/DMAc solutions used for SEC/MALLS. In contrast, more accurate cellulose concentrations of the LiCl/DMAc solutions are obtained using freeze-dried samples, which is significant for evaluation of complete dissolution in LiCl/DMAc from mass loss values (caused by, for example, incomplete dissolution) in SEC/MALLS analysis. Soaking of cellulose samples including some regenerated cellulose fibers in dimethylsulfoxide followed by solvent-exchange to DMAc has been reported for complete dissolution in LiCl/DMAc for SEC/MALLS analysis (Silbermann et al. 2017). However, the mass recovery ratios of the starting cellulose materials in the LiCl/DMAc solutions subjected to SEC/MALLS analysis were not taken into account, and no data for water-activated or freeze-dried cellulose samples were provided. In this study, one softwood bleached sulfite pulp (SBSP) sample, which was prepared by acid sulfite pulping and subsequent bleaching and used as the dissolving pulp for production of viscose rayon and some cellulose derivatives, was also applied to dissolution in 8% (w/w) LiCl/DMAc and subsequent SEC/MALLS analysis as a reference (Mendes et al. 2021).
Materials and methods
Samples
The viscose rayon and Tencel fibers were commercial products. Bemliese (or cuprammonium nonwoven) was kindly provided by Asahi Kasei Co., Ltd. (Miyazaki, Japan). The regenerated samples were cut into short lengths of 3–5 mm with scissors. The SBSP was dissolving pulp fibers produced from softwood chips by acid sulfite pulping and subsequent bleaching processes (Nippon Paper Co. Ltd., Japan). This SBSP contained 96% glucose, 1.5% xylose, and 0.9% mannose as neutral sugars (Ono et al. 2018). The 1 M cupriethylenediamine hydroxide solution (Cu(EDA)2(OH)2) is a commercial product (Sigma Aldrich, USA). LiCl, DMAc, and the other chemicals and solvents were laboratory grade (FUJIFILM Wako Pure Chemical, Co., Tokyo, Japan) and used as received.
Dissolution of the cellulose samples in 8% (w/w) LiCl/DMAc
The three regenerated cellulose samples (viscose rayon, Tencel, and Bemliese) and SBSP were dissolved in 8% (w/w) LiCl/DMAc according to the procedures shown in Fig. 1. The first procedure was followed by the EDA-activation method commonly used in our laboratory (Ono et al. 2016a, b, 2020). After vacuum drying at 40 °C for 1 day, the cellulose sample (20 mg on dry mass) was soaked in 100% EDA (5 mL), and the mixture was stirred with a magnetic stir bar overnight. Solvent exchange from EDA to methanol (MeOH, 35 mL) was then performed by centrifugation, and the cellulose/MeOH mixture was shaken overnight. This treatment was repeated twice with fresh MeOH (35 mL each). The mixture was then solvent exchanged from MeOH to DMAc (35 mL) by centrifugation and shaking the cellulose/DMAc mixture overnight. This treatment was repeated again with fresh DMAc. After centrifugation of the mixture to remove excess DMAc, 8% (w/w) LiCl/DMAc (5 g) was added to the cellulose sample, and the mixture was stirred at ~ 23 °C for 1 or 2 weeks.

Scheme for dissolution of the three regenerated cellulose samples and one dissolving pulp in 8% (w/w) LiCl/DMAc by three activation methods for SEC/MALLS analysis of the cellulose samples
The second procedure was followed by the conventional water-activation method (Bikova and Treimanis 2002; Dupont 2003; Henninges et al. 2014) with slight modification. The vacuum-dried sample (20 mg on dry mass) was soaked in water (20 mL), and the mixture was stirred overnight. Solvent exchange from water to ethanol (EtOH, 35 mL) was then performed by centrifugation, and the mixture was shaken overnight. This treatment was repeated twice with fresh EtOH (35 mL each). Solvent exchange from EtOH to DMAc was then performed by centrifugation, and the cellulose/DMAc mixture was stirred overnight. This treatment was repeated again with fresh DMAc. After centrifugation of the mixture to remove excess DMAc, 8% (w/w) LiCl/DMAc (5 g) was added to the cellulose sample, and the mixture was stirred at ~ 23 °C for 1, 2, or 3 weeks.
In the third procedure, the three regenerated cellulose samples were dissolved in 8% (w/w) LiCl/DMAc according to the following freeze-drying method. The vacuum-dried sample (50 mg on dry weight) was soaked in water (20 mL), and the mixture was stirred overnight. Solvent exchange from water to EtOH (35 mL) was then performed, and the mixture was shaken for 3 h. Solvent exchange from EtOH to tert-butyl alcohol (t-BuOH) was then performed, and the mixture was shaken for 3 h. This treatment was repeated again with fresh t-BuOH followed by freeze drying for 5 days. The freeze-dried sample (8 mg) was dispersed in 8% (w/w) LiCl/DMAc (2 g), and the mixture was stirred at ~ 23 °C for 1, 2, or 3 weeks.
