
Abstract
Chromophores, colored substances of rather high stability that reduce brightness, are present in all kinds of cellulosic products, such as pulp, fibers, aged cellulosic material, and even in very low concentrations in highly bleached pulps. Thus, they are the prime targets of industrial pulp bleaching. In this study, the three cellulosic key chromophores 2,5-dihydroxy-1,4-benzoquinone (DHBQ, 1), 5,8-dihydroxy-1,4-naphthoquinone (DHNQ, 2), and 2,5-dihydroxyacetophenone (DHAP, 3) were bleached with ozone at pH 2 resembling industrial conditions (Z-stage). Bleaching kinetics were followed by UV/Vis spectrophotometry. The chemical structures of the degradation products were analyzed using NMR spectroscopy as well as GC/MS and confirmed by comparison with authentic reference compounds. The main, stable intermediates in the ozonation reactions have been elucidated by employing ozone not in excess but roughly stoichiometric amounts: mesoxalic acid (4) from DHBQ (1), rhodizonic acid (5) from DHNQ (2), and hydroxy-[1,4]-benzoquinone (8) from DHAP (3). As the final products at a large excess of ozone, a complex mixture of carboxylic acids (C1 to C4) was obtained, with the C4 acids being formed by subsequent condensation of smaller fragments (malonic acid and mesoxalic acid) rather than directly as ozonation products. At shorter reaction times and lower ozone excess, some aldehydes and ketones (C2 and C3) were contained in addition. The mixture of the degradation products was not completely stable but tended to undergo further changes, such as decarboxylation and condensation reactions. The reaction mechanisms of degradation by ozone, intermediate formation and re-condensation are described and discussed.
Graphic abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Chromophores in cellulosic products, such as pulp and paper, are responsible for yellowing and brightness revision. The translation of “chromophore” from Ancient Greek literally means “carrier of color” (Rosenau et al. 2011). In wood-derived cellulosic materials, chromophores mainly originate from residual lignin and hemicelluloses and their degradation products. Pulping and bleaching processes aim at reducing or eliminating them. In lignin-free or highly bleached cellulosics, such as bacterial celluloses, cotton, highly bleached pulps, historic or aged rag papers, they stem from polysaccharides and their degradation products. The reason for re-formation of chromophores from polysaccharides is the substantial number of processing steps during pulping and bleaching, which might damage cellulose oxidatively, and natural as well as artificial aging processes (thermal, chemical, or irradiation stress) which cause the introduction of oxidized groups along the cellulose chain, followed by condensation reactions to chromophoric moieties (see for instance: Potthast et al. 2005; Yoneda et al. 2008; Henniges et al. 2013). Already in the 1970s studies had shown that carbohydrates can form a plethora of aromatic and quinoid compounds under acidic as well as alkaline conditions. These complex product mixtures are sometimes described as “Theander products” (Popoff and Theander 1976, 1978; Popoff et al. 1976; Olsson et al. 1977; Forsskahl et al. 1976; Theander and Westerlund 1980).
Defined residual chromophoric structures in cellulosics have become analytically accessible through the CRI (chromophore release and identification) method (Rosenau et al. 2004), their amount being marginal and ranging from ppm (chromophore mixtures) down to the single-digit ppb range for isolated compounds. It should be pointed out again that the method is intrinsically limited to lower chromophore concentrations and not able to analyze pulps with high contents of chromophores, such as those in pulps during bleaching. The human eye is very sensitive in the range of 500–600 nm, which covers the colors green and yellow, and therefore even the tiniest amounts of chromophores in cellulosic materials can be visually recognized as a yellow tint (Sharpe et al. 2011; Bowmaker and Dartnall 1979). The CRI method has been applied to different cellulosic pulps from various bleaching sequences, bacterial celluloses, cotton materials and aged pulps (i.e., cellulose I substrates), lyocell and viscose fibers (i.e., cellulose II products) as well as some cellulose derivatives, in which defined chromophores have been isolated (Rosenau et al. 2004, 2007, 2011, 2014). These carbohydrate-derived chromophores in cellulosic materials generally belong to three compound classes: hydroxy-[1,4]-benzoquinones, hydroxy-[1,4]-naphthoquinones, and hydroxyacetophenones (Korntner et al. 2015).
The cellulosic key chromophores offer some interesting chemical characteristics which are the reason why they are almost ubiquitous (Hosoya et al. 2013a, b, 2015a). First, they possess strongly delocalized double bonds due to exceptionally strong resonance stabilization which renders them the most successful survivors of bleaching treatments. Most bleaching agents act primarily on double bonds but are less effective when such localized double bonds are not present. Second, the key chromophores represent minima on the thermodynamic C–H–O maps which means they are easily formed in condensation reactions from multi-component mixtures, such as those formed upon carbohydrate degradation. The key chromophores are thus not only the last chromophores to remain upon bleaching, but also the first ones to be regenerated upon aging (Korntner et al. 2015; Hettegger et al. 2019). They usually exhibit pronounced color changes upon pH variation in aqueous solutions, or when dissolved in organic media of different polarities (Krainz et al. 2009).
Ozone is a highly reactive allotrope of oxygen and has been known since 1785 when Martinus van Marum observed its formation upon electric discharge in oxygen gas. Its pungent smell gave it the name (Ancient Greek oζειν = to smell). In 1865 its structure was determined to be O3. The ozone molecule is V-shaped and consists of three oxygen atoms with two equally long O–O bonds (Hollemann et al. 1985). It is one of the strongest known oxidants (standard potential E0 = 2.07 V) and is surpassed in oxidizing power by only a few chemicals, such as singlet oxygen (2.42 V), hydroxyl radicals (2.86 V), and fluorine (2.87 V) (Cotton et al. 1999). At the same time, ozone is regarded as a “traceless” and “clean” chemical, which explains its manifold applications in industry. The highly toxic gas shows a pale blue hue when concentrated. While surface-near ozone is responsible for some of the typical negative “smog” effects, stratospheric ozone is imperative for terrestrial life as it wards off cosmic irradiation.
Since the 1980s ozone is applied in the pulp and paper industry for bleaching purposes (so-called “Z-stages”), especially in ECF (elemental chlorine-free), and TCF (totally chlorine-free) bleaching sequences. The chemistry of ozone bleaching cannot be summarized here. For this purpose, we would like to refer to review articles and some key publications (Sjöström 1993; Sixta 2006; Ek et al. 2009; Suess 2010; Pouyet et al. 2014; Chirat et al. 1997; Chirat and Lachenal 1994, 1997; Lachenal et al. 2009). Suffice it to point out few aspects that are also directly relevant to the experiments performed in this work. The stability of ozone in water is rather low and is influenced by the respective conditions, such as pH, presence of organic matter, and temperature (von Gunten 2003). Especially at high temperatures and in alkaline media ozone becomes less stable and tends to decompose fast, generating reactive oxygen species (Streng 1961). Depending on the conditions, the half-life time of ozone in water ranges from hours to only seconds. Hence, ozone bleaching is carried out at low pH (2–3), at temperatures not far exceeding room temperature (30–60 °C) and mostly in medium consistency (10–15% cellulose) or high consistency (30–35% cellulose) processes where the presence of liquid water is limited (e.g. Hruschka 1986, Patt et al. 1991, Soteland 1978), resulting in a short diffusion path through the thin immobile hydrate film on the fibers, which enables a quick reaction of the dissolved O3 molecule. The risk of oxidative damage of cellulose (DP loss) in the Z-stage increases with time because reactive moieties in lignin are more and more consumed so that unwanted parallel reactions (oxidation of carbohydrates, ozone degradation, radical formation) become more prominent (Chirat and Lachenal 1997; Gierer 1997; Reitberger et al. 1999).
Ozone, as a strong electrophile, attacks sites with high electron density, especially under the acidic conditions of pulp bleaching. It reacts quite selectively with olefinic and aromatic double bonds to give aldehydes, ketones, and acids (Criegee 1975; Sjöström 1993; Reitberger et al. 1999; Ragnar 1999a, b; Hendrickx and Vinckier 2003; Epstein and Donahue 2010). This reaction, called ozonolysis, has been a standard method for the structure elucidation of organic compounds until superseded by NMR-based and MS-based methods. The reaction proceeds according to the well-known Criegee mechanism via primary and secondary ozonides (Criegee 1975; Kuczkowski 1992), see Scheme 1. Apart from these ionic pathways, also undesired homolytic (radical) reactions are possible which are triggered mainly by the presence of transition metal ions or higher pH (Ek et al. 2009; Reitberger et al. 1999; Sixta 2006).

