
Abstract
The thermal stability of Ru–Re NPs on γ-alumina support was studied in hydrogen at 800 °C and in air at 250–400 °C. The catalysts were synthesized using Cl-free and Cl-containing Ru precursors and NH4ReO4. Very high sintering resistance of Ru–Re NPs was found in hydrogen atmosphere and independent of Ru precursors and Re loading, the size of them was below 2–3 nm. In air, metal segregation occurred at 250 °C, leading to formation of RuO2 and highly dispersed ReOx species. Ruthenium agglomeration was hindered at higher Re loading and in presence of residual Cl species. Propane oxidation rate was higher with the Ru(N)–Re catalysts than with Ru(N) and that containing Cl species. The Ru(N)–Re (3:1) catalyst exhibited the highest activity and the lowest activation energy (91.6 kJ mol−1) what is in contrast to Ru(Cl)–Re (3:1) which had the lowest activity and the highest activation energy (119.3 kJ mol−1). Thus, the synergy effect was not observed in Cl-containing catalysts.
Graphic Abstract

Similar content being viewed by others

Explore related subjects
Find the latest articles, discoveries, and news in related topics.Avoid common mistakes on your manuscript.
1 Introduction
The thermal stability of supported metal nanoparticles (NPs) at high temperatures is a very important issue determining their applications as catalysts in the various industrial processes. Numerous literature data indicate that particle sintering is one of the most critical mechanisms causing catalyst deactivation what led to lower catalytic activity with time [1, 2]. Thermally-induced deactivation of the supported metal-based catalysts results from loss of catalytic surface area caused by the growth of the metal crystallite size, as well as from reducing of support area due to support collapse and of catalytic surface area due to pore collapse on crystallites of the active metal phase. The sintering process of metal species can be described by the two main mechanisms, the Ostwald ripening or particle migration and coalescence [3,4,5]. Generally, catalysts atoms become mobile already at temperatures between one-third and one-half of the melting point, i.e. at the so-called Hutting and Tamman temperatures, respectively. Atoms at defects would exhibit mobility usually at Hutting temperature while atoms from the bulk would start to exhibit mobility at Tamman temperature, which leads to atomic migration and agglomeration. Many other factors affect also the sintering process, for example, metal particle size, the loading or metal dispersion, the interaction between the metal and support, or the atmosphere and time in which the catalyst is heated [6]. Also, the presence of residue chloride on the surface of the metal catalysts (mainly from chloride-based precursors) may cause severe sintering of the metal particles.
In the past years, supported bimetallic nanoparticles have attracted increasing attention because they frequently exhibit much better deactivation resistance, catalytic activity, and selectivity in comparison to the monometallic systems [7, 8]. Higher thermal stability of the bimetallic catalysts frequently results from the introduction of a second, higher melting point metal. For example, Cao and Veser reported that alloying the Pt nanoparticles with Rh, which has a melting point about 200 °C higher than Pt, caused strong improvement of the thermal stability of the metal particles, which was correlated with the composition, i.e. higher Rh content resulting in better thermal stability [9]. Weckhuysen et al. showed also that the addition of Pd to Ru catalysts results in the improvement of stability of the metal particles due to the prevention of sintering [10].
Bimetallic catalysts based on ruthenium and rhenium have become very interesting because they can be used in several important reactions such as selective hydrogenation of amides [11], hydrogenolysis of glycerol [12, 13], the selective hydrogenation of dimethyl terephthalate [14] or for the hydrodeoxygenation of fatty acid esters to alkanes [15]. The Ru–Re catalysts showed an excellent catalytic performance because synergistic interaction between two metals significantly enhanced hydrogen adsorption capacity. However, a survey of literature teaches that, although a growing number of contributions deal with the bimetallic Ru–Re catalysts, no information is available concerning the resistance towards the sintering of these materials in hydrogen at high temperature. Such knowledge is important since Ru–Re catalysts can be attractive for catalytic processes of great industrial interest such as ammonia synthesis, which is carried out at high temperature and pressure [16, 17] or in the Fischer–Tropsch process [18]. Ayvali et al. found that dissociation of CO at the metallic surface of small RuRe NPs was easier than with pure Ru and Re (as the result of a synergic effect) and therefore, high reactivity towards CO dissociation validates the use of bimetallic structures in the Fischer–Tropsch process [18]. The recently reported studies for Re-based bimetallic catalysts, for example for the Re–Ni/Al2O3 catalysts, indicate that Re promoter could effectively suppress the Ni0 sintering process [19]. Also, for the bimetallic Co–Re particles supported on Al2O3 superior sintering resistance was reported by Mom et al. [20]. The authors attribute this behaviour to a higher adatom detachment barrier for the bimetallic nanoparticles due to the increased stability of both Co and Re in the bimetallic particles concerning their pure phases.
As mentioned above, despite growing interest in catalytic applications, the supported Ru–Re catalysts have been explored very light so far, in fact from sintering in hydrogen atmosphere at high temperatures and oxidation point of view not at all. The oxidation mechanism is well understood in the monometallic Ru [21] and Re catalysts [22, 23], however, how Re addition affects the oxidation mechanism and the sintering process in bimetallic Ru–Re catalysts is not known. Previous studies suggest the sintering of Ru particles dispersed on γ-Al2O3 is strongly prompted by the exposure to air or oxygen at temperatures ≥ 250 °C, Recently, we reported the effect of Re on ruthenium dispersion in the Ru–Re/γ-Al2O3 catalysts prepared from Cl-free and Cl-containing Ru precursors [24]. It was found that the addition of Re and reduction of the bimetallic Ru–Re catalysts in hydrogen at 500 °C led to significant improvement of ruthenium dispersion, mainly in the Cl-free samples. It should be noted that the structure characterization of supported Ru–Re catalysts still presents a difficulty since pure Ru and Re have the same hcp crystal structure and the bulk binary Ru–Re system has an isomorphous phase diagram, showing complete Ru–Re solubility [25]. The catalytic performance of the Cl-free bimetallic Ru–Re/γ-Al2O3 catalysts reduced at 500 °C was evaluated in the propane [26, 27] and methane oxidation [28]. A synergic effect between Ru and Re was found which led to higher activity and good stability in repeated cycles. The light alkanes, emitted mainly by natural gas-fueled vehicles and gas power plants, have a greenhouse effect much greater than carbon dioxide and should be eliminated from the atmosphere.
The aim of this study is investigating the effect of rhenium and Ru precursors (chlorine-free and chlorine-contaminated) on the thermal stability of bimetallic Ru–Re NPs treated in hydrogen at high temperature of 800 °C and in oxidizing atmosphere up to 400 °C using a similar bimetallic catalyst family as reported previously [24], and then testing their catalytic activity in propane oxidation. To the best of our knowledge, there is no research on the thermally induced sintering of supported Ru–Re NPs in H2 and O2 atmosphere. Such studies are important since Ru–Re catalysts could be applied in catalytic high-temperature hydrogen involving reactions or in environmental technologies (soot and VOCs removal in oxidative conditions). We have employed H2 and O2 chemisorption, X-ray diffraction (XRD), high-resolution electron microscopy (HRTEM), and high-angle annular dark-field STEM (HAADF-STEM) as the main characterization tools to study the morphological evolution of bimetallic catalysts microstructure and the sintering phenomenon of Ru–Re NPs deposited on γ-Al2O3. The influence of residual chlorine on the catalytic performance of the bimetallic Ru(Cl)–Re/γ-Al2O3 catalysts has not been studied for any catalytic reaction. The presence of chlorine in the catalysts can strongly modify their adsorptive and catalytic properties.