SEC/MALLS analysis
The cellulose solutions in 8% (w/w) LiCl/DMAc obtained by the processes described in the previous section were diluted with fresh DMAc to prepare cellulose solutions in ~ 1% (w/v) LiCl/DMAc. Each solution was passed through a 0.45-µm poly(difluoroethylene) disposable filter (Millex, Merck Millipore, Tokyo, Japan) and then subjected to SEC/MALLS analysis with 1% (w/v) LiCl/DMAc as the eluent (Ono et al. 2016b, 2022a, c). KD-806 M and KD-G columns (Shodex, Tokyo, Japan) were used as the SEC and guard columns, respectively. A MALLS detector (DOWN HELEOS-II, λ = 658 nm; Wyatt Technologies, USA) and a refractive index detector (RID-10A, Shimadzu, Japan) were set in a high-pressure liquid chromatograph system. ASTRA software (version 6.1, Wyatt Technologies, USA) was used for data acquisition and the analyses. The number- and mass-average molar masses (Mn and Mw, respectively) of the cellulose samples were calculated using the value of 0.131 mL/g as the specific refractive index increment (dn/dc) (Ono et al. 2016a).
Viscosity-average degrees of polymerization of the cellulose samples
The freeze-dried cellulose sample (0.04 g) was soaked in water (10 mL), and the mixture was stirred for 10 min. The 1 M Cu(EDA)2(OH)2 solution (10 mL) was added to the cellulose/water mixture, and the solution was stirred until the cellulose sample was completely dissolved in 0.5 M Cu(EDA)2(OH)2 (20 mL). The viscosity ratio of the solution ηrel was measured using a Cannon–Fenske-type capillary viscometer. The limiting viscosity number [η] (or intrinsic viscosity) was calculated from the viscosity relative inclement ηsp (or specific viscosity) using the Schulz–Blaschke equation (Schulz and Dinglinger 1941): [η] = ηsp/c(1 + 0.28ηsp), where c is the cellulose concentration (g/mL). The DPv value of the sample was calculated from [η] using the Mark–Houwink–Sakurada equation: [η] = 0.909 × DPv0.9 (Marx 1955; Isogai et al. 1989a, b).
Solid-state 13C-NMR analysis
Each of the air-dried cellulose samples was set in a ZrO2 sample rotor, and the solid-state 13C NMR spectrum was obtained by a NMR system (JNM-ECA II 500, JEOL, Tokyo, Japan) equipped with a cross-polarization (CP) and magic-angle sample-spinning probe (Funahashi et al. 2017; Ono et al. 2022a, b, c) under the following conditions: a sample spinning rate of 15 kHz, a proton 90° pulse time of 2.5 μs, and a relaxation delay of 5 s. CP transfer was achieved using a ramped amplitude sequence for a CP contact time of 2 ms. Each spectrum was acquired by 12,000 scans for 16 h.
Results
Solid-state 13C-NMR spectra of the cellulose samples
The solid-state 13C-NMR spectra of the three regenerated cellulose samples and SBSP are shown in Fig. 1. Viscose rayon, Tencel, and Bemliese exhibited typical NMR patterns of low-crystallinity cellulose II samples. The crystalline C1 carbon peaks appeared at 105 and 107 ppm, and the crystalline C4 carbon peaks appeared at 88 and 89 ppm (Horii et al. 1982; Zhao et al. 2007; Östlund et al. 2013; Idström et al. 2016). The C6-OH carbon atoms of the three regenerated cellulose samples exhibited broad single peaks at ~ 63 ppm, showing that they had gauche–trans and gauche–gauche conformations, which correspond to the C6-OH groups of cellulose II and amorphous structures, respectively (Horii et al. 1983; Larsson PT 2005; Funahashi et al. 2017). The NMR spectrum of SBSP showed the typical pattern of wood chemical pulp (Zhou et al. 2020), and the crystallinity of cellulose I measured from the peak areas of the crystalline and amorphous C4 carbon atoms (C4cry and C4amo, respectively, in Fig. 2) was 56%. The relative peak areas of C6/C1 for the four samples in Fig. 2 were 0.78–0.83 (Yang et al. 2018; Zhou et al. 2020; Ono et al. 2021, 2022c).

Solid-state13C-NMR spectra of the three regenerated cellulose samples (viscose rayon, Tencel, and Bemliese) and dissolving pulp (SBSP)
Each of the regenerated cellulose samples had a small peak (Cʹ) at ~ 97 ppm, which is the same as those of the C1 carbon atoms of the reducing ends (Dudley et al. 1983; Isogai et al. 1989a, b; Moulthrop et al. 2005; Yuan et al. 2022). However, the peak ratios of (C1 + Cʹ)/C1ʹ (or the DPn values calculated from the 13C peak ratios) were 25, 40, and 42 for viscose rayon, Tencel, and Bemliese, respectively, which are not plausible because the DPn values of the regenerated cellulose samples determined by SEC/MALLS were much higher than 170, as described in the following section. Some solid-state 13C NMR spectra of viscose rayon samples in the literature exhibit the same small peaks at ~ 97 ppm (Horii et al. 1982; Ibbett et al. 2007; Zhao et al. 2007; Li et al. 2012; Shanshan et al. 2012; Mori et al. 2012; Wei et al. 2018; Zhang et al. 2018; From et al. 2020; Fadavi et al. 2021), whereas other regenerated cellulose samples do not show the corresponding small peaks (Newman and Hemmingson 1994; Ago et al. 2004; Jin et al. 2007; Duchemin et al. 2007; Östlund et al. 2013; Idström et al. 2016). The small peak at ~ 97 ppm appears in the NMR spectra of some dried regenerated cellulose samples (Nomura et al. 2020), whereas mercerized cellulose does not show such a peak (Maunu et al. 2000).