Stepwise mechanism of the ozonolysis of an olefin (R1, R2, R3 = alkyl, H) in an aqueous medium (“Criegee mechanism”)
The dominant Criegee mechanism is a sequence of ionic reactions, [2 + 3]-cycloadditions (1,3-dipolar additions) and fragmentations. The first step consists of a [2 + 3]-cycloaddition of the electron-deficient ozone with an (electron-rich) olefinic bond (which can also be a localized double bond in an aromatic system). This produces a 1,2,3-trioxolane, the primary ozonide, which undergoes fragmentation (reverse 1,3-dipolar addition) to a carbonyl compound and a highly reactive carbonyl-O-oxide, the actual Criegee intermediate. In another [2 + 3]-cycloaddition a 1,2,4-trioxolane, the secondary ozonide, is formed, which is even stable enough to be isolated in some cases (Criegee 1975; Kuczkowski 1992, Epstein and Donahue 2010). In an aqueous medium, the secondary ozonide hydrolyzes to form two carbonyl fragments and hydrogen peroxide. If the carbonyl compounds are aldehydes, the formed H2O2 will almost immediately oxidize them to the respective carboxylic acids. External addition of H2O2 can thus enforce complete conversion to acids. Note that secondary literature does not always reflect some details of the mechanism in aqueous media quite correctly because data are just transferred from the classical ozonation in organic medium: the simultaneous formation of hydrogen peroxide is often neglected, the hydrolysis of the secondary ozonide is an acid-catalyzed process and not triggered by hydroxy anions, and the product carbonyls are in equilibrium with their hydrates.
The behavior of the three cellulosic key chromophores 2,5-dihydroxy-[1,4]-benzoquinone (DHBQ, 1), 5,8-dihydroxy-[1,4]-naphthoquinone (DHNQ, 2) and 2,5-dihydroxyacetophenone (DHAP, 3), see Scheme 2, towards common bleaching agents has been studied and also their behavior under conditions simulating industrial bleaching has been studied in detail. Both hydrogen peroxide in alkaline medium (P stage bleaching; for DHBQ: Hosoya and Rosenau 2013, for DHNQ: Zwirchmayr et al. 2017, for DHAP: Zwirchmayr et al. 2018) and chlorine dioxide in acidic medium (D stage bleaching; for DHNQ: Hosoya et al. 2018, for DHBQ: Guggenberger et al. 2020) have been covered. In this account, we would like to complete the picture and add a study on the behavior of the three key chromophores towards ozone as used industrially in the Z-bleaching stages. The experiments in an aqueous medium at acidic pH were complemented with tests of salt effects and ozonation reactions in organic solvents for comparison.

Chemical structures of the cellulosic key chromophores DHBQ (1), DHNQ (2), and DHAP (3) used in the ozone bleaching experiments
Results and discussion
Under classical ozone bleaching conditions the behavior of ozone is much more complex than upon controlled application in synthesis or structural analysis. Other reactions than the Criegee pathway involve charge transfer mechanisms due to the strong electron-deficient character of ozone, formation of reactive oxygen species of which the hydroxyl radicals are most detrimental to cellulose during bleaching (Chirat and Lachenal 1994, 1997, Ragnar et al. 1999a, b; Hoigne and Bader 1976, Pouyet et al. 2014). Industrial bleaching with ozone is carried out at a pH of 2–3 (Ek et al. 2009; Hruschka 1986; Patt et al. 1991), which was also chosen for our experiments to mimic conditions of industrial Z-stages. In a strongly acidic medium, the conversion of ozone into reactive oxygen species (ROS) is minimized, while it is pronounced in less strongly acidic or neutral and especially alkaline media, with hydroxyl radicals (·OH) being the most “dangerous” species with regard to unwanted cellulose oxidation and eventually chain degradation. For practical reasons—and because it was known that the presence of pulp might influence the reaction rate, but not the reaction mechanisms per se (Krainz et al. 2009)—the first set of reactions was carried out in aqueous solution (see experimental section) before working with a simulated medium-consistency setup (12% of pulp). Concentrations of the chromophores were set so that the reactions could be conveniently monitored by UV/Vis analysis, i.e., with decolorization occurring in about 10 min. The experiments were performed at room temperature, otherwise the reactions (= decoloration) would have proceeded too rapidly to be well followed from a practicability point of view. Due to the limited solubility of the chromophores in acidic media (while it is excellent in alkaline media) the concentration range for the chromophores dissolved in water was limited anyway. In solid form, the key chromophores are substances with intense color, which translates into their acidic solutions and changes pronouncedly upon transition into alkaline medium (Fig. 1).

The three cellulosic key chromophores DHBQ (1), DHNQ (2), and DHAP (3) as neat solids and in acidic and alkaline aqueous solution (approx. 1 mM)
In some initial tests, the reactions with the chromophores were carried out in the presence of a trap selective for hydroxyl radicals, 5-(2-propyl)-γ-tocopherol (Rosenau et al. 2006), in an amount equimolar to the employed chromophores. This trap can be used under drastic conditions, such as in concentrated alkali during viscose production, and its products can be easily separated even from very complex media due to its high lipophilicity. From these experiments, the contribution of hydroxyl radicals was seen to be minor in the first half of the reaction time (no trapping product during the first 5 min), but to increase significantly in the second half, so that about 10–20% of the trap reacted. This demonstrated that the radical processes involving ·OH, although not prominent in the initial step of the reaction, were well involved in later stages, so that the complexity of the final products (see below) must also be contributed, at least in part, to the involvement of hydroxyl radicals. Additionally, the experiments were conducted with oxygen (in the absence of ozone) which gave no conversion at all, so that a direct conversion through oxygen can be excluded, although its involvement or activation in later stages by ozone and ozone-derived reactive species can of course not be ruled out.
Aliquots for analysis were withdrawn about every minute and the changes were followed by UV/Vis absorption spectrophotometry. Apart from special experiments with near-stoichiometric amounts of O3—employed to better identify reaction intermediates (see below)—ozone was generally present in large excess. Generally, the visible light part of the UV/Vis spectra (λ = 400–700 nm) was recorded. The products of chromophore degradation were assumed to be compounds containing carbonyl and carboxyl moieties (Ahn et al. 2019) so that the range below approx. 400 nm would always be highly absorbing before and still after the reaction and thus would not allow reasonable monitoring of the changes. In the UV/Vis spectra of DHBQ (1) and DHAP (3) no distinct peaks in the range of 400–700 nm were observed (Figs. 2 and 6), and the absorption wavelength of 400 nm was chosen as a reference point. It still lies in the visible range of the light spectrum and coincides with yellow color. The spectrum of DHNQ (2) showed a broad absorption between 400 and 700 nm with two maxima and a shoulder (Fig. 3).