2 Experimental
2.1 Catalyst Preparation and Treatment
The synthesis method of bimetallic Ru–Re/γ-Al2O3 (co-impregnation) and monometallic Ru and Re catalysts (incipient wetness impregnation) was reported in previous work [24]. Aqueous solutions of Ru(NO)(NO3)3 or RuCl3 and NH4ReO4 (all Alfa Aesar) were used as metal precursors and γ-Al2O3 (Puralox) as support material. After impregnation, the Ru-based samples were dried overnight in air (110 °C) and then reduced in H2 flow (with a heating rate of 10 °C min−1) at 500 °C for 5 h, while the monometallic Re samples were reduced at a higher temperature of 550 °C. Next, after cooling to room temperature it were sintered in H2 flow at 800 °C also for 5 h. To test the thermal stability of the catalysts in an oxidizing atmosphere, the sintered catalysts were heated in static air at 250, 300, and 400 °C for 1 h at the rate of 5 °C min−1. According to the Ru precursor, the catalysts containing ruthenium metal are named Ru(N) or Ru(Cl).
2.2 Catalyst Characterization
The catalysts were characterized following the procedures partly described in previous studies [24, 26]. X-ray diffraction (XRD) patterns were conducted on a PAN analytical XPert Pro X-ray diffractometer with CuKα radiation. TEM images and SAED patterns of the catalysts were obtained with Philips CM20 SuperTwin microscope. High-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) measurements of the selected samples were carried out with a FEI Titan3 G2 60–300 microscope at 300 kV using a HAADF detector and energy dispersive X-ray spectroscopy with FEI ChemiSTEM technology (at Technische Universität Graz, Austria). The Ru/Re atomic ratios and chlorine contents for the reduced and oxidized samples were measured by energy dispersive spectroscopy EDS (EDAX Pegasus XM4 spectrometer installed on FEI NovaNanoSEM 230 microscope). Inductively Coupled Plasma Optical Emission Spectroscopy (ICP-OES) analyses were performed by using a Thermo Scientific ICAP 7000 apparatus.
The textural properties samples were measured by nitrogen adsorption–desorption at liquid nitrogen temperature on a Micromeritics ASAP 2020 C instrument. Before analysis the catalyst sample was degassed for 4 h at 300 °C. The ruthenium dispersion was determined by a H2 chemisorption experiment, while the extent of Ru and Re oxidation was estimated by the O2 uptake measurements using a conventional gas volumetric system employed earlier [24, 26]. Experimental details are described in the Electronic Supporting Information (ESI). The amount of irreversibly bound hydrogen was taken to calculate the ruthenium dispersion by assuming a 1:1 stoichiometry of H: Ru [29].
2.3 Catalytic Measurements
Catalytic tests for the oxidation of propane were carried out at atmospheric pressure in a fixed-bed quartz microreactor (i.d. = 6 mm) and at the temperature range of 120–270 °C. The catalyst weight was fixed at 50 mg and before the catalytic test, sample was brought into a contact for 20 min at RT with the gas mixture (2500 vppm of propane in the synthetic air) with a total flow rate of 50 cm3 min−1. Next, the catalyst was activated in the gas mixture from RT to 200 °C (heating rate 5 °C min−1) and stabilized at this temperature for 1 h. The volumetric flow rates of the gas mixture were controlled by a mass flow controller (MKS). The reaction temperature was monitored with a thermocouple protected by a quartz tube inserted in the center of the catalyst bed. Next, after cooling the sample to 120 °C, the conversion of propane was measured as a function of temperature in a typical heating cycle run. Analyses were performed at each reaction temperature until a steady-state was obtained, and at least three analyses were taken and data averaged. The effluent gases were analyzed online with a gas chromatograph (Perkin-Elmer ARNEL Clarus 500) equipped with Elite Plot-Q chromatographic column and a flame ionization detector. Propane conversion was calculated based on inlet and outlet measured concentration. The conversion data were reproducible within 5% accuracy. After a first catalytic run, the catalyst sample was cooled down to 120 °C and the process was repeated similarly. The measured rate of propane oxidation per gram of the catalyst, along with measurements of Ru dispersion, was applied to calculate turnover frequency (TOF), defined as moles of propane converted per surface metal atom per second (s−1).
3 Results and Discussion
3.1 Structure Characterization of the Ru–Re Catalysts Reduced at 500 °C
The morphological characteristics and dispersion data of the Ru–Re/γ-Al2O3 and monometallic (Ru, Re) catalysts reduced at 500 °C were reported in a previous work [24] and the main textural and ICP-OES results are presented in the ESI material (Table S1). In the bimetallic catalysts, the Ru content was 5 wt.%, while the Re content amounted to 1, 3, or 9.2 wt.%, which corresponds to the Ru/Re atomic ratio of 9:1, 3:1, and 1:1, respectively. On the Cl-free bimetallic Ru(N)–Re (9:1), Ru(N)–Re (3:1) and Ru(N)–Re (1:1) catalysts the Ru dispersion (H/Ru) amount to 0.57, 0.74, and 0.83, respectively and it was always higher than that on the monometallic Ru(N) catalyst (0.52). The mean metal particle size estimated by HRTEM decreased from 1.3 nm for the Ru(N)–Re (9:1) to 1.1 nm for the Ru(N)–Re (3:1) and 0.9 nm for the Ru(N)–Re (1:1) catalyst and these values were smaller than that obtained for the monometallic Ru(N) sample (1.4 nm). On the Cl-containing bimetallic Ru(Cl)–Re (3:1) catalyst the Ru dispersion was much lower (H/Ru = 0.21) and close to the Ru(Cl) sample (H/Ru = 0.20). Characterization results showed that all the Ru–Re catalysts consist primarily of RuRe bimetallic nanoparticles with sizes below 2–3 nm, while the larger NPs in the catalyst at the Ru/Re atomic ratio of 1:1 were monometallic Re [24].
3.2 Overall Characterization of the Ru–Re Catalysts Sintered in H2 at 800 °C
The metal contents of Ru and Re, measured by the ICP-AES method, were approximately the same as in samples reduced at 500 °C (Table S1). The textural data for the catalysts treated in H2 at 800 °C are given in Table 1. The alumina support undergoes small textural changes since the BET surface area (168 m2 g−1) is only 6% lower than in the bare support (178 m2 g−1). Also, minor textural changes occur for the Re/γ-Al2O3 catalysts. However, loss of the BET surface area of all the Ru-based catalysts was higher after treatment at 800 °C i.e. about 14% relative to the samples reduced at 500 °C (see Table S1). The decline of SBET is mainly caused by the changes in the pore structure of the catalysts. The total pore volumes of the catalysts treated at 800 °C (Table 1) are about 7% lower as compared to that reduced at 500 °C. Probably part of the metal species is located inside the pores or some alumina pores are blocked by larger metal particles that can grow on the alumina surface during hydrogen treatment at 800 °C. Some increase of the mean pore diameter may suggest that this blockage effect is more severe for the smallest pores of the given catalyst (Table 1). All the catalysts are mesoporous and their mean pore diameter is in the range of 7.8–8.2 nm. As shown in Table 1, the Re surface densities are in the range of 0.19–1.75 Re atoms nm−2 and these values are below the monolayer capacity of 2.3 Re atoms nm−2 [30].