Some TEMPO-oxidized cellulose samples without post-oxidation with NaClO2 or post-reduction with NaBH4 (Shinoda et al. 2012) show the corresponding small peaks at ~ 97 ppm in their solid-state 13C-NMR spectra (Follain et al. 2010; Biliuta et al. 2010; Cao et al. 2012; Li et al. 2017; Lin et al. 2017). These TEMPO-oxidized cellulose samples contain small amounts of C6-aldehydes formed as intermediates (Isogai et al. 2011, 2018; Isogai 2022). Thus, it is possible that the Cʹ peak at ~ 97 ppm for the regenerated cellulose samples can be ascribed to hydrated C6-aldehydes, which are formed by partial oxidation of the C6-OH groups during the dissolution, aging, spinning/regeneration, and/or drying process in the commercial production system. The extended 13C-NMR spectra of the cellulose samples are shown in Fig. S1 in the Electronic Supplementary Material. Although the three regenerated cellulose samples exhibited no clear C=O peaks owing to carboxy, aldehyde, and/or ketone groups in the region 170–235 ppm, the intensity-magnitude spectra indicated the presence of small C=O peaks at 197–198 ppm. The presence of C=O groups in regenerated cellulose samples has been reported by Potthast et al. (2003).
SEC/MALLS analysis of the regenerated cellulose samples and SBSP
The three regenerated cellulose samples were pretreated by the three methods (EDA soaking, water soaking, and freeze drying) (Fig. 1), and the pretreated samples were stirred in 8% (w/w) LiCl/DMAc for 1–3 weeks. Freeze-dried wood chemical pulps, such as SBSP, are insoluble in 8% (w/w) LiCl/DMAc, and thus only the EDA- and water-soaking treatments were applied to SBSP before stirring in 8% (w/w) LiCl/DMAc. All of the EDA- and water-pretreated samples visually dissolved in 8% (w/w) LiCl/DMAc within 1 week. The freeze-dried Tencel and Bemliese samples visually dissolved in 8% (w/w) LiCl/DMAc, whereas freeze-dried viscose rayon in 8% (w/w) LiCl/DMAc was slightly cloudy even after stirring the mixture for 2 weeks, probably because of the presence of a small amount of insoluble particles (Siller et al. 2014).
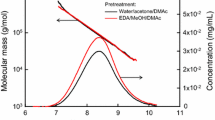
The SEC/MALLS results of viscose rayon are shown in Fig. 3a, Figs. S2–S4, and Tables S1–S3. The molar mass plots for the three pretreatments agreed well (Fig. 3a). The peak areas of the SEC-elution patterns roughly corresponded to the sample masses injected into the SEC/MALLS system. The SEC-elution peak area of the viscose rayon sample pretreated by freeze drying was smaller than those pretreated by EDA and water soaking (Fig. 3a). The lower calculated mass value of the freeze-dried viscose rayon (42 µg, Table S3) than those of EDA-soaked and water-soaked viscose rayon (48 µg, Tables S1 and S2) indicates some mass loss. This was probably caused by filtration of insoluble particles present in the 8% (w/w) LiCl/DMAc solution. However, the other molar mass parameters, such as Mn, Mw, DPw, and Mw/Mn, were similar for the three pretreated viscose rayon samples. Furthermore, no significant differences in the molar mass plots and SEC-elution patterns were observed between the viscose rayon samples stirred in 8% (w/w) LiCl/DMAc for 1 and 2 weeks, showing that stirring the pretreated viscose rayon sample in 8% (w/w) LiCl/DMAc for 1 week is sufficient to obtain constant SEC/MALLS data.

Molar mass plots and SEC-elution patterns of a viscose rayon, b Tencel, c Bemliese, and d SBSP dissolved in 8% (w/w) LiCl/DMAc following the three activation methods and stirring for 1 week before dilution to 1% (w/v) LiCl/DMAc
The SEC/MALLS results of Tencel are shown in Fig. 3b, Figs. S5–S7, and Tables S4–S6. The molar mass plots for the three pretreatments agreed well, and the peak area of the SEC-elution pattern of freeze-dried Tencel was smaller than those of Tencel pretreated by the other two methods (Fig. 3b). However, the molar mass parameters of the Tencel samples, except for the calculated mass values (Tables S4–S6), were similar for the samples pretreated by the three methods. No significant differences in the molar mass plots or SEC-elution patterns were observed for the Tencel samples stirred in 8% (w/w) LiCl/DMAc for different times. Stirring the pretreated Tencel sample in 8% (w/w) LiCl/DMAc for 1 week was sufficient to obtain constant SEC/MALLS analysis data.