The reaction of ozone with DHBQ (1, c = 4.38 mM) in water. Left: bleaching kinetics (absorption at 400 nm); right: VIS spectra at different reaction times

The reaction of ozone with DHNQ (2, c = 0.91 mM) in water. Left: bleaching kinetics (absorption at 400 nm and 517 nm); right: VIS spectra at different reaction times
Ozone treatment of DHBQ
The color of the aqueous solution of DHBQ (1) was orange, which was represented by a broad peak at 400 nm. Upon ozone addition, the discoloration was complete within 5–6 min. The VIS spectra did not indicate any intermediates or parallel processes (Fig. 2). Logarithmization (ln) gave a good linear fit, which pointed to a typical first-order kinetics for the decoloration, i.e., the consumption of the chromophore. Whether the process in fact follows rather a pseudo-first-order kinetics, with ozone as the excess reagent whose concentration can be assumed to be constant, and which elementary, rate-determining step is described by this kinetics, cannot be answered from this data.
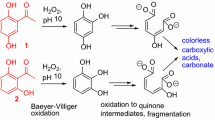
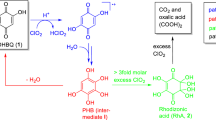
From the literature (Bernatek and Straumsgård 1959), the intermediates of the ozonation of benzo-[1,4]-quinone (para-benzoquinone) were known. Assuming a similar reaction also for DHBQ and hoping to be able to isolate some intermediates, we also conducted the ozonation of DHBQ in a way that ozone was not present in large excess or even deficient. For this purpose, the ozone concentration in an aliquot of the aqueous solution was determined and then DHBQ was added in a certain stoichiometric ratio. Apart from the non-reacted starting material, only one major product, accounting for about 80% of the DHBQ having reacted, was found, namely mesoxalic acid (4a). In the presence of water, this compound forms the respective ketohydrate, i.e., the 2,2-dihydroxy compound (4), while the 2-keto form is present in mesoxalate esters. The absence of products longer than C3, even at lower ozone input, seemed to indicate that the reaction of ozone with the “one-sided” mono-ozonation product to form the “two-sided” bis-ozonation product was clearly favored over the reaction of ozone with the starting material DHBQ (1). The primary product of ozonation, formyloxoacetic acid, is a strong reductant that is immediately oxidized to 4 by ozone, the ozonation side product H2O2, or air, or otherwise fragmented by losing either C=O (decarbonylation) or CO2 (decarboxylation). While the 2-keto form of mesoxalic acid (4a) is rather unstable undergoing similar CO-loss or CO2-loss already above 40 °C, the hydrate form (4) is completely stable even in boiling water because of strong stabilization by intramolecular hydrogen bonds in an entropically favored tetracyclic structure (rounded box in Scheme 3). In the presence of excess ozone, mesoxalic acid (4) produces initially oxalic acid and CO2, the oxalic acid slowly being further oxidized into two equivalents of CO2.

Ozonation of DHBQ (1) with about the double stoichiometric amount of ozone, affording mesoxalic acid (4a) as the main product, present as ketohydrate (4) in water
Ozone treatment of DHNQ
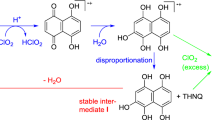
DHNQ (2) gave a dark red aqueous solution with two broad absorption maxima between 450 and 550 nm (Fig. 3). It is poorly water-soluble in acidic and neutral medium, but very well soluble in alkaline medium. Although the concentration used was lower than for DHBQ (1) and DHAP (3), the color was more intense than for the other two chromophores. Decoloration by ozone proceeded quickly within minutes. After about 180 s it became noticeable that absorption was not uniformly decreased over the whole wavelength range but declined more strongly at 515 nm than at 400 nm (Fig. 3). At this point, the initially red color of DHNQ changed into a light-yellow. These observations were indicative of the formation of a chromophoric intermediate, which was more stable towards ozone than the original chromophore and was more slowly degraded to colorless final products.
Attempts to isolate the intermediate were unsuccessful at first: both NMR spectroscopy (1H) and GC/MS provided no conclusive results. However, a seemingly unimportant observation provided the central clue: when neutralizing the bleached and colorless chromophore solution of DHNQ with NaOH it darkened again into yellow. Evidently, chromophoric structures were still present after the bleaching procedure, but they were colorless at pH 2 and became only visible in alkaline solution. Such a pH-dependent color change is very common: the deprotonation at higher pH increases the electron density in the system and shifts the absorption wavelengths to higher values. However, when a drop of Pb(OAc)2 solution was added into the bleached solution, an immediate dark, almost black discoloration occurred. This was indicative of the presence of rhodizonic acid, which in the presence of water exists as a dihydrate (5a), while the anhydrous form (5c) can only be obtained under special drying conditions (Scheme 4). In the form of the rhodizonate dianion (C6O62−, 5b), rhodizonic acid gives yellow solutions with alkali metal ions, red ones with earth alkali ions (Ca2+, Ba2+), and dark precipitates with some transition metal ions (Fe, Cu, Mn, etc.) and main group metal ions (e.g. Pb, Sb, Bi) (Feigl and Suter 1942; Chalmers and Telling 1967). Most characteristic is the extremely sensitive reaction with Pb2+ that leads to a bluish-black precipitate (5b-Pb in Scheme 4, right): rhodizonic acid has been used in criminology for the detection of gunshot traces even until our days (Werner et al. 2019).

Chemical structures of rhodizonic acid hydrate (5a), rhodizonate anion (5b), anhydrous rhodizonic acid (5c), lead rhodizonate (5b-Pb), and visual appearance of a lead rhodizonate precipitate
With this knowledge, the failure of 1H NMR and GC/MS to report the intermediate was understandable. The rhodizonate dianion does not possess any acidic protons, and it cannot be derivatized by common trimethylsilylating agents. However, 13C NMR spectroscopy was able to provide a convincing proof of its presence and structure (Fig. 4). The three resonances in acidic aqueous solution (left) changed to a single resonance in alkaline medium (right): rhodizonic acid is present as its dihydrate in an acidic aqueous medium but forms rhodizonate in alkaline medium, which has six magnetically equivalent carbon atoms. Note that—by contrast to its mirror-symmetric, non-aromatic parent compound rhodizonic acids—the rhodizonate dianion C6O62− is an aromatic, C6-centrosymmetric, very stable structure. The reaction of rhodizonic acid with excess ozone provides a mixture of C1–C4 carboxylic acids (see below).

13C NMR spectra, formulae, and visual appearances of the aqueous solution of rhodizonic acid (5) (left) and sodium rhodizonate (5a-Na) (right)
Interestingly, the intermediacy of rhodizonate has also been confirmed in the bleaching of DHBQ (1) with ClO2 (Guggenberger et al. 2020). Also in this case, the intermediate rhodizonate was more stable towards the bleaching agent than the starting chromophore. In the case of DHNQ-bleaching with ozone the intermediacy of DHBQ, and then the formation of rhodizonate from DHBQ as a subsequent process, can be excluded: no rhodizonate was detected upon ozone bleaching of DHBQ (see above) so that rhodizonic acid can be excluded as a follow-up product of DHBQ. When starting from DHNQ, it was obviously formed by another path which did not involve DHBQ.
For further confirmation of the intermediate structure, we repeated the ozone treatment of DHNQ under similar conditions, but in organic solution (acetone), which slowed down the reaction rate and rendered the rhodizonic acid much better observable. Figure 5 (left) displays typical curves of the kinetics of the intermediate formation of rhodizonic acid and illustrates the fact that this intermediate was less quickly degraded than the starting material DHNQ and was still present when the latter was already completely consumed (Fig. 5, right).