3.2.1 XRD Characterization of the Catalysts Sintered in Hydrogen at 800 °C
XRD patterns of the monometallic and bimetallic catalysts heated in H2 at 800 °C are presented in Figs. 1, 2 and 3. The pattern of the 1% Re/γ-Al2O3 catalyst (Fig. 1, trace a) shows diffraction peaks at 2Θ of 37.6°, 45.7°, 62°, and 66.6° corresponding only to the structure of the γ-Al2O3 (JCPDS 29-0063). A very weak peak at 2Θ of 42.9° is visible in the pattern of the 3% Re catalyst that could be ascribed to metallic Re i.e. to the Re(101) crystal planes. For the 9.2% Re catalyst, this peak is much more intense and additionally weak and broad peaks at 2Θ of 37.58°, 75.2°, and 82.1°, corresponding to the rhenium (JCPDS 71-6589), are visible (Fig. 1, trace c). Only for this sample the mean crystallite size of Re metal could be calculated from the line broadening of the most intense Re(101) reflection (dav = 9 nm). After reduction at 550 °C very weak reflection at 2Θ of 42.9° was also seen but the calculation of the crystallite size of Re was impossible [24]. The XRD results indicate that low-loaded Re catalysts are resistant to sintering due to the well-known strong interaction of Re with γ-alumina, but with an increase of the Re loading its interaction with the support weakens and treatment at 800 °C leads to some sintering of the Re metal [22, 23].

XRD patterns recorded from the monometallic Re/γ-Al2O3 catalysts sintered in H2 at 800 °C with Re content of 1 wt.% (a), 3 wt.% (b), and 9.2 wt.% (c). All peaks from pattern a are ascribed only to γ-Al2O3 structure (JCPDS 29-0063)

XRD patterns of the catalysts prepared from Ru(NO)(NO3)3, sintered in H2 at 800 °C: monometallic 5% Ru(N)/γ-Al2O3 (a) and bimetallic 5% Ru(N)–Re/γ-Al2O3 with Re content of 1 wt.% (b), 3 wt.% (c), and 9.2 wt.% (d)

XRD patterns of the catalysts prepared from RuCl3, sintered in H2 at 800 °C: monometallic 5% Ru(Cl)/γ-Al2O3 (a), and bimetallic 5% Ru(N)-3%Re/γ-Al2O3 (b)
The XRD patterns of the Cl-free Ru(N) and Ru(N)–Re catalysts sintered in hydrogen at 800 °C are presented in Fig. 2. The pattern of the 5% Ru(N) catalyst (trace a) shows the diffraction peak at around 2θ = 44° which could be ascribed to (101) planes of the hexagonal Ru (JCPDS 06-0663). The mean crystallite size of Ru calculated from this reflection is 12 nm, suggesting a large agglomeration and sintering of the Ru phase. After reduction at 500 °C reflections from Ru were not observed (Ru particles below 2–3 nm) [24]. The addition of a small amount of Re (1%) to the Ru(N) catalyst did not change the XRD pattern and the mean crystallite size of Ru is the same (12 nm). However, this reflection disappears after the addition of 3% Re and only γ-Al2O3 reflections are visible (Fig. 2, trace c), i.e. identical as in the sample reduced at 500 °C [24]. This result evidence very good Ru dispersion and that the Ru–Re phase forms very small bimetallic NPs or the metal phase is amorphous. The formation of small Ru–Re NPs may affect also the strength of metal–support interactions. Therefore, the addition of an appropriate amount of Re to the 5% Ru(N) catalyst (Ru/Re atomic ratio of 3:1) inhibits the sintering of the Ru phase at high temperature. Further increase of the Re loading to 9.2% (Ru/Re atomic ratio of 1:1) leads to the appearance of sharp, intensive peaks at 2Θ of 37.6°, 40.5°, 42.9°, 56.4°, 67.9°, 75.2°, 82.1°, and 83.7°, which could be ascribed to metallic rhenium (JCPDS 71-6589), (Fig. 2, trace d). The mean crystallite size of Re is 15 nm, which is larger than in the monometallic 9.2% Re catalyst (9 nm). Probably, part of added rhenium, which is not included in the formation of bimetallic Ru–Re NPs, has a weaker interaction with γ-Al2O3 and agglomerates during the high-temperature treatment in hydrogen. Determination of XRD reflections from metallic Ru is difficult for this catalyst because they may partly overlap with rhenium reflections. It is also possible that all Ru atoms are included in the formation of small bimetallic Ru–Re NPs and sintering of the ruthenium phase was inhibited.
The XRD patterns of the chlorine-containing Ru(Cl) and Ru(Cl)–Re (3:1)/γ-Al2O3 catalysts sintered at 800 °C are presented in Fig. 3. In opposite to the Cl-free Ru(N) catalyst, in the pattern of the monometallic Ru(Cl) catalyst only very weak reflection at 2θ of 43°–44° is hardly detectable and can be ascribed to the Ru phase (trace a). Probably, the presence of residual Cl ions (from the Ru precursor) limits the aggregation of the ruthenium phase or the small Ru particles are in stronger interaction with the support surface. For the bimetallic Ru(Cl)–Re (3:1) catalyst diffraction peaks due to metallic Ru or Re were not visible (trace b). Thus, in both chlorine-contaminated samples, metal particles are highly dispersed similarly as in the bimetallic Cl-free Ru(N)–Re catalyst with the same Ru/Re atomic ratio (3:1).
3.2.2 TEM and STEM-HAADF Characterization of the Ru–Re Catalysts Sintered at 800 °C
TEM characterization were conducted to identify the morphology of the studied catalysts. Figure 4 shows representative TEM images and corresponding particle size distribution of the Re/γ-Al2O3 catalysts sintered in hydrogen at 800 °C. In the TEM image of the 1% Re catalyst, dark small particles with a size of 1–4 nm, uniformly distributed on the support are visible (Fig. 4a). At the Re loading of 3%, in addition to the smallest nanoparticles, few larger nanoparticles (up to 7 nm) with a crystalline structure are also visible (Fig. 4b). The HRTEM image in the inset shows a 6 nm crystallite with lattice fringes of distances 0.21 nm what corresponds to the (101) lattice planes of Re metal. In SAED pattern (not shown), weak diffraction spots from Re were also present. The size of Re crystallites in the 9.2% catalyst sometimes exceeded 12 nm (Fig. 4c), and thus they were detected by XRD (Fig. 1, pattern c). The mean particle size of the 1, 3, and 9.2% Re catalysts was 1.8, 2.1, and 2.5 nm, respectively. With the increase of the Re loading, distribution of sizes is also significantly widened from 1–4 nm to 1–7 nm and 1–13 nm, respectively. For comparison, the mean particle size of the 3 and 9.2% Re catalysts reduced at 550 °C was smaller (1.6 and 1.7 nm, respectively), while for the 1% Re sample metal particles were not observed [24].