The SEC/MALLS results of Bemliese are shown in Fig. 3c, Figs. S8–S10, and Tables S7–S9. The molar mass plot was the same line regardless of the pretreatment (Fig. 3c). The SEC-elution patterns of Bemliese pretreated by EDA and water soaking were similar. However, the peak area and SEC-elution pattern for the freeze-dried Bemliese sample were different from those pretreated by EDA and water soaking. Not only the calculated mass values, but also the other molar mass parameters of the freeze-dried Bemliese were different from those of the EDA- and water-pretreated samples (Tables S7–S9). The Mn, Mw, and DPw values of freeze-dried Bemliese were higher than those of EDA- and water-pretreated Bemliese, whereas the calculated mass values were lower. Thus, the SEC/MALLS data obtained for the EDA- and water-pretreated samples were regarded as being constant and more accurate than those of the freeze-dried sample.
The SEC/MALLS data of SBSP are shown in Fig. 3d and Table S10. The molar mass plot was the same regardless of the pretreatment and stirring time, and the SEC-elution patterns were almost the same (Fig. 3d). All of the molar mass parameters were similar for the samples pretreated by the two methods (Table S10).
Discussion
As described in the previous section, the molar mass parameters, including the calculated mass values, of EDA- and water-pretreated viscose rayon, Tencel, Bemliese, and SBSP were similar. Consequently, EDA pretreatment resulted in almost no mass loss of the low-molar-mass fractions in the three regenerated cellulose samples and SBSP used in this study. Furthermore, the SEC/MALLS data for the cellulose samples obtained after stirring the EDA- and water-pretreated samples in 8% (w/w) LiCl/DMAc for 1 week can be regarded as being reproducible and constant for analytical studies of their molar masses and molar mass distributions.
The representative molar mass plots and SEC-elution patterns of the three regenerated cellulose samples and SBSP pretreated by EDA soaking are shown in Fig. 4a, in which the peak-top heights of the SEC-elution patterns were adjusted to be similar. All of the SEC-elution patterns showed mostly single peaks without additional low-molar-mass peaks owing to, for instance, hemicelluloses (Ono et al. 2020, 2022a, b). The hemicellulose molecules originally present in the softwood chips were mostly removed by the acid sulfite pulping and subsequent bleaching processes for production of SBSP, differing from softwood and hardwood bleached kraft pulps (Ono et al. 2017, 2018). The peak-top elution volume increased in the order of SBSP < Bemliese < Tencel < viscose rayon, showing that their molar masses decreased in the opposite order of SBSP > Bemliese > Tencel > viscose rayon. All of the molar mass plots were roughly on the same line and the molar mass decreased with increasing SEC-elution volume. This showed that all of the cellulose molecules in the four samples were dispersed in 1% (w/v) LiCl/DMAc at the individual molecular level without forming any aggregates, and they were suitably separated by the SEC column depending on their sizes.

a Molar mass plots and SEC-elution patterns of the three regenerated cellulose samples and SBSP and b the corresponding double logarithmic plots (or conformation plots)
Double logarithmic plots, or conformation plots, of the four cellulose samples shown in Fig. 4a are shown in Fig. 4b. All of the plots were roughly on the same line, and the slopes were 0.58–0.62, showing that all of the cellulose molecules in the four samples had linear random-coil conformations in 1% (w/v) LiCl/DMAc without any branched or shrunk structures. This is reasonable as pure β-(1 → 4)-linked glucans. We have reported that softwood holocellulose samples, softwood bleached kraft pulps (SBKPs), 17.5% NaOH-extracted softwood holocellulose and SBKP samples (i.e., α-cellulose samples prepared from softwood holocellulose and SBKP samples, respectively), and dilute acid-hydrolyzed SBKP show conformation plots with slope values of < 0.45. These results indicate that some cellulose molecules in the high-molar-mass fractions of these samples have branched structures with glucomannan through lignin molecules or lignin fragments (Ono et al. 2017, 2018; Ono and Isogai 2020). Differing from the above softwood-originating samples, SBSP did not have branched structures as in the cases of hardwood kraft pulps and cotton, bacterial, tunicate, and algal cellulose samples (Ono et al. 2016b, 2017, 2018). This is the reason why SBSP is used as dissolving pulp fibers for production of regenerated cellulose samples and cellulose derivatives.
The molar mass parameters of the four cellulose samples are summarized in Table 1. The average values for the viscose rayon and Tencel samples were calculated from those obtained for the EDA-pretreated, water-pretreated, and freeze-dried samples. For Bemliese, the average values were calculated from those of only the EDA- and water-pretreated samples. The values obtained for freeze-dried Bemliese were excluded because they were clearly different from those obtained for the EDA- and water-pretreated samples, as described in the previous section (Tables S7–S9 in the Electronic Supplementary Material).