The reaction of ozone with DHNQ (2, c = 2.09 mM) in acetone. Left: bleaching kinetics showing the consumption of DHNQ (black, adsorption at 517 nm) and the intermediacy of rhodizonic acid (red, absorption at 400 nm; right: VIS spectra at different reaction times, RhA: rhodizonic acid
Ozone treatment of DHAP
The solution of the chromophore DHAP (3) in water at pH 2 showed a yellow-green color with maximum absorption at 410 nm. The consumption of DHAP upon reaction with ozone followed a kinetics (Fig. 6), of which the curve had the typical shape of a starting material being in fast, dynamic equilibrium with an intermediate from which the actual product is formed (A ↔ B → C). It was logical to assume that the species in equilibrium are DHAP (3) and its corresponding ortho-quinone methide 6 (Scheme 5), which is quite common in the chemistry of DHAP and its derivatives (Hosoya et al. 2015a). In acidic medium, the formation of 6 from 3 is facilitated by pre-organization due to the strong intramolecular hydrogen bond and strain-induced bond localization effects (cf. also Rosenau et al. 2005). Indeed, when ozonation was carried out with sub-stoichiometric or roughly equivalent amounts of ozone, the main reaction products (approx. 60% of converted 3) found were hydroxy-[1,4]-benzoquinone (8) and acetic acid, which is indicative of the ozonation of the exocyclic double bond in ortho-quinone methide 6 as the first reaction step, followed by a Criegee-type cleavage of that bond. The primarily obtained 4-hydroxy-[1,2]-benzoquinone (7) tautomerizes immediately into the stable 2-hydroxy-[1,4]-benzoquinone (8), with the former acetyl moiety being cleaved off as acetic acid (Scheme 5).

The reaction of ozone with DHAP (3, c = 4.10 mM) in water. Left: bleaching kinetics (absorption at 400 nm); right: VIS spectra at different reaction times

Proposed mechanism of the first reaction step in ozonation of 2,5-dihydroxyacetophenone (3). The compounds 2-hydroxy-[1,4]-benzoquinone (8) and acetic acid are the main products when ozone is not applied in excess but near-stoichiometric amounts
It should be noted at this point that the reaction of ozone with hydroxyacetophenones is strongly dependent on the OH-substitution pattern at the aromatic ring, both with regard to reactivity (kinetics) and final degradation products. The constitutional isomer 2,6-dihydroxyacetophenone reacted rather differently from DHAP (data not shown, see Budischowsky 2019), from which it differs only in the position of one phenolic hydroxy group.
Analysis of the final degradation products of the key chromophores
GC/MS and NMR methods were used to investigate the composition of the ozonation mixtures, which was far from trivial. The analysis of product mixtures of polar organic compounds, as frequently encountered in the chemistry of renewable resources and biorefineries, is generally difficult because of their complexity, their instability (compositional changes over time, follow-up reactions, possible microbial infestation), the likely need for extraction and derivatization steps before chromatographic separation and analysis. The generally extremely low concentrations in experiments with chromophores represent another principal challenge.
Fortunately, we had at our disposal some robust procedures for the analysis of complex mixtures of acids, hydroxyacids, hydroxyketones, and mono-/disaccharides and compounds alike by extraction-derivatization-GC/MS sequences, developed in previous studies. Extensive optimization work has been invested into establishing the optimum derivatization and GC/MS conditions, and the ethoximation/silylation sequence has been demonstrated to give superior results for those extremely complex multi-component mixtures (Becker et al. 2013a, b). Especially for compounds containing aldehyde/keto functions in combination with hydroxy or carboxy groups (e.g. short-chain ketoacids, hydroxyaldehydes, but also monosaccharides and sugar acids) OH-derivatization by trimethylsilylation alone was deficient, and additional derivatization of the carbonyl moiety was required for smooth GC analysis. The effectiveness and reliability of the approach in tackling the composition of multi-component biorefinery product mixtures have been well proven in previous work, for example, the analysis of viscose spinning baths (Liftinger et al. 2015) or formose reaction mixtures (Zweckmair et al. 2014) or irradiated cellulosic pulps (Henniges et al. 2013).
Scheme 6 shows the structures of the identified ozonation/degradation products of the cellulosic key chromophores (1–3). Interestingly, these compounds were obtained from all three chromophores with only minor differences in composition (Table 1). All products—apart from one exception—are short-chain (< C4) carboxylic acids or hydroxyacids. In the order of increasing chain lengths, the following compounds have been identified: formic acid (9), acetic acid (10), glycolic acid (11), ethylene glycol (1,2-ethanediol, 12), oxalic acid (13), malonic acid (14), lactic acid (15), tartronic acid (16), mesoxalic acid hydrate (4), maleic acid (17) and 2,3-oxirandicarboxylic acid (2,3-epoxysuccinic acid, 18). These compounds, accounting for about 85% of the reaction products (total peak/integral area), were identified directly as the free acids by NMR and as their respective trimethylsilyl derivatives in GC/MS. The only non-acid, ethylene glycol 12, probably survived further oxidation during ozonation in the form of its rather stable (five-membered) dioxolans or (six-membered) dioxans, which are formed readily with keto and 1,2-diketo compounds, respectively. The detected maleic acid (17) is unlikely to have survived ozonation as such and is rather a condensation product formed after completion of the actual bleaching (see below).

Products of the reaction of the three key chromophores with excess ozone, identified by GC/MS and NMR. For correlation between products and starting materials see Table 1
NMR spectroscopy is obviously not the first method that comes to mind when multi-component mixtures are to be studied, such as the mixture of compounds in Scheme 6. Moreover, the special chemical structure of compounds similar to those in Table 1 implies that from 1H NMR spectra (with H–D exchange of acidic hydroxy groups) only very little spectral information is obtained because the compounds only yield singlets, and no couplings or splitting patterns can be used for structure elucidation. In fact, none of the compounds in Scheme 6 gives any more complex peak pattern than a simple singlet in the 1H spectra (apart from oxalic acid and mesoxalic acid having no signal at all, and lactic acid). In the present ozonation case, however, this apparent disadvantage turned into an advantage: due to the absence of multiplets there was no spectral overlap, and spiking with authentic samples allowed fast and easy compound identification and peak assignment, which was additionally confirmed by 2D techniques (HSQC and HMBC for carbons bound to the detected proton directly or indirectly, respectively). This straightforward NMR approach (“one peak–one compound”) works of course only, if the composition of the mixture is generally known. If this is the case, then it is a very fast and reliable approach to compare reaction mixtures, see Fig. 7. Under comparable measurement conditions (room temperature, solvent, and concentration), the approach provided the same information as GC—with some trade-offs in accurate quantifiability, but on the other hand great advantages in simplicity and speed. In our case, the results from GC/MS analysis were fully in agreement with the NMR outcome in all cases.