Representative TEM images and corresponding particle size distribution of the Re/γ-Al2O3 catalysts sintered in H2 at 800 °C with Re content of 1 wt.% (a), 3 wt.% (b), and 9.2 wt.% (c)
Monometallic Ru(N)/γ-Al2O3 catalyst undergoes larger sintering in hydrogen at 800 °C as can be seen from the TEM image, and the Ru particle size distribution presented in ESI in Fig. S1. The majority of the Ru particles have a size below 3 nm. However, frequently larger Ru crystallites with size even above 20 nm, were visible. The HRTEM image (inset in Fig. S1) shows the 10 nm crystallite with lattice fringes of distances 0.205 nm what corresponds to the (101) lattice planes of Ru metal. The presence of a few large Ru crystallites causes that the mean crystallite size calculated from the XRD (12 nm) is much higher than that from the TEM size distribution (2.3 nm). Opposite, in the Cl-containing Ru(Cl) catalyst (Fig. 5a) only small particles with size of 1–3 nm were observed in agreement with XRD data (Fig. 3). The mean particle size of Ru is slightly lower than that in the Ru(N) sample and amount to 1.9 nm.

TEM images recorded for the 5% Ru(Cl)/γ-Al2O3 (a) and 5% Ru(Cl)-3% Re/γ-Al2O3 (b) catalysts after heating in H2 at 800 °C
Figure 6 shows representative TEM images and corresponding particle size distribution of the bimetallic Ru(N)–Re/γ-Al2O3 catalysts sintered at 800 °C. It is evident that the addition of 1% of Re did not change the morphology and the distribution of particle sizes (Fig. 6a) and the mean particle size (2.7 nm) is only slightly higher as compared to the Ru(N) catalyst (2.3 nm). Combined the XRD and TEM results of the monometallic Ru(N) catalyst (Figs. 2, S1), the larger metal particles observed in the HRTEM image of the Ru(N)-1%Re catalyst (inset to Fig. 6a) could be due to the aggregation of Ru0 crystal particles rather than rhenium crystallites, although the lattice fringes of distances 0.21 nm may correspond both to metallic Ru(101) or Re(101) crystal planes. Also, the fast Fourier transform (FFT) pattern from this crystalline particle (inset in Fig. 6a) contains spots corresponding to 0.21, 0.22, or 0.24 nm lattice fringes, which can be assigned to the hexagonal structure either metallic Re or Ru. The particle sizes of the Ru(N)-1%Re catalyst ranges mainly between 0.6 and 2 nm and few larger nanoparticles up to a size of 24 nm are detected. The size distribution is much wider than 0.6–3 nm obtained for the catalyst reduced at 500 °C for which dav was 1.3 nm [24]. For this catalyst (Ru/Re atomic ratio of 9:1) performing the EDS analysis was possible. Figure 7 shows the HAADF-STEM images and EDS spectra with calculations of the atomic composition of the selected areas in images. From these measurements, it appears that larger particles with sizes above 10 nm consist mainly of Ru atoms. In such large crystallites, the EDS analysis indicates a very small amount of Re atoms (4.1 at%), and probably Re atoms are located on the surface of Ru particle (Fig. 7a). The EDS spectrum from the area where small nanoparticles are below 2–3 nm (Fig. 7b), leads to the conclusion that small NPs consist of both Ru and Re atoms, and EDS analysis indicates that the metal composition of Ru:Re in the particles is 78:22 molar ratio. The obtained Ru:Re atomic ratio is slightly different than the value of 9:1 expected from the synthesis of this catalyst. Probably, the amount of rhenium was insufficient to formation only Ru–Re NPs, and excessive Ru atoms agglomerate to large Ru crystallites during the high-temperature treatment in hydrogen (Figs. 6a, 7a).

Representative TEM images and corresponding particles size distribution of the bimetallic 5% Ru(N)–Re/γ-Al2O3 catalysts sintered in H2 at 800 °C with Re content of 1 wt.% (a), 3 wt.% (b), and 9.2 wt.% (c)

HAADF-STEM images and EDS spectra recorded for the 5% Ru(N)-1% Re/γ-Al2O3 catalyst sintered in H2 at 800 °C: from larger (ca.15 nm) particle (a) and from the region with small (2–3 nm) particles (b)
The TEM images obtained for the Ru(N)-3%Re/γ-Al2O3 catalyst sintered in hydrogen at 800 °C contain only small particles (0.6–3 nm) with an average size of 1.3 nm and a very narrow size distribution (0.6 nm standard deviation) (Fig. 6b), and thus they were not detected by XRD (Fig. 2). Similar TEM results were obtained previously for the catalyst reduced at 500 °C [24]. It is evident that this catalyst is very stable under high-temperature treatment and probably all particles are bimetallic Ru–Re (with the molar composition close to 3:1). Similar TEM images were obtained for the chlorine-containing bimetallic Ru(Cl)-3%Re catalyst for which the average size of metal particles is 1.5 nm and also a very narrow size distribution is visible (Fig. 5b). The number of metallic particles with a size below 1.5 nm is significantly greater (63%) than in the monometallic Ru(Cl) catalyst (24%, Fig. 5a). Therefore, it is obvious that both in the Cl-free and Cl-contaminated samples, Re promoter at a sufficient amount could effectively suppress the Ru sintering process. Our attempts to obtain HAADF-STEM images (with good quality) and EDS spectra of a single particle in the Ru(N)–Re (3:1) catalyst sintered at 800 °C were unsuccessful since such small bimetallic NPs were found to be less stable under the electron beam (about 98% particles below 2 nm). Similar difficulties in obtaining STEM-HAADF images of the small core–shell type RuRe/PVP NPs were reported by Ayvali et al. [18]. Release of atoms from the surface of the RuRe NPs during the analysis was observed, leading to the presence of defects on the outer shell of the nano-objects and isolated atoms on the grid. HAADF-STEM images and EDS spectra of the Ru(N)–Re (3:1) catalyst reduced at 500 °C had better quality and are shown in ESI in Fig. S2. All observed RuRe particles in this catalyst have the atomic composition of Ru:Re = 3:1.
In the TEM image of the bimetallic Ru(N)-9.2% Re/γ-Al2O3 catalyst treated in hydrogen at 800 °C, besides small, evenly dispersed nanoparticles, also larger particles with size up to 25 nm are visible (Fig. 6c). The SEAD pattern shows strong diffraction spots that can be assigned to the Re or Ru structure (inset to Fig. 6c). However, considering the XRD results (Fig. 2) and characterization data obtained for analogous system reduced at 500 °C [24], one can assume that large particles consist of Re atoms, while the smallest ones are bimetallic Ru–Re NPs. Therefore, at high Re loading, only part of the Re atoms is incorporated into the formation of bimetallic Ru–Re NPs.
Summarizing, the particle size distribution of the bimetallic 5%Ru(N)-Re/γ-Al2O3 catalysts sintered in hydrogen at 800 °C reveal that despite the different amounts of Re the resulting Ru–Re bimetallic NPs have the more or the same size (85–98% particles below 2 nm), (Fig. 6). Additionally, the presence of residual chlorine (from the metal precursor) does not affect the size of the Ru–Re NPs being formed (Fig. 5b). In the samples reduced at 500 °C, also about 90% bimetallic Ru–Re nanoparticles have particle size below 2 nm and very narrow size distribution (0.7–0.4 nm standard deviation) was observed [24]. Therefore, TEM data indicate that the amount of Re precursor introduced in the synthesis of bimetallic Ru–Re catalysts and the high-temperature treatment in hydrogen has marginal influence on the particle size of the formed Ru–Re bimetallic NPs. A small fraction of the larger particles in the bimetallic Ru(N)–Re/γ-Al2O3 catalyst with low Re content (1 wt.%) contains nearly pure Ru metal, as shown by HAADF-STEM, while at the highest Re content (9.2 wt.%) the separated Re large particles have been formed as well and thus, large broadening of the particle size distribution (standard deviation 4.2–3.6 nm) was observed for these samples (Fig. 6). The results clearly indicate that small bimetallic Ru–Re NPs are much more stable than pure Ru or Re particles. If we assume that the step limiting the sintering of nanoparticles is the dissociation of the metal–metal bonds (Ostwald ripening mechanism), the observed behaviour suggests that the reduced sintering of the bimetallic Ru–Re NPs (with sizes up to 2–3 nm) could be attributed to the stronger Ru–Re bonds compared to those of Ru–Ru and Re–Re. This statement means that there is a favourable interaction between Ru and Re (as the result of a synergic effect), which is also evidenced by the study of solid solutions over the entire composition range in the Ru–Re binary alloy phase diagram [31]. It could be noted that formation energy ∆Eform of the binary RuRe alloy amount to − 176 (meV atom−1). The large negative value of Eform confirms also the chemical stability and strong interaction between the two metals [32]. Interaction between small bimetallic Ru–Re NPs and alumina support (via the oxygen groups) may be also stronger than Ru or Re in the monometallic samples.