When the Mark–Houwink–Sakurada equation, [η] = 0.909 × DPv0.9, was used, the average DPv value was similar to that of the average DPw value for each regenerated cellulose sample. However, the average DPv value of SBSP (610) was much lower than the DPw value (1810) determined by SEC/MALLS. The DPv values of SBSP were almost unchanged for the SBSP/Cu(EDA)2(OH)2 solutions stirred for 10, 20, and 40 min. This clear discrepancy between the DPv and DPw values for SBSP may have been caused by the stability of SBSP in the alkaline 0.5 M Cu(EDA)2(OH)2 solution. The SBSP sample may have contained some chemical structures susceptible to depolymerization by the alkaline Cu(EDA)2(OH)2 solution. Generally, the C=O groups in cellulosic pulps cause depolymerization by β-alkoxy elimination under alkaline conditions, although the presence of such C=O groups could not be detected in the solid-state 13C-NMR spectrum of SBSP (Figs. 2 and S1). Thus, a small amount of C=O groups present randomly along each cellulose molecule of SBSP may have caused the low DPv value.
Although the solid-state 13C-NMR spectra of the regenerated cellulose samples indicated the presence of C=O groups as small peaks at ~ 97 ppm (Fig. 2), there were no significant differences between the DPw and DPv values (Table 1). This is probably because the C=O groups susceptible to the alkaline Cu(EDA)2(OH)2 solution are located close to both ends of each cellulose chain in the regenerated cellulose samples, whereas the C=O groups in SBSP are located more randomly along each cellulose chain. However, this is speculative, and further studies are needed to clarify the reason for the discrepancy between the DPv and DPw values for SBSP.
Conclusions
The three commercial regenerated cellulose samples, viscose rayon, Tencel, and Bemliese, exhibited similar solid-state 13C-NMR spectra to low-crystallinity cellulose II. The 13C-NMR spectra of the three samples had small peaks at ~ 97 ppm, which may be ascribed to the hydrated C6-aldehyde groups formed during the commercial fiber production processes. The three regenerated cellulose samples were dissolved in 8% (w/w) LiCl/DMAc by EDA-soaking, water-soaking, and freeze-drying pretreatment and subsequent stirring of the pretreated samples in 8% (w/w) LiCl/DMAc for 1 week. Based on the calculated mass values obtained by SEC/MALLS, almost all of the cellulose molecules in the EDA- and water-pretreated regenerated cellulose samples were dissolved in 8% (w/w) LiCl/DMAc and subjected to SEC/MALLS without significant mass loss. However, 10%–20% of the freeze-dried samples were not subjected to SEC/MALLS, probably because of incomplete dissolution in 8% (w/w) LiCl/DMAc. The average Mn and Mw values calculated from the SEC/MALLS data were almost the same as those obtained for the EDA- and water-pretreated samples. The average DPw values were calculated to be 340, 530, 880, and 1810 for viscose rayon, Tencel, Bemliese, and SBSP, respectively. The conformation plots of the samples had slopes of 0.58–0.62, showing that all of the cellulose molecules in the four samples were dissolved in 1% (w/v) LiCl/DMAc, forming linear random-coil conformations. For the regenerated cellulose samples, the DPw values determined by SEC/MALLS were similar to the corresponding DPv values. However, for SBSP, the DPv values were lower than the DPw values, indicating the inaccuracy of the DPv values of SBSP.
References
Abu-Rous M, Varga K, Bechtold T, Schuster KC (2007) A new method to visualize and characterize the pore structure of TENCEL (Lyocell) and other man-made cellulosic fibres using a fluorescent dye molecular probe. J Appl Polym Sci 106:208–32091. https://doi.org/10.1002/app.26722
Ago M, Endo T, Hirotsu T (2004) Crystalline transformation of native cellulose from cellulose I to cellulose II polymorph by a ball-milling method with a specific amount of water. Cellulose 11:163–167. https://doi.org/10.1023/B:CELL.0000025423.32330.fa
Bikova T, Treimanis A (2002) Problems of the MMD analysis of cellulose by SEC using DMA/LiCl: a review. Carbohydr Polym 48:23–28. https://doi.org/10.1016/S0144-8617(01)00207-7
Biliuta G, Fras L, Strnad S, Harabagiu V, Coseri S (2010) Oxidation of cellulose fibers mediated by nonpersistent nitroxyl radicals. J Polym Sci Part a: Polym Chem 48:4790–4799. https://doi.org/10.1002/pola.24270
Borbély E (2008) Lyocell, the new generation of regenerated cellulose. Acta Polytech Hungarica 5:11–18
Cao X, Ding B, Yu J, Al-Deyab SS (2012) Cellulose nanowhiskers extracted from TEMPO-oxidized jute fibers. Carbohydr Polym 90:1075–1080. https://doi.org/10.1016/j.carbpol.2012.06.046