Stacked 1H NMR spectra (D2O) of the ozonation product mixtures of DHBQ (bottom), DHNQ (middle), and DHAP (top)
In a—rather approximate—mass balance, the final degradation products accounted for 30–45% of the chromophore mass introduced. The apparent gap could be closed—at least to some extent—by attempting to detect evolved carbon dioxide, which is the final oxidation product of any organic compound. This was done in a closed vessel providing sufficient volume (2 L) so that ozone was present in large excess for ozonation of each chromophore in aqueous solution (100 mL). After alkalization, the CO2 formed was determined as BaCO3. The mass of CO2 accounted for 33%, 50%, and 47% of the introduced mass of the initial chromophores 1, 2, and 3, respectively, showing that a significant amount of the starting material was converted into CO2 already after a short time of 10 min. It should be noted that not all of the CO2 is necessarily formed directly by ozone-oxidation of organic compounds. Decarboxylation reactions are also likely to contribute significantly, especially since α-ketoacids, β-ketoacids as well as 1,3-dicarboxylic acids, which are very prone to decarboxylation, are typical ozonation products and are prominently represented in the product mixture.
Experiments with different salts as additives (Na2SO4, K2SO4, MgSO4, and CaCl2) were carried out to assess if their presence would influence the bleaching process in any way. Previous studies revealed that degradation of DHBQ (1) was influenced by metal cations during hydrogen peroxide bleaching due to complex stabilization (Hosoya et al. 2015b). No such effect was observed during bleaching with ozone, and the kinetics were unaffected by salts present. This seems reasonable as at the low pH none of the hydroxy groups would be deprotonated and interact with cations.
In a later set of experiments, a cellulosics pulp was enriched in chromophores by soaking and treated with ozone in a simulated medium-consistency (12%) bleaching. In this case, the reaction kinetics were not followed, but the final reaction products were extracted and analyzed. The product distribution, superimposed by some products of carbohydrate oxidation, was similar to that in Table 1.
Analysis of intermediates in the ozonation of the key chromophores
When ozonation was carried out with little excess of ozone or stoichiometric amounts, the product spectrum was completely different from that after the completion of ozonation. Besides the main reaction intermediates already described (see above), the compounds given in Scheme 7 and Table 2 were obtained (along with traces of some of the final ozonation products (Table 1) which are not listed here). It should be noted that the compounds in Scheme 7 are intermediates that are consumed in later stages of the reaction and disappear—they are converted into those compounds shown in Scheme 6 by excess O3 and longer reaction times. Interestingly—but on the other hand also expected from the general mechanism of the ozonation reaction—all of the products 19–23 in Scheme 7 contain a carbonyl (keto or aldehyde) function, mostly in masked form as the respective hydrate or hemiacetal/hemiketal. Carbonyl compounds are the initial products of double bond cleavage by ozonation. The products 19–23 were detected as their trimethylsilylated ethoxime derivatives in GC/MS and directly in NMR, accounting for about 55% of the products (total peak area besides the main intermediates 4, 5, and 8). This lower value is due to the fact that in an aqueous medium hydroxyaldehydes and formylcarboxylic acids are not present as a single species, but undergo very complex dynamic equilibria involving hydrates, hemialdals, hemiacetals, etc., each compound being able to react with itself or with other species. At low concentrations, as in our case, the ketohydrates dominated, but also some dimers were detected. For instance, glycolaldehyde (20) was in equilibrium with its hydrate (20a) and its dimer (20b), a bis(hemiacetal), and glyoxal (21) was even found as monohydrate (21a), dihydrate (21b), two dimers (21c and 21d) and an addition compound with glycolaldehyde (21 + 20a) (Scheme 7).

Concerning subsequent reactions, such as condensations or oxidations, the exact nature of these hydrates, dimers, and oligomers formed from one species and their ratio in aqueous solution is often inconsequential as they react exactly like the monomer, quasi as monomer “equivalent”, cf. the chemical behavior of paraformaldehyde that reacts as formaldehyde equivalent. For analytical purposes, however, the complexity of the hydrate-hemiacetal mixtures and their changeability due to dynamic equilibria in an aqueous solution poses a huge challenge. In the present case, with highly diluted solutions, mainly hydrates and dimers were present. However, in concentrated solutions, a multitude of hydrates, dimers, and oligomers exists (Whipple 1970), some of them still of unknown structure, even for such a seemingly simple compound as glyoxal.
Stability of the ozonation product mixtures
The composition of the mixtures of ozonation intermediates (Scheme 7 and Table 2) was highly changeable. Already after one hour, a visible color change to yellow occurred which continued to intensify. After one day, none of the initially found compounds could be detected. After two days, some precipitate of fine brownish flakes appeared. Since some of the initially formed compounds are among the most reactive carbonyl compounds at all, it was clear that a plethora of condensation processes took place, leading to obviously chromophoric compounds of higher molecular weight. These condensation reactions will not be discussed in detail here but will be reported elsewhere in connection with the re-formation of chromophores from low-molecular-weight chromophore degradation products.
It was interesting to observe that the composition of the final aqueous ozonation product mixture was also not stable over time. When analyzed again after several days, the concentration of formic (9), acetic (10), and glycolic acid (11) had significantly increased, while the concentration of malonic (14), tartronic (16), and mesoxalic acid (4) had decreased so much that they were barely detectable. This decrease was certainly in large part due to decarboxylation, a typical reaction of 1,3-dicarboxylic acids (Scheme 8, top), but not exclusively. Fumaric acid (23) appeared as a prominent new product. It could not have formed by cis–trans-isomerization of maleic acid because the concentration of the latter did not decrease. The only reasonable path of formation was the condensation of mesoxalic acid with malonic acid in a favored Knoevenagel-type aldol condensation process. The initially formed α,α′-disubstituted bis(β-ketocarboxylic acid) decarboxylates immediately, even much faster than the starting materials, to afford fumaric acid (Scheme 8, bottom). This sequence was easily confirmed in test experiments with pure authentic compounds. The statement on the instability of the ozonation product mixtures refers only to aqueous solutions; the organic solutions of the ethoxime/trimethylsilyl derivatives as used for GC/MS were stable over weeks without changes.