3.3 Hydrogen Chemisorption Studies
Hydrogen chemisorption at 100 °C was used to determine ruthenium dispersion in the monometallic Ru and bimetallic Ru–Re catalysts sintered at 800 °C. Determination of metal dispersion in the Ru-containing catalysts by hydrogen chemisorption is a widely used method ([29], and herein cited) but it cannot be easily applied for Re or bimetallic Re-containing catalysts. It is known that rhenium does not induce dissociative hydrogen chemisorption at RT but may adsorb atomic hydrogen at higher temperatures [22, 24, 33]. Chądzynski and Kubicka found that the formation of monolayer of chemisorbed hydrogen on Re/γ-Al2O3 catalysts can be obtained by saturation with H2 from 500 °C to RT [33]. Previously we reported that hydrogen chemisorption at 100 °C on the Re/γ-Al2O3 catalysts was negligible (below 5–10 µmol g−1 cat−1) [24], therefore in this study we assume that Re does not contribute to the amount of adsorbed hydrogen. Chemisorption data are summarized in Table 2 and compared with some results presented for the catalysts reduced at 500 °C [24]. The H2 uptake depended strongly on the Ru precursor, but regardless of the metal precursor used, it was always higher for the bimetallic Ru–Re catalysts than for the monometallic Ru samples.
For the Ru(N)–Re samples sintered at 800 °C, the H2 uptake increases with the Re loading, and a particularly large increase is visible for the samples with the Ru/Re atomic ratio of 9:1 and 3:1). With a further increase in Re content (9.2%), only small changes in the H2 uptake occurred (Table 2) in full agreement with XRD and TEM data, which evidence that part of Re metal, which does not chemisorb hydrogen, was not alloyed with the ruthenium phase. At the same Ru loading (5 wt.%) chemisorption of hydrogen is about two times higher for the bimetallic Ru(N)–Re (3:1) and Ru(N)–Re (1:1) catalysts (~ 160 µmol g−1 cat−1) as compared to the monometallic Ru(N) catalyst (84 µmol g−1 cat−1). The H2 uptake values are, however, lower for the Ru(N) or Ru(N)–Re catalysts by about 34 or 15–20%, respectively, compared to the same catalysts reduced at 500 °C (Table 2), indicating that bimetallic samples are more resistant to sintering than monometallic Ru(N) catalyst.
The chemisorption data of the Ru(Cl) and Ru(Cl)–Re (3:1) catalysts should be considered as that of chlorine modified Ru surface. Evidently, hydrogen uptake both on the mono- and the bi-metallic sample is significantly lower as compared to the Cl-free catalysts (Table 2). Our previous and literature data [24, 29, 34] evidenced that chlorine had a negative effect on the H2 or CO chemisorption on the Ru-based catalysts prepared from RuCl3. However, opposite to the Cl-free catalysts, H2 uptakes on the sintered Cl-containing samples are much higher than on the same samples reduced at 500 °C. Particularly large increase is visible for the bimetallic Ru(Cl)–Re (3:1) catalyst, for which the H2 uptake is nearly 100% higher. The better H2 chemisorption properties could be attributed to the chlorine removal from the ruthenium surface during the hydrogen treatment at temperature as high as 800 °C. From SEM–EDS analyses shown in Table 3, it is evident that about 46 or 39% of the initial content of chlorine is eliminated from the Ru(Cl) or Ru(Cl)–Re catalysts by increasing temperature treatment in hydrogen from 500 to 800 °C. However, it must be considered that even under such conditions, chlorine was not completely eliminated from the catalyst surface. In many reports, it was found that it is very difficult to eliminate residual chlorine from the Ru/Al2O3 catalysts based on metal chlorides [29, 35]. Narita et al. [36] reported that for the Ru/Al2O3 catalysts derived from RuCl3, ·large amounts of chlorine remained on the catalyst surface after reduction at ~ 320 °C and chlorine was partitioned between the support and the metal. This residual chlorine was observed to inhibit hydrogen chemisorption. When the reduction temperature was increased to ~ 700 °C also increase of H2 chemisorption was observed evidencing remove the chlorine from the catalyst surface.
Metal dispersion (H/Ru) and the average particle size estimated from H2 chemisorption were compared with the results obtained by TEM and XRD (Table 2). Metal dispersion of the bimetallic Ru(N)–Re and Ru(Cl)–Re catalysts is higher than the monometallic Ru(N) and Ru(Cl) catalysts for which H/Ru = 0.33 or 0.27, respectively. The rise of the Re content to 3 wt.% in the Ru(N)–Re samples causes a large increase in metal dispersion (H/Ru = 0.63, i.e. by about 90%) but the further increase to 9.2 wt.% almost does not change the metal dispersion (H/Ru = 0.65). After reduction at 500 °C, the bimetallic Ru(N)–Re catalysts were more dispersed and the highest dispersion (H/Ru = 0.83) was obtained for the catalyst with a Ru/Re atomic ratio of 1:1 [24]. Only in a bimetallic Ru(N)–Re (3:1) catalyst, the average particle size as determined by chemisorption and TEM is consistent. In the samples with the Ru/Re atomic ratio of 9:1 and 1:1, chemisorption and TEM results converge, but the XRD data are evidently too high. TEM data (Fig. 6) show clearly that the reason is the presence of a large population of very small particles (85–98% particles below 2 nm) and these particles were overlooked by the XRD. It also means that this fraction of the metal particles is resistant to sintering in hydrogen at 800 °C. Dispersion of bimetallic Ru(Cl)–Re (3:1) catalyst is lower (H/Ru = 0.40) than that of the chlorine-free sample at the same composition (H/Ru = 0.63) but much higher than in the Ru(Cl) sample (H/Ru = 0.27), as well as in the samples reduced at 500 °C (H/Ru = 0.20–0.21). Evidently, the positive influence of Re addition and the high-temperature treatment at 800 °C on the Ru dispersion is visible. TEM data also show that the Ru(Cl)–Re (3:1) catalyst had the mean particle size slightly smaller than the Ru(Cl) catalyst. Since chlorine leads to the blockage of the Ru sites for hydrogen chemisorption some discrepancies between the mean particle size based on H2 chemisorption and TEM data are observed (Table 2).