Duchemin BJCZ, Newman RH, Staiger MP (2007) Phase transformations in microcrystalline cellulose due to partial dissolution. Cellulose 14:311–320. https://doi.org/10.1007/s10570-007-9121-4
Dudley RL, Fyfe CA, Stephenoson PJ, Deslandes Y, Hamer GK, Marchessault RH (1983) High-resolution carbon-13 CP/MAS NMR spectra of solid cellulose oligomers and the structure of cellulose II. J Am Chem Soc 105:2469–2472. https://doi.org/10.1021/ja00346a059
Dupont AL (2003) Cellulose in lithium chloride/N, N-dimethylacetamide, optimization of a dissolution method using paper substrates and stability of the solutions. Polymer 44:4117–4126. https://doi.org/10.1016/S0032-3861(03)00398-7
Fadavi F, Abdulkhani A, Zadeh ZE, Hamzeh Y (2021) Influence of swelling media on the crystallinity and accessibility of lyocell fibers: An FTIR and 13C NMR analysis. J Nat Fiber 19:6365–6376. https://doi.org/10.1080/15440478.2021.1916675
Fink HP, Weigle P, Ganster J (2001) Structure formation of regenerated cellulose materials from NMMO-solutions. Prog Polym Sci 26:1473–1524. https://doi.org/10.1016/S0079-6700(01)00025-9
Follain N, Marais MF, Montanari S, Vignon MR (2010) Coupling onto surface carboxylated cellulose nanocrystals. Polmer 51:5332–5344. https://doi.org/10.1016/j.polymer.2010.09.001
From M, Larsson PT, Andreasson B, Medrohho B, Svanedal I, Edlund H, Norgren M (2020) Tuning the properties of regenerated cellulose: effects of polarity and water solubility of the coagulation medium. Carbohydr Polym 236:116068. https://doi.org/10.1016/j.carbpol.2020.116068
Funahashi R, Okita Y, Hondo H, Zhao M, Saito T, Isogai A (2017) Different conformations of surface cellulose molecules in native cellulose microfibrils revealed by layer-by-layer peeling. Biomacromolecules 18:3687–3694. https://doi.org/10.1021/acs.biomac.7b01173
Henninges U, Vejdovszky P, Siller M, Jeong MJ, Rosenau T, Potthast A (2014) Finally dissolved! Activation procedures to dissolve cellulose in DMAc/LiCl prior to size exclusion chromatography analysis – A review. Curr Chromatogr 1:52–68. https://doi.org/10.2174/2213240601666131118220030
Horii F, Hirai A, Kitamaru R (1982) Solid-state high-resolution 13C-NMR studies of regenerated cellulose samples with different crystallinities. Polym Bull 8:163–170. https://doi.org/10.1007/BF00263023
Horii F, Hirai A, Kitamaru R (1983) Solid-state 13C-NMR study of conformations of oligosaccharides and cellulose - conformation of CH2OH group about the exo-cyclic C-C bond. Polym Bull 10:357–361. https://doi.org/10.1007/BF00281948
Ibbett RN, Domvoglou D, Fasching M (2007) Characterisation of the supramolecular structure of chemically and physically modified regenerated cellulosic fibres by means of high-resolution Carbon-13 solid-state NMR. Polymer 48:1287–1269. https://doi.org/10.1016/j.polymer.2006.12.034
Idström A, Schantz S, Sundberg J, Chmelka BF, Gatenholm P, Nordstierna L (2016) 13C NMR assignments of regenerated cellulose from solid-state 2D NMR spectroscopy. Carbohydr Polym 151:480–487. https://doi.org/10.1016/j.carbpol.2016.05.107
Isogai A, Usuda M, Kato T, Uryu T, Atalla RH (1989a) Solid-state CP/MAS 13C NMR study of cellulose polymorphs. Macromolecules 22:3168–3172. https://doi.org/10.1021/ma00197a045
Isogai A, Mutoh N, Onabe F, Usuda M (1989b) Viscosity measurements of cellulose/SO2-amine-dimethylsulfoxide solution. Sen’i Gakkaishi 45:299–306. https://doi.org/10.2115/fiber.45.7_299
Isogai A, Saito T, Fukuzumi H (2011) TEMPO-oxidized cellulose nanofibers. Nanoscale 3:71–85. https://doi.org/10.1039/c0nr00583e
Isogai A, Hänninen T, Fujisawa S, Saito T (2018) Catalytic oxidation of cellulose with nitroxyl radicals under aqueous conditions. Prog Polym Sci 86:122–148. https://doi.org/10.1016/j.progpolymsci.2018.07.007
Isogai A (2022) TEMPO-catalyzed oxidation of polysaccharides. Polym J 54:387–402. https://doi.org/10.1038/s41428-021-00580-1
Jin H, Zha C, Gu L (2007) Direct dissolution of cellulose in NaOH/thiourea/urea aqueous solution. Carbohydr Res 342:851–858. https://doi.org/10.1016/j.carres.2006.12.023
Kreze T, Malej S (2003) Structural characteristics of new and conventional regenerated cellulose fibers. Text Res J 73:675–684. https://doi.org/10.1177/0040517503073008
Kuchtoá G, Herink P, Herink T, Chýlková J, Mikulášek P, Dušek L (2023) From lab-scale to pilot-scale treatment of real wastewater from the production of rayon fiber. Process Saf Environ 171:834–846. https://doi.org/10.1016/j.psep.2023.01.059
Larsson PT, Westlund PO (2005) Line shapes in CP/MAS 13C NMR spectra of cellulose I. Spectrochim Acta Part A 62:539–546. https://doi.org/10.1016/j.saa.2005.01.021