Top: decarboxylation of 1,3-dicarboxylic acids (R1, R2 = H, OH, alkyl); bottom: formation of fumaric acid (23) from two ozonation intermediates in an aldol condensation / double decarboxylation sequence
Conclusions
The key chromophores 2,5-dihydroxy-1,4-benzoquinone (DHBQ, 1), 5,8-dihydroxy-1,4-naphthoquinone (DHNQ, 2), and 2,5-dihydroxyacetophenone (DHAP, 3) were bleached to complete decoloration with ozone in an aqueous solution at room temperature within 5–6 min. The main intermediates of the degradation reactions, formed upon incomplete conversion, namely mesoxalic acid from DHBQ, rhodizonic acid from DHNQ, and hydroxy-[1,4]-benzoquinone from DHAP, were identified. They were accompanied by a complex and changeable mixture of C1-C3 carbonyl compounds (aldehydes, hydroxyaldehydes, and hydroxyketones). Upon complete conversion, a colorless, rather complex mixture of C1-C4 carboxylic acids, ketoacids, and hydroxyacids was obtained (Table 1). In addition, more than 40% of the starting mass was converted to carbon dioxide. The final product mixtures were also not completely stable but showed slow condensation and decarboxylation of the contained 1,3-dicarboxylic acids (e.g. malonic, tartronic, mesoxalic acid). All products were unambiguously identified by NMR spectroscopy, GC/MS, and comparison with authentic samples (spiking) and comprehensively analytically characterized.
From the results, it seems important that industrial ozone bleaching be carried out at the lowest acceptable pH, which of course must be reconciled with the acid-hydrolytic chain cleavage of cellulose which is strongly accelerated with increasing acidity. Lower pH not only increases the stability of ozone and reduces the formation of reactive oxygen species, as they dominate under neutral or alkaline conditions, but they also promote decarboxylation of 1,3-dicarboxylic acids. These ozonation products are highly methylene-active compounds and therefore tend to undergo condensation reactions and chromophore generation, whereas this property is largely lost after decarboxylation (cf. acetic acid vs. malonic acid). It is equally crucial that ozone is present in large excess relative to the chromophores, otherwise, mainly carbonyl-containing compounds rather than carboxyl-containing compounds are obtained, which drastically increases the tendency to re-condensation and new chromophore formation. The issue of needing a high ozone/chromophore ratio may be even more relevant for industrial settings—with the inherent challenges of ozone diffusion, accessibility, and local O3 availability—than for model experiments with dissolved chromophores in aqueous solution. In general, careful removal of ozonation/degradation products by pulp washing is important. This applies especially to aldehyde or ketone intermediates, but also to the carboxyl-rich end product mixtures, as they can also condense and produce higher-Mw material and possible chromophores. While bleaching of the key chromophores with ClO2 or H2O2 (D or P bleaching stage, respectively) produced simple product mixtures consisting mainly of CO2, acetic acid, and oxalic acid, the product mixtures of ozonation (Z bleaching stage) are more complex and may undergo re-condensation. If they are not completely removed from the pulp, problems with brightness stability are to be expected, which emphasizes the need for thorough washing steps.
Materials and methods
General
Commercial chemicals from Sigma-Aldrich (Schnelldorf, Germany) were of the highest grade available and were used without further purification. Distilled water was used for all aqueous solutions. The key chromophores 2,5-dihydroxy-1,4-benzoquinone (DHBQ 1), 5,8-dihydroxy-1,4-naphthoquinone (DHNQ 2), and 2,5-dihydroxyacetophenone (DHAP 3) were available from earlier work but are also commercially available and were used as received. For O3 generation (with O2 as carrier gas), an Anseros Ozone Generator COM-AD-02 was used.
Analytical instruments
GC/MS analysis was performed on an Agilent 7890A gas chromatograph coupled with an Agilent 5975C triple-axis mass selective detector (MSD; Agilent Technologies, Santa Clara, CA, USA). A DB-5 ms column (30 m × 0.25 mm i.d. × 0.25 μm film thickness; J&W Scientific, Folsom, CA, USA). An Agilent 6560 QTOF mass spectrometer equipped with an Agilent G1607A dual Jetstream coaxial ESI interface was used, injection volume: 5 µL, sheath gas temperature: 150 °C, sheath gas flow rate: 12 L/min, nebulizer gas pressure: 20 psi, MS capillary voltage: 4 kV, nozzle voltage: 2 kV, fragmentor: 275 V, scanning mass range: 50 to 1700 m/z with a TOF acquisition rate of 2.8 spectra/s.
For paper-spray MS, the paper tips (isosceles triangle, a = 10 mm) were cut and connected to 4 kV capillary voltage. Whatman filter paper No 1 (Wagner & Munz GmbH, Vienna, Austria) was used as the model sample matrix. The aqueous sample solution was used as a spray solvent. After applying 30 µL of the spray solvent onto the paper surface, mass spectra were recorded in positive or negative ion mode with a Thermo LTQ-MS (LTQ XL™ Linear Ion Trap Mass Spectrometer, Thermo Fisher Scientific, Waltham, Massachusetts, USA) equipped with an ESI ion source. Injection volume: 5 µL. Where applicable, a splitter with a ratio of 1:2 was used to divert the flow for better ESI spray quality. MS settings: Spray voltage: 6 kV, sheath gas pressure: 5 psi, auxiliary gas: 2 a.u., transfer capillary temperature: 275 °C, scan range: m/z 300 to 1000. In the case of tandem-MS investigations, an isolation width of ± 0.5 m/z was selected. Thermo Fisher Scientific Xcalibur software (Thermo Xcalibur 2.2 SP1 build 48) was used for compound identification.
All NMR spectra were recorded on a Bruker (Rheinstetten, Germany) Avance II 400 instrument (resonance frequencies: 400.13 MHz for 1H and 100.63 MHz for 13C) equipped with a 5 mm observe broadband probe head (BBFO) with z-gradients at room temperature with standard Bruker pulse programs. The samples were dissolved in 0.6 mL of the NMR solvent CDCl3 or D2O (99.8% D, Euriso-top, Saint-Aubin, France). Chemical shifts are given in ppm, referenced to residual solvent signals (CDCl3: 7.26 ppm for 1H, 77.0 ppm for 13C) or DSS (1H and 13C at 0 ppm) for D2O. 1H NMR data were collected with 32 k complex data points and apodized with a Gaussian window function (lb = -0.3 Hz and gb = 0.3 Hz) prior to Fourier transformation. The 13C-jmod spectra with WALTZ16 1H decoupling were acquired using 64 k data points. Signal-to-noise enhancement was achieved by multiplication of the FID with an exponential window function (lb = 1 Hz). All two-dimensional experiments were performed with 1 k × 256 data points, while the number of transients and the sweep widths were optimized individually. HSQC experiments were acquired using adiabatic pulse for inversion of 13C and GARP-sequence for broadband 13C-decoupling, optimized for 1J(CH) = 145 Hz.
UV/Vis spectra were recorded on a LAMBDA 45 UV/Vis spectrophotometer (PerkinElmer, Waltham, MA, USA), quartz glass cuvettes (l = 1 cm) were used for all measurements. The speed of scanning was 480 nm per minute in the range of 400 to 700 nm.
General ozonation procedure
To improve chromophore solubility in water, a slurry of the chromophore (up to 200 mg) in acetonitrile was prepared and water (pH 2, H2SO4) was added to a volume of 300 mL. The chromophore solution was transferred into a sealable 500 mL glass reactor (Schott, Mainz, Germany). A Teflon tubing carried the gas stream from the generator to the reactor. Ozone was directly injected into the solution through a glass frit and well distributed by stirring. Two additional Teflon tubes were attached to the reactor, one as a gas outlet and one for sampling during the reaction. The ozone generator was set to a level of 50% with a gas flow of 50 L/h. The ozone concentration was 0.021 g O3/L and the output 10.5 g O3/h. The amount of ozone that was produced at the selected generator adjustments was determined by iodometric titration and verified by the instrument's datasheet. Constant ozone generation was obtained after the specified warm-up time. Experiments were carried out at room temperature and were typically stopped when the solution was decolorized. The solution was degassed by applying reduced pressure for 1 min and neutralized with NaOH (0.1 M). One aliquot of the solution was used for direct analysis. An aliquot of this sample (0.4 mL) was mixed with D2O (0.4 mL) for NMR studies. For GC/MS, the solution (100 mL) was extracted with chloroform (5 × 5 mL), the combined organic phases were dried over anhydrous MgSO4 and concentrated at room temperature to 5 mL. Another aliquot of the reaction solution was freeze-dried and further analyzed, a third one was adjusted to pH 10 (0.1 M NaOH) and then analyzed by NMR (mixing 0.4 mL of this sample with 0.4 mL of D2O). In the case of the sample at pH 10, no GC/MS analysis was performed because the products, mostly carboxylic acids, were present as dissociated salts and thus not extractable into organic media.