3.4 Characterization of the Ru–Re Catalysts Treated in Oxygen Atmosphere
3.4.1 Oxygen Adsorption Measurements
Oxygen adsorption results are shown in Table 4. The O2 uptake at RT on the sintered Re/γ-Al2O3 catalysts is low and corresponded to the dissociative chemisorption [22]. At 250 °C, the O2 uptake is much higher indicating partial oxidation of the Re NPs. At 300–400 °C, a larger increase of the O2 uptake is observed only for the 9.2% Re catalyst. For all samples the measured O2 uptakes even at 400 °C are lower than that needed for the total oxidation of Re to Re2O7. Bare et al. [37] found that in the Re/γ-Al2O3 catalysts reduced at 500–700 °C, rhenium is present as a mixture of species: unreduced Re(VII) species, Re nanoclusters, and isolated Re atoms. The presence of a mixture of various oxidation states is ascribed to the strong oxophilicity of rhenium.
The O2 uptake on the sintered 5% Ru(N) catalyst already at RT is much higher and indicates that only 12% of Ru is in the metallic state. At 250 °C, oxidation of Ru is complete and some excess O2 consumption is associated probably with its adsorption to RuO2 or formation of RuO4. The O2 uptake at RT on the Ru(N)–Re catalysts containing 1, 3, or 9.2% Re, is higher by 10%, equal, or lower by 23% respectively, than the sum of the values for the monometallic (Re, Ru) samples. The results indicate blocking of the low-temperature oxidation of the Ru phase by the rhenium species existed on the Ru–Re particle surface, well visible when the amount of Re increase in the bimetallic catalysts. The literature data show that in the Ir–Re/SiO2 [38], Rh–Re/SiO2 [39] and Pt–Re/SiO2 catalysts [40] the surface of the metallic Ir, Rh, and Pt is partially covered by two-dimensional ReOx clusters and that the size of this monolayer increases with the Re loading [40]. At 250–400 °C, the O2 uptakes on samples containing ≤ 3% of Re indicate the complete oxidation of Ru and Re, although some excess of oxygen adsorption associated with the RuO2 phase was noticed only for the Ru(N)–Re (9:1) sample. The addition of 9.2% of Re to the Ru(N) catalyst causes an increase of the O2 uptake with temperature but even at 400 °C, the O2 uptake is much lower (990 μmol O2 g−1 cat−1) than the theoretical value for the oxidation of Re to Re2O7 and Ru to RuO2 (1341 μmol O2 g−1 cat−1) and also lower than the sum of the experimental values for the 5% Ru(N) and 9.2% Re catalysts (1253 μmol O2 g−1 cat−1). Changes in the adsorptive properties evidently depend on the Ru/Re atomic ratio and may be related to the Ru–Re interaction, which not only partly limits oxygen adsorption at low temperature but also for high Re loading limits oxidation of metals at elevated temperatures. Part of the Re metal, which was not alloyed with the Ru phase, may be also more difficult to the total oxidation at the used oxidation conditions. (Table 4).
On the Cl-containing 5% Ru(Cl) catalyst, the O2 uptake at RT is about three times lower than that on the 5% Ru(N) catalyst and about 2.7 times lower on the bimetallic Ru(Cl)–Re (3:1) catalyst compared to the Ru(N)–Re (3:1). Considering the TEM and H2 chemisorption results, such a significant suppression in O2 adsorption at RT confirms a large blockage of the Ru sites by the residual chlorine species. In the case of Ru(N) sample, measured O/Ru ratio at RT is close to 1.75, while for the Ru(Cl) sample close to 0.73, indicating that the formation of RuOClx (instead of RuO2) is possible. The formation of the oxychloride species (RuOClx) on the surface of the ruthenium particle was also reported by Mazzieri et al. for the Ru/γ-Al2O3 catalysts prepared from the RuCl3 precursor [41]. At 250–400 °C, the O2 uptake is also lower by 15–12% for both Cl-containing catalysts as compared to that on the corresponding Cl-free samples (Table 4).
3.4.2 XRD and TEM Studies of the Catalysts Oxidized at 250–400 °C
The XRD data of the partially or fully oxidized catalysts may correspond to the state of Ru and Re during the activation or oxidation step in the Ru–Re/γ-Al2O3 catalysts used in environmental technologies such as VOCs and soot removal in oxidative conditions. XRD patterns of all the Ru and Ru–Re samples oxidized at 250 °C (not shown) were very similar to those presented earlier i.e. for the Cl-free catalysts reduced at 500 °C and next oxidized at 250 °C [26] and contained peaks from the γ-Al2O3 and RuO2 oxide (JCPDS 88-0322). Therefore, in the Re-modified samples metal segregation occurs already at this temperature. Only for the sintered Ru(N)–Re (1:1) catalyst, which contained large Re crystallites (15 nm, Fig. 2), additional weak Re(101) peak appeared in the XRD pattern suggesting that large Re particles were not fully oxidized in agreement with the O2 uptake data (Table 4). With increase oxidation temperature no evidence of any rhenium or Re oxide phase was found for all the bimetallic Ru–Re catalysts. Thus, we conclude the absence of ReOx peaks reflect the formation of highly dispersed, non-crystalline Re(+ 7) oxide phases (oxidation state predicted by phase diagrams) as a result of high temperature treatment in air. We previously reported that the oxidation process of Re metal does not lead to the formation of a stable bulk Re oxide since at elevated temperatures volatile Re2O7 oxide can be formed, which above 150–200 °C sublimes and subsequently adsorbs over the alumina surface as isolated ReO4 groups [22, 23]. Treatment at higher temperatures accelerates oxidation of rhenium causing that the concentration of the ReO4 species over the support surface is higher [22]. It must be noted that calculated surface Re densities (Table 1, 0.19–1.9 Re atoms nm−2) are below the monolayer coverage (2.3 Re atoms nm−2 [30]), hence loss of Re by volatilization of the Re2O7 oxide should be avoided. In fact, the EDS analyses confirmed that after the oxidation treatment of the catalysts at 400 °C, the content of Re and Ru is similar to that in the reduced samples (Table S1). In this study the O2 uptake measurements (Table 4) showed the expected Re enrichment of the Ru–Re particle surface even at low temperature. The standard enthalpies of formation of the Ru and Re oxide species are—305 kJ mol−1 for RuO2 and 1240 kJ mol−1 for Re2O7. Therefore, the ReOx oxide species grow on the surface of Ru–Re NPs due to migration of Re atoms from the bulk to the surface. At higher temperature Re2O7 species sublimes and next adsorbs on γ-Al2O3 as ReO4 species, while Ru atoms agglomerated to RuO2 oxide particles. It is also possible that part of rhenium species is associated with the ruthenium phase, and there was an enrichment of ReOx species at the peripheral interface of the RuO2 particles and the γ-Al2O3 and additionally, some rhenium species may also exist as a thin film on top of the RuO2 oxide particles. Ma et al. [19] studied the role of ReOx species in the Ni–Re/Al2O3 catalyst for amination reaction using a combination of various experimental and theoretical methods and found that rhenium oxide species prefer to atomically disperse on the surface of NiO in calcined Ni–Re/Al2O3 catalyst. Our XRD results clearly show that metal segregation in the bimetallic Ru–Re catalysts leads to RuO2 agglomeration and transformation of the Re phase into a highly dispersed species. The segregation process of Pt and Re during the oxidation step of the regeneration procedure of the Pt–Re/γ-Al2O3 catalysts was also reported [42]. The summary of the mean crystallite size of RuO2 oxide, estimated from the line broadening of the most intense (110) peak of RuO2, is presented in Fig. 8. For the monometallic Ru(N) and bimetallic Ru(N)–Re (9:1) catalyst with the lowest Re content (1 wt.%), in which there were separated larger Ru crystallites (12 nm, Fig. 2), the mean crystallite size of RuO2 increases from about 14–13 nm at 250 °C to 20–18 nm at 400 °C. Much smaller sizes of RuO2 crystallites and their changes with oxidation temperature were visible for the monometallic Ru(Cl) i.e. from 6.7 nm at 250 °C to 9.5 nm at 400 °C, and for the bimetallic Ru–Re catalysts containing higher amounts of Re (from 7–8 nm at 250 °C to 10–11 nm at 400 °C). Evidently, adding more rhenium to the Cl-free or Cl-containing Ru(Cl)/γ-Al2O3 catalyst causes smaller aggregation of the Ru phase. Probably, Re species strongly bonded to the alumina surface as isolated ReO4 groups, at higher concentration worked as a stabilizer, just like as a nanoglue, against agglomeration of the ruthenium phase. The residual Cl species partly located also on the γ-alumina surface additionally hinder aggregation of the RuO2 particles. As shown in Fig. 8, at 400 °C smaller sizes of RuO2 crystallites were observed in the bimetallic Ru(Cl)–Re (3:1) catalyst (9 nm) as compared to the Cl-free catalyst at the same composition (10.5 nm).