Li R, Zhang L, Xu M (2012) Novel regenerated cellulose films prepared by coagulating with water: structure and properties. Carbohydr Polym 87:95–100. https://doi.org/10.1016/j.carbpol.2011.07.023
Li Z, Sathitsuksanoh N, Zhang W, Goodell B, Renneckar S (2017) Towards an understanding of cellulose microfibril dimensions from TEMPO-oxidized pulp fiber. ACS Symp Ser 1251:55–73. https://doi.org/10.1021/bk-2017-1251.ch003
Li H, Legere S, He Z, Zhang H, Li J, Yang B, Zhang S, Zhang L, Zheng L, Ni Y (2018) Methods to increase the reactivity of dissolving pulp in the viscose rayon production process: a review. Cellulose 25:3733–3753. https://doi.org/10.1007/s10570-018-1840-1
Lin F, You Y, Yang X, Jiang X, Lu Q, Wang T, Huang B, Lu B (2017) Microwave-assisted facile synthesis of TEMPO-oxidized cellulose beads with high adsorption capacity for organic dyes. Cellulose 24:5025–5040. https://doi.org/10.1007/s10570-017-1473-9
Marx M (1955) Viskosimetrische molekulargewichtsbestimmung von cellulose in kupfer-äethylendiamin. Makromol Chem 14:157–176. https://doi.org/10.1002/macp.1955.020160118
Maunu S, Liitiä T, Kauliomäki S, Hortling B, Sundquist J (2000) 13C CPMAS NMR investigations of cellulose polymorphs in different pulps. Cellulose 7:147–159. https://doi.org/10.1023/A:1009200609482
Mendes ISF, Prates A, Evtuguin DV (2021) Production of rayon fibres from cellulosic pulps: state of the art and current developments. Carbohydr Polym 273:118466. https://doi.org/10.1016/j.carbpol.2021.118466
Mori T, Chikayama E, Tsuboi Y, Ishida N, Shisa N, Noritake Y, Moriya S, Kikuchi J (2012) Exploring the conformational space of amorphous cellulose using NMR chemical shifts. Carbohydr Polym 90:1197–1203. https://doi.org/10.1016/j.carbpol.2012.06.027
Moulthrop JS, Swatloski RP, Moyna G, Rogers RD (2005) High-resolution 13C NMR studies of cellulose and cellulose oligomers in ionic liquid solutions. Chem Commun 2005:1557–1559. https://doi.org/10.1039/B417745B
Newman RH, Hemmingson J (1994) Carbon-13 NMR distinction between categories of molecular order and disorder in cellulose. Cellulose 2:95–110. https://doi.org/10.1007/BF00816383
Nomura S, Kugo Y, Erata T (2020) 13C NMR and XRD studies on the enhancement of cellulose II crystallinity with low concentration NaOH posttreatments. Cellulose 27:3553–3563. https://doi.org/10.1007/s10570-020-03036-6
Ono Y, Ishida T, Soeta H, Saito T, Isogai A (2016a) Reliable dn/dc values of cellulose, chitin, and cellulose triacetate dissolved in LiCl/N, N-dimethylacetamide for molecular mass analysis. Biomacromol 17:192–199. https://doi.org/10.1021/acs.biomac.5b01302
Ono Y, Tanaka R, Funahashi R, Takeuchi M, Saito T, Isogai A (2016b) SEC–MALLS analysis of ethylenediamine-pretreated native celluloses in LiCl/N, N-dimethylacetamide: softwood kraft pulp and highly crystalline bacterial, tunicate, and algal celluloses. Cellulose 23:1639–1647. https://doi.org/10.1007/s10570-016-0948-4
Ono Y, Saito T, Isogai A (2017) Branched structures of softwood celluloses: proof based on size-exclusion chromatography and multi-angle lase-light scattering. Nanocellul. Prep. Prop Appl. 1251:151–169. https://doi.org/10.1021/bk-2017-1251.ch008
Ono Y, Funahashi R, Saito T, Isogai A (2018) Investigation of stability of branched structures in softwood cellulose using SEC/MALLS/RI/UV and sugar composition analysis. Cellulose 25:2667–2679. https://doi.org/10.1007/s10570-018-1713-7
Ono Y, Isogai A (2020) Analysis of celluloses, plant holocelluloses, and wood pulps by size-exclusion chromatography/multi-angle laser-light scattering. Carbohydr Polym 251:117045. https://doi.org/10.1016/j.carbpol.2020.117045
Ono Y, Takeuchi M, Isogai A (2021) TEMPO/NaBr/NaClO and NaBr/NaClO oxidations of cotton linters and ramie celluloses. Cellulose 28:6035–6049. https://doi.org/10.1007/s10570-021-03944-1
Ono Y, Takeuchi M, Isogai A (2022a) Changes in neutral sugar composition, molar mass and molar mass distribution, and solid-state structures of birch and Douglas fir by repeated sodium chlorite delignification. Cellulose 29:2119–2129. https://doi.org/10.1007/s10570-022-04448-2
Ono Y, Takeuchi M, Zhou Y, Isogai A (2022b) Characterization of cellulose and TEMPO-oxidized celluloses prepared from Eucalyptus globulus. Holzforschung 76:169178
Ono Y, Takeuchi M, Kimura S, Puangsin B, Wu CN, Isogai A (2022c) Structures, molar mass distributions, and morphologies of TEMPO-oxidized bacterial cellulose fibrils. Cellulose 29:4977–4992. https://doi.org/10.1007/s10570-022-04617-3