Variations of the general ozonation procedure
(A) For detection of the intermediates (see Table 2), ozonation was stopped at shorter reaction times, when about 2 or 5 molar eq. of ozone (relative to the contained chromophore) were generated (the amount having actually reacted evidently being smaller). The solution work-up and analysis were carried out as described above. Half of the volume was not neutralized and used for conversion into the corresponding O-benzyl oximes. For that purpose, O-benzylhydroxylamine hydrochloride (1.59 g, 10 mmol) was added to the acidic product mixture solution (200 mL). The mixture was stirred for 1 h at r.t., heated to 80 °C for 2 h, cooled to r.t., neutralized with 1 M NaOH, and freeze-dried. The waxy residue was used for GC/MS and NMR analysis.
(B) For detection and approximate quantitation of evolved carbon dioxide, the gas outlet was connected to a bubbler filled with 0.1 M NaOH, in which the gas stream was well distributed in the solution by means of a sintered frit and additional stirring. After completion of ozonation, the gas supply was changed from the O2/O3 stream to nitrogen and the apparatus was flushed for 5 min. Aqueous Ba(OH)2 (0.1 M) was added until no further white precipitate (BaCO3) appeared. The precipitate was collected by filtration, dried, and weighed. Note that air cannot be used for flushing instead of nitrogen, because it contains CO2. The NaOH used for trapping must be free of carbonate, which needs to be tested beforehand. Ba(OH)2 should not be present right from the beginning but be added later as the formed precipitate sticks to the frit so that determination of the yield would be grossly inaccurate.
GC/MS analysis
(A) The degradation products formed during the ozone treatment were derivatized by a combined ethoximation/trimethylsilylation approach (according to Becker et al. 2013a, b). For this purpose, 10 mg of the freeze-dried residue or 1 mL of the chloroform extract were used. To the residue or extract, 200 µL of the first derivatization reagent (containing 40 mg of ethoxyamine hydrochloride and 1 mg of methyl-α-D-galactopyranoside (internal standard) in 1 mL of pyridine) was added. The mixture was vortexed, sealed, and kept at 70 °C for 1 h. After cooling to r.t., 200 µL of the second derivatization agent (containing 1.5 mg of 4-dimethylaminopyridine in 1 mL of pyridine) and 200 µL of N,O-bis(trimethylsilyl)-trifluoroacetamide (containing 10% trimethylchlorosilane) were added. The solution was mixed by vortexing, closed tightly, and heated to 70 °C for 2 h. After cooling to r.t., the mixture was diluted with 600 µL of ethyl acetate and analyzed.
(B) For identification of the ketone/aldehyde intermediates the mixture of the O-benzyloximes was dissolved in 1 mL of chloroform and analyzed by GC/MS or paper-spray-MS. Each 0.2 µL aliquots were injected by a CTC-PALxt autosampler that was controlled by Chronos software v.3.5 (Axel Semrau, Spockhövel, Germany). The NIST/Wiley 2008 database was used for compound identification.
NMR data of identified degradation products
Multiplicity is given as singlet (s), doublet (d), quartet (q), or double intensity (d.i.). Identity of the final stable degradation products (4, 9–18) and the main intermediates (4, 5, 8) was confirmed by spiking the solution with authentic samples, the identity of the minor intermediates (19–23) by comparison with an equimolar mixture of the authentic compounds.
Data availability
Data available from the authors upon request.
Code availability
Not applicable.
References
Ahn K, Zaccaron S, Zwirchmayr NS, Hettegger H, Hofinger H, Bacher M, Henniges U, Hosoya T, Potthast A, Rosenau T (2019) Yellowing and brightness reversion of celluloses: CO or COOH, who is the culprit? Cellulose 26:429–444
Becker M, Zweckmair T, Forneck A, Rosenau T, Potthast A, Liebner F (2013a) Evaluation of different derivatisation approaches for GC/MS analysis of carbohydrates in complex matrices of biological and synthetic origin. J Chromatogr A 1281:115–126
Becker M, Liebner F, Rosenau T, Potthast A (2013b) Ethoximation-silylation approach with expanded retention index (ERI) for mono- and disaccharide analysis by GC/MS. Talanta 115:642–651
Bernatek E, Straumsgård KA (1959) Ozonolysis of p-Benzoquinone II. Acta Chim Scand 13:178–186
Bowmaker JK, Dartnall HJA (1979) Visual pigments of rods and cones in a human retina. J Physiol 298:501–511
Budischowsky D (2019) Ozone bleaching of cellulosic chromophores. Master Thesis. Institute of Chemistry of Renewable Resources, Department of Chemistry, University of Natural Resources and Life Sciences, Vienna
Chalmers RA, Telling GM (1967) A reassessment of rhodizonic acid as a qualitative reagent. Mikrochim Acta 55:1126–1135
Chirat C, Lachenal D (1994) Effect of ozone on pulp components; application to bleaching of kraft pulps. Holzforschung 48(1):133–139
Chirat BC, Lachenal D (1997) Effect of hydroxyl radicals on cellulose and pulp and their occurrence during ozone bleaching. Holzforschung 51:147–154
Chirat C, Lachenal D, Angelier R, Viardin MT (1997) DZ and ZD bleaching: fundamentals and application. J Pulp Paper Sci 23(6):289–292
Cotton FA, Wilkinson G, Murillo CA, Bochmann M (1999) Advanced Inorganic Chemistry, 6th edn. Wiley, New York
Criegee R (1975) Mechanism of ozonolysis. Angew Chem Int Ed Engl 14(11):745–752
Ek M, Gellerstedt G, Henriksson G (2009) Pulp and paper chemistry and technology, vol 2: paper products physics and technology, De Gruyter, Berlin
Epstein SA, Donahue NM (2010) Ozonolysis of cyclic alkenes as surrogates for biogenic terpenes: primary ozonide formation and decomposition. J Phys Chem 114:7509–7515
Feigl F, Suter HA (1942) Analytical use of sodium rhodizonate. Ind Eng Chem Anal Ed 14:840–842
Forsskahl I, Popoff T, Theander O (1976) Formation of Aromatic Compounds from Carbohydrates. II. Reaction of D-xylose and D-glucose in slightly alkaline, aqueous solution. Carbohydr Res 48(1):13–21
Gierer J (1997) Formation and involvement of superoxide (O2−·/HO2·) and hydroxyl (OH·) radicals in TCF bleaching processes: a review. Holzforschung 51(1):34–46
Guggenberger M, Hettegger H, Zwirchmayr N S, Hosoya T, Bacher M, Zaccaron S, Böhmdorfer S, Reiter H, Spitzbart M, Dietz T, Eibinger K, Mahler A K, Winter H, Röder T, Potthast A, Rosenau T (2020) Degradation of the cellulosic key chromophore 2,5-dihydroxy-[1,4]-benzoquinone (DHBQ) under conditions of chlorine dioxide pulp bleaching: formation of rhodizonate as secondary chromophore. A combined experimental and theoretical study. Cellulose 27(7):3623–3649
Hendrickx MFA, Vinckier C (2003) 1,3-Cycloaddition of ozone to ethylene, benzene, and phenol: a comparative ab initio study. J Phys Chem 107:7574–7580
Henniges U, Hasani M, Potthast A, Westman G, Rosenau T (2013) Electron beam irradiation of cellulosic materials—opportunities and limitations. Materials 6(5):1584–1598
Hettegger H, Amer H, Zwirchmayr NS, Bacher M, Hosoya T, Potthast A, Rosenau T (2019) Pitfalls in the chemistry of cellulosic key chromophores. Cellulose 26:185–204
Hoigné J, Bader H (1976) The role of hydroxyl radical reactions in ozonation processes in aqueous solutions. Water Res 10(5):377–386
Hollemann A F, Wiberg E, Wiberg N (1985) Lehrbuch der Anorganischen Chemie. 91.–100., verbesserte und stark erweiterte Auflage, de Gruyter, Berlin
Hosoya T, Rosenau T (2013) Degradation of 2,5-dihydroxy-1,4-benzoquinone by hydrogen peroxide under moderately alkaline conditions resembling pulp bleaching: a combined kinetic and computational study. J Org Chem 78(22):1194–11203