The mean crystallite sizes of RuO2 oxide, estimated from the XRD results—Sherrer calculations from line broadening of the most intense (110) peak of RuO2 at 2Θ near 28°,—after oxidation of the catalysts at 250, 300 and 400 °C
Representative HRTEM images and SAED patterns (insets) of the Ru(N) and bimetallic Ru(N)–Re catalysts oxidized at 250 °C are shown in Fig. 9. This oxidation temperature was chosen for the study of changes in the morphology of the catalysts because the activity tests were always performed below 280 °C. In line with the XRD results, all the SAED patterns contains strong reflections which could be ascribed to the RuO2 oxide structure. The HRTEM images of the Ru(N) and Ru(N)–Re (9:1) are very similar (Fig. 9a, b) and show that the morphology of the RuO2 crystallites is rodlike (as characteristic for the bulk RuO2 structure) with particle dimensions up to 6 nm × 25 nm. Also, in agreement with XRD data, HRTEM image of the Ru(N)–Re (3:1) reveals that RuO2 crystallites are smaller (Fig. 9c) and for the Ru(N)–Re (1:1) only sparse RuO2 particles and more irregular are visible (Fig. 9d). Moreover, in the HRTEM images of the bimetallic samples with Re content of 3 wt.% and 9.2 wt.% very large number of very small particles/clusters (∼1 nm and smaller) uniformly distributed on alumina is visible (Fig. 9c, d). Probably, some of these very small particles are related to the oxidized forms of rhenium species [22]. The above XRD and TEM data show that even at moderate temperatures Ru–Re bimetallic NPs are not stable in the oxidative atmosphere as opposed to the reductive atmosphere, at which even at 800 °C small Ru–Re bimetallic NPs supported on γ-Al2O3 are very stable.

Representative HRTEM images and SAED patterns (insets) of the monometallic Ru(N) catalyst (a), and bimetallic Ru(N)–Re (9:1) (b), Ru(N)–Re (3:1) (c), and Ru(N)–Re (1:1) (d) after oxidation in air at 250 °C for 1 h
4 Catalytic Activity of the Catalysts in Propane Oxidation
Catalytic measurements were performed on the selected catalysts sintered in H2 at 800 °C and next activated under propane/air mixture at 200 °C for 1 h. Such a procedure is very advantageous for obtaining a catalyst, which could be stable under used O2-rich reaction conditions. TEM studies showed that the morphology of the catalysts already changes during their oxidation at 200 °C (Fig. S3). Namely, RuO2 particles were not detected only in the Ru(N)–Re (1:1) catalyst but in samples with lower Re content (Fig. S3b, c) few small particles with lattice fringes of distances 0.32 nm that correspond to RuO2 oxide were visible. However, the majority of the metal particles oxidized at 200 °C were so small that they were hardly observable in HRTEM images. To evaluate the catalytic performance and stability of mono- and bimetallic Ru–Re catalysts, the oxidation of propane was performed usually in three successive cycles i.e. after the first catalytic test the sample was cooled down to 120 °C and the second and next third test was carried out at the same reaction conditions.
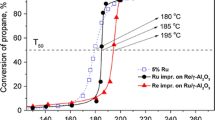
Figure 10 presents the propane conversion as a function of reaction temperature over the Cl-free and Cl-contaminated Ru and bimetallic Ru–Re/γ-Al2O3 catalysts (the first test), while the temperatures required to achieve different degrees of conversion (50 or 95%) in all catalytic tests are summarized in Table 5. The reaction products were only CO2 and water. In the first catalytic test, both Cl-free Re-modified catalysts exhibited a catalytic performance slightly better than the monometallic Ru(N) catalyst. For example, the light-off temperature of the bimetallic Ru(N)–Re (3:1) catalyst (T50% = 215 °C) was lower than that of Ru(N) catalyst (T50% = 223 °C, Table 5). It should be noted that monometallic 3% Re/γ-Al2O3 catalyst sintered at 800 °C was completely inactive in the reaction temperature range (up to 300 °C) in agreement with previous work [26]. However, at higher reaction temperatures (> 240 °C) the bimetallic Ru(N)–Re catalysts possess a catalytic performance similar to the Ru(N) catalyst (Fig. 10). It should be noted that the catalytic performance of the Cl-free Ru(N)–Re (9:1) and Ru(N)–Re (3:1) catalysts reduced at 500 °C was better (T50% = 160 °C or 173 °C, respectively) [26], than those reported here i.e. after sintering in hydrogen at 800 °C. Differences should be explained not only by the lower Ru dispersion in the samples sintered at 800 °C (Table 2) but also by different experimental conditions (the propane concentration in the flow was two times higher, mass of the catalyst was 100 mg and the total flow rates was 100 ml min−1 [26]). The catalytic performance of the Cl-containing Ru(Cl) and especially bimetallic Ru(Cl)–Re (3:1) catalyst is markedly lower than that using Cl-free catalysts at the same composition. In the first catalytic test the light-off temperature of the monometallic Ru(Cl) catalyst (T50% = 234 °C) and bimetallic Ru(Cl)–Re (3:1) catalyst (T50% = 257 °C) was higher by 11 °C or 42 °C, respectively, than that of Ru(N) or Ru(N)–Re (3:1) catalyst (Table 5). Moreover, the temperature at which 95% conversion is attained is also significantly higher for the Cl-containing samples. Additionally, larger differences in T95% occurred for the bimetallic Ru(Cl)–Re catalyst (277 °C) as compared to the Ru(Cl) sample (260 °C), indicating that oxidation of propane over the bimetallic Ru(Cl)–Re catalyst is more difficult. Thus, a poisoning effect of the residual chlorine precursors remaining on the surface of the bimetallic Ru(Cl)–Re catalyst is greater compared to monometallic Ru(Cl) despite the fact that these catalysts have the same Ru loading (5 wt.%), similar ruthenium particle size (HRTEM and XRD data) and higher dispersion of Ru–Re species (Table 2). On the other hand, as shown in Table 3, the content of chlorine species in the bimetallic Ru(Cl)–Re (3:1) catalyst sintered at 800 °C i.e. before the first catalytic test, was higher (2.34 wt.%) as compared to the Ru(Cl) sample (1.95 wt.%). Chlorine ions likely prevent, to some extent, the effective interaction between ruthenium and rhenium species, which is clearly seen in the bimetallic Ru(N)–Re catalysts based on the chlorine-free precursors. Literature data frequently reported that chlorine poison the active sites on the supported Pt and Pd catalysts for the combustion of alkanes [43,44,45]. The poisoning effect of chlorine has also been observed in the light alkane’s oxidation over the monometallic Ru/γ-Al2O3 and Ru/CeO2 catalysts prepared from RuCl3 [46, 47].

Light-off curves taken from the propane conversion over the Cl-free and Cl-contaminated Ru and bimetallic Ru–Re/γ-Al2O3 catalysts in the first cycle. Experimental conditions: mass of the catalyst: 50 mg, feed composition: 2500 vppm C3H8 in the synthetic air, total flow rate 50 cm3 min−1, pressure: atmospheric
In the second and third tests, all the Cl-free catalysts are only slightly less active, which is evidenced by the small shift of the corresponding light-off curves toward higher temperatures, and Fig. S4 in ESI shows the conversion curves with the example of the 5% Ru(N)-1% Re catalyst. As shown in Table 5, differences in T50% values between the first and third test are also very small (only 3–7 °C) and they are nearly not affected by the composition of the catalysts. Moreover, for a given Cl-free catalyst (mono- or bi-metallic), the temperature at which 95% conversion is achieved is the same in all catalytic tests, indicating a very good recycling potential of the samples sintered at 800 °C for the propane total oxidation. We previously reported that stability of the Ru(N)/γ-Al2O3 catalyst reduced at 500 °C in the oxidation of propane was much lower than bimetallic Ru(N)–Re/γ-Al2O3 catalysts reduced also at 500 °C [26, 27]. Namely, during the catalytic process the Ru(N) catalyst has undergone more significant structural alternation (formation of a less active large RuO2 crystallites) as compared to the bimetallic Ru(N)–Re samples [26]. Debecker et al. showed that RuO2-based catalysts, which are very active in propane oxidation, should be used at reaction temperature less than 250–300 °C, where the structural changes are limited [48]. Similarly, as in our study, the authors found that the reproducibility of the catalytic data obtained for the nano-RuOx/TiO2 catalyst was very good i.e. the three light off curves obtained when the temperature was increased in the step mode were almost superimposed (up to 300 °C). Currently, much attention is paid to Ru-based catalysts for their stability and deactivation processes in the VOCs and CO oxidation [26,27,28, 48,49,50,51,52]. Some authors, studying active species and structural deactivation of various Ru-based catalysts, have reported that under oxidizing reaction conditions, the metallic Ru surface converts to a catalytically very active thin ruthenium oxide layer RuOx, which next may transform into a less active oxide phase [46]. As shown in Fig. 11, the chlorine-containing bimetallic Ru(Cl)–Re (3:1) catalyst exhibits interesting catalytic performance because it is more active in the second test than in the first one. The T50% value in the second test is about 14 °C lower (243 °C) than in the first test (257 °C). Much smaller differences in T50% values are seen for the monometallic Ru(Cl) sample, but at higher reaction temperature improvement in catalytic performance is more visible (Table 5). However, in the second test bimetallic Ru(Cl)–Re (3:1) catalyst, despite the greater improvement in catalytic performance, is still less active than the monometallic Ru(Cl) and much less active than the most active Cl-free bimetallic catalyst at the same composition. The better catalytic performance of both chlorine-containing catalysts in the second test can be explained by the further removal of deactivating chlorine species from the ruthenium surface, although a large part of the chlorine still remains on the catalyst surface (Table 3). We suppose that chlorine removal from catalysts is preferred by the presence of water vapor generated during the propane oxidation reaction in the O2-rich atmosphere.

Light-off curves taken from the propane conversion over the Cl-contaminated Ru(Cl) and bimetallic Ru(Cl)–Re/γ-Al2O3 catalysts in the two consecutive cycles. Experimental conditions as in Fig. 10
Differences in activity of the studied catalysts depending on the Ru precursor used are more evident in the Arrhenius plots presented in Fig. 12. The specific reaction rate used in the Arrhenius plots is expressed in micromoles of propane converted on 1 g of the catalyst per second (µmol C3 g−1 s−1). It is clear that chlorine-containing catalysts are much less active than those without chlorine. From the Arrhenius plots, the apparent activation energies Eapp, were calculated and summarized in Table 5. As it can be seen the activation energies for the chlorine-free Ru(N) and Ru(N)–Re (9:1) catalysts are nearly the same, 105.7 and 104.8 kJ mol−1, respectively, and are in good agreement with the literature data for propane oxidation over Ru/Al2O3 [53]. However, Eapp is significantly lower (91.6 kJ mol−1) for the most active bimetallic Ru(N)–Re (3:1) catalyst suggesting that the addition of more rhenium (3 wt.%) lowers the propane activation barrier and facilitates the cleavage of the C–H bond in propane molecule, which is the rate-determining step. Similar decreasing of Eapp from 104.6 to 90.4 kJ mol−1 was reported recently by Liao et al. [54] for the oxidation of propane over V2O5 promoted Pt/SiO2 catalysts. Taylor et al. also found that addition of tungsten to Pd/TiO2 catalyst significantly improved catalyst performance for the total oxidation of propane [55]. For the oxidation of propane on the Cl-containing catalysts activation energy is much higher (~ 119.3 kJ mol−1), in good agreement with their lower activity, and does not depend on the composition of the catalysts. Table 5 shows the specific reaction rate (µmol C3 gcat−1 s−1) values determined at 200 °C i.e. in the kinetic region (with propane conversion less than 15%). It can be seen that the specific reaction rate is the highest for the Cl-free bimetallic Ru(N)–Re (3:1) catalyst (0.47 µmol C3 gcat−1 s−1) and it is about 60% higher compared to Ru(N) sample (0.29 µmol C3 gcat−1 s−1). As expected, the large decrease in the reaction rate is observed for the Cl-containing catalysts. The Ru(Cl) sample exhibits about 40% lower specific reaction rate as compared to Ru(N) sample but the bimetallic Ru(Cl)–Re (3:1) catalyst exhibits about five times lower specific reaction rate (0.09 µmol C3 gcat−1 s−1) as compared to chlorine-free Ru(N)–Re (3:1) catalyst (0.47 µmol C3 gcat−1 s−1). To compare the intrinsic activity, kinetic-controlled turnover frequencies (TOFs) at 200 °C were calculated based on the Ru dispersions (H/Ru, Table 2). The results shown in Table 5 indicate that the monometallic Ru(N) catalyst (with dispersion H/Ru = 0.33) gives the highest TOF value of 1.73 × 10–3 s–1, and the bimetallic Ru(N)–Re catalysts (with dispersion H/Ru = 0.47 or 0.63, respectively) possess slightly smaller TOF values of 1.64 × 10–3 s–1 or 1.53 × 10–3 s–1, whereas the Ru(Cl)–Re (3:1) catalyst (with dispersion H/Ru = 0.40) give the lowest TOF of 0.45 × 10–3 s–1.

Arrhenius-type plots for propane oxidation over the monometallic Cl-free and Cl-contaminated Ru and bimetallic Ru–Re/γ-Al2O3 catalysts
To try to explain activity results, the used monometallic Ru(N) and Ru