Östlund Å, Idström A, Olsson C, Larsson PT, Nordstierna L (2013) Modification of crystallinity and pore size distribution in coagulated cellulose films. Cellulose 20:1657–1667. https://doi.org/10.1007/s10570-013-9982-7
Potthast A, Röhrling J, Rosenau T, Borgards A, Sixta H, Kosma P (2003) A novel method for the determination of carbonyl groups in cellulosics by rluorescence labeling. 3. Monitoring oxidative processes. Biomacromolecules 4:743–749. https://doi.org/10.1021/bm025759c
Sayyed AJ, Deshmukh NA, Pinjari DV (2019) A critical review of manufacturing processes used in regenerated cellulose fibres: viscose, cellulose acetate, cuprammonium, LiCl/DMAc, ionic liquids, and NMMO based lyocell. Cellulose 26:2913–2940. https://doi.org/10.1007/s10570-019-02318-y
Schulz GV, Dinglinger A (1941) Molekulargewichtsbestimmungen an einer Reihe von Polymethacrylsäuremethylestern nach verschiedenen Methoden [osmotisch, viscosimetrisch und durch Fällungstitration]. J Prakt Chem 158:136–162. https://doi.org/10.1002/prac.19411580113
Shanshan G, Jianqin W, Zhengwei J (2012) Preparation of cellulose films from solution of bacterial cellulose in NMMO. Carbohydr Polym 87:1020–1025. https://doi.org/10.1016/j.carbpol.2011.06.040
Shinoda R, Saito T, Okita Y, Isogai A (2012) Relationship between length and degree of polymerization of TEMPO-oxidized cellulose nanofibrils. Biomacromol 13:842–849. https://doi.org/10.1021/bm2017542
Silbermann S, Weilach C, Kliba G, Fackler K, Potthast A (2017) Improving molar mass analysis of cellulose samples with limited solubility. Carbohydr Polym 178:302–310. https://doi.org/10.1016/j.carbpol.2017.09.031
Siller M, Ahn K, Pircher N, Rosenau T, Potthast A (2014) Dissolution of rayon fibers for size exclusion chromatography: a challenge. Cellulose 21:3291–3301. https://doi.org/10.1007/s10570-014-0356-6
Veit D (2022) Cellulosic man-made fibers. In: Fibers: history, production, properties, market. Springer Nature Switzerland, Switzerland, pp. 1831–882. https://doi.org/10.1007/978-3-031-15309-9
Wei X, Wang Y, Li J, Wang F, Chang G, Fu T, Zhou W (2018) Effects of temperature on cellulose hydrogen bonds during dissolution in ionic liquid. Carbohydr Polym 201:387–391. https://doi.org/10.1016/j.carbpol.2018.08.031
Yamamoto M, Kuramae R, Yanagisawa M, Ishii D, Isogai A (2011) Light-scattering analysis of native wood holocelluloses totally dissolved in LiCl-DMI solutions: high probability of branched structures in inherent cellulose. Biomacromolecules 12:3982–3988. https://doi.org/10.1021/bm201211z
Yang H, Wang T, Oehme D, Petridis L, Hong M, Kubicki JD (2018) Structural factors affecting 13C NMR chemical shifts of cellulose: a computational study. Cellulose 25:23–36. https://doi.org/10.1007/s10570-017-1549-6
Yuan S, Tyufekchiev MV, Timko MT, Schmidt-Rohr K (2022) Direct quantification of the degree of polymerization of hydrolyzed cellulose by solid-state NMR spectroscopy. Cellulose 29:2131–3144. https://doi.org/10.1007/s10570-022-04433-9
Zhang X, Xiao N, Wang H, Liu C, Pan X (2018) Preparation and characterization of regenerated cellulose film from a solution in lithium bromide molten salt hydrate. Polymers 10:614. https://doi.org/10.3390/polym10060614
Zhao H, Kwak JH, Wang Y, Franz JA, White JM, Holladay JE (2007) Interactions between cellulose and N-methylmorpholine-N-oxide. Carbohydr Polym 67:97–103. https://doi.org/10.1016/j.carbpol.2006.04.019
Zhou Y, Ono Y, Takeuchi M, Isogai A (2020) Changes to the contour length, molecular chain length, and solid-state structures of nanocellulose resulting from sonication in water. Biomacromolecules 21:2346–2355. https://doi.org/10.1021/acs.biomac.0c00281
Acknowledgments
We thank Edanz for editing a draft of this manuscript.
Funding
Open access funding provided by The University of Tokyo. This study was supported in part by the New Energy and Industrial Technology Development Organization (NEDO), Japan.
Author information
Authors and Affiliations
Contributions
All of the authors contributed to conception, design, the experiments, and the analyses. All of the authors read and approved the final manuscript. The manuscript was approved by all of the authors for publication.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence this study.
Ethical approval and consent to participate
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ono, Y., Hou, G., Chitbanyong, K. et al. Molar masses and molar mass distributions of commercial regenerated cellulose materials and softwood dissolving pulp determined by SEC/MALLS. Cellulose 30, 8221–8233 (2023). https://doi.org/10.1007/s10570-023-05414-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-023-05414-2