Hosoya T, French AD, Rosenau T (2013a) Chemistry of 2,5-dihydroxy-[1,4]-benzoquinone, a key chromophore in aged cellulosics. Mini Rev Org Chem 10(3):302–308
Hosoya T, French AD, Rosenau T (2013b) Chemistry of 5,8-dihydroxy-[1,4]-naphthoquinone, a key chromophore in aged cellulosics. Mini Rev Org Chem 10(3):309–315
Hosoya T, French AD, Rosenau T (2015a) Chemistry of 2,5-dihydroxyacetophenone, a key chromophore in cellulosic substrates. Mini Rev Org Chem 12(1):88–95
Hosoya T, Henniges U, Potthast A, Rosenau T (2015b) Effects of inorganic salts on the degradation of 2,5-dihydroxy-[1,4]-benzoquinone as a key chromophore in pulps by hydrogen peroxide under basic conditions. Holzforschung 69(6):685–693
Hosoya T, Zwirchmayr N S, Klinger K M, Reiter H, Spitzbart M, Dietz T, Eibinger K, Kreiner W, Mahler A K, Winter H, Röder T, Potthast A, Elder T, Rosenau T (2018) Chromophores in cellulosics XVIII. Degradation of the cellulosic key chromophore 5,8-dihydroxy-[1,4]-naphthoquinone under conditions of chlorine dioxide pulp bleaching: a combined experimental and theoretical study. Cellulose 25(9):4941–4954
Hruschka A (1986) Zellstoffbleiche mit Sauerstoff und Ozon. Wochenbl Pap 17:681–686
Korntner P, Hosoya T, Dietz T, Eibinger K, Reiter H, Spitzbart M, Röder T, Borgards A, Kreiner W, Mahler A K, Winter H, French A D, Henniges U, Potthast A, Rosenau T (2015) Chromophores in lignin-free cellulosic materials belong to three compound classes. Chromophores in cellulosics, XII. Cellulose 22(2):1053–1062
Krainz K, Potthast A, Suess U, Dietz T, Nimmerfroh N, Rosenau T (2009) Effects of selected key chromophores on cellulose integrity upon bleaching. Holzforschung 63(6):647–655
Kuczkowski RL (1992) The structure and mechanism of formation of ozonides. Chem Soc Rev 21:79–83
Lachenal D, Pipon G, Chirat C (2009) Final pulp bleaching by ozonation: chemical justification and practical operating conditions. J Pulp Paper Sci 35(2):53–56
Liftinger E, Zweckmair T, Schild G, Eilenberger G, Böhmdorfer S, Rosenau T, Potthast A (2015) Analysis of degradation products in rayon spinning baths. Holzforschung 69(6):695–702
Olsson K, Pernemalm P A, Popoff T, Theander O (1977) Formation of aromatic compounds from carbohydrates: V. Reaction of D-glucose and methylamine in slightly acidic, aqueous solution. Acta Chem Scand B Org Chem Biochem 31(6):469–474
Patt R, Hammann M, Kordsachia O (1991) The role of ozone in chemical pulp bleaching. Holzforschung 45:87–92
Popoff T, Theander O (1976) Formation of aromatic compounds from carbohydrates. IV. Chromones from reaction of hexuronic acids in slightly acidic, aqueous solutions. Acta Chem Scand B 30:705–710
Popoff T, Theander O W (1978) Formation of aromatic compounds from carbohydrates. VI. Reaction of dihydroxyacetone in slightly acidic, aqueous solution. Acta Chem Scand B 32(1):1–7.
Popoff T, Theander O, Rømming C, Foltmann B, Taticchi A, Anthonsen T (1976) Formation of aromatic compounds from carbohydrates. Part III. Reaction of D-glucose and D-fructose in slightly acidic. Aqueous Solution Acta Chem Scand B 30:397–402
Potthast A, Rosenau T, Kosma P, Saariaho AM, Vuorinen T, Sixta H (2005) On the nature of carbonyl groups in cellulosic pulps. Cellulose 12(1):43–50
Pouyet F, Chirat C, Potthast A, Lachenal D (2014) Formation of carbonyl groups on cellulose during ozone treatment of pulp: consequences for pulp bleaching. Carbohydr Polym 109:85–91
Ragnar M, Eriksson T, Reitberger T, Brandt P (1999a) A new mechanism in the ozone reaction with lignin like structures. Holzforschung 53:423–428
Ragnar M, Eriksson T, Reitberger T (1999b) Radical formation in ozone reactions with lignin and carbohydrate model compounds. Holzforschung 53(3):292–298
Reitberger T, Eriksson T, Ragnar M, Brandt P (1999) Radical formation in ozone bleaching. In: 10th international symposium on wood and pulping chemistry (ISWPC), Yokohama, Japan 1999, vol I, pp 302–307
Rosenau T, Potthast A, Milacher W, Hofinger A, Kosma P (2004) Isolation and identification of residual chromophores in cellulosic materials. Polymer 45:6437–6443
Rosenau T, Ebner G, Stanger A, Perl S, Nuri L (2005) From a theoretical concept to biochemical reactions: strain induced bond localization (SIBL) in oxidation of vitamin E. Chem Eur J 11(1):280–287
Rosenau T, Potthast A, Möslinger R, Kosma P (2006) Confirmation of the presence of hydroxyl radicals during pre-ripening of alkali cellulose. J Wood Chem Technol 26(1):1–11
Rosenau T, Potthast A, Kosma P, Suess HU, Nimmerfroh N (2007) Isolation and identification of residual chromophores from aged bleached pulp samples. Holzforschung 61(6):656–661
Rosenau T, Potthast A, Krainz K, Yoneda Y, Dietz T, Shields ZPI, French AD (2011) Chromophores in cellulosics, VI. First isolation and identification of residual chromophores from aged cotton linters. Cellulose 18(6):1623–1633
Rosenau T, Potthast A, Krainz K, Hettegger H, Henniges U, Yoneda Y, Rohrer C, French AD (2014) Chromophores in cellulosics, XI: Isolation and identification of residual chromophores from bacterial cellulose. Cellulose 21(4):2271–2283
Sharpe LT, Stockman A, Jagla W, Jägle H (2011) A luminous efficiency function, VD65* (λ), for daylight adaptation: a correction. Color Res Appl 36(1):42–46
Sixta H (2006) Handbook of Pulp, vol 1. Wiley-VCH, Weinheim
Sjöström E (1993) Wood chemistry: fundamentals and applications. Academic Press, San Diego
Soteland N (1978) Bleaching of chemical pulps with oxygen and ozone. Norsk Skogind 32:199–204
Streng AG (1961) Tables of ozone properties. J Chem Eng Data 6(3):431–436
Suess HU (2010) Pulp bleaching today. Walter de Gruyter, Berlin
Theander O, Westerlund E (1980) Formation of aromatic compounds from carbohydrates. VIII. Reaction of D-erythrose in slightly acidic. Aqueous Solution Acta Chim Scand B 34:701–705
von Gunten U (2003) Ozonation of drinking water: part I. Oxidation kinetics and product formation. Water Res 37(7):1443–1467
Werner D, Gassner AL, Marti J, Christen S, Wyss P, Weyermann C (2019) Comparison of three collection methods for the sodium rhodizonate detection of gunshot residues on hands. Sci Justice 9:004
Whipple EB (1970) Structure of glyoxal in water. J Am Chem Soc 92(24):7183–7186
Yoneda Y, Mereiter K, Jäger C, Brecker L, Kosma P, Rosenau T, French A (2008) Van der Waals versus hydrogen-bonding forces in a crystalline analog of cellotetraose: cyclohexyl 4′-O-cyclohexyl β-d-cellobioside cyclohexane solvate. J Am Chem Soc 130(49):16678–16690
Zweckmair T, Böhmdorfer S, Bogolitsyna A, Rosenau T, Potthast A, Novalin S (2014) Accurate analysis of formose reaction products by LC-UV: an analytical challenge. J Chromatogr Sci 52(2):169–175
Zwirchmayr NS, Hosoya T, Henniges U, Gille L, Bacher M, Furtmüller P, Rosenau T (2017) Degradation of the cellulosic key chromophore 5,8-dihydroxy-[1,4]-naphthoquinone by hydrogen peroxide under alkaline conditions. J Org Chem 82(21):11558–11565
Zwirchmayr NS, Henniges U, Bacher M, Hosoya T, Reiter H, Spitzbart M, Dietz T, Eibinger K, Kreiner W, Mahler AK, Winter H, Röder T, Potthast A, Elder T, Rosenau T (2018) Degradation of the cellulosic key chromophores 2,5- and 2,6-dihydroxyacetophenone by hydrogen peroxide under alkaline conditions. Chromophores in cellulosics, XVII. Cellulose 25(7):3815–3826
Acknowledgments
The authors would like to thank the Austrian Biorefinery Center Tulln (ABCT) for financial support.
Funding
Open access funding provided by University of Natural Resources and Life Sciences Vienna (BOKU). The financial support by the Austrian Biorefinery Center Tulln (ABCT) is gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
D.B., N.S.Z. and T.R. contributed to the study conception and design. Material preparation, data collection and analysis were performed by all authors. The first draft of the manuscript was written by D.B. and T.R. and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval
Not applicable.
Consent to participate
All celluloses and chromophores agreed to participate in the experiments.
Consent for publication
All authors agreed to the publication in the submitted form.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Budischowsky, D., Zwirchmayr, N.S., Hosoya, T. et al. Degradation of cellulosic key chromophores by ozone: a mechanistic and kinetic study. Cellulose 28, 6051–6071 (2021). https://doi.org/10.1007/s10570-021-03909-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-021-03909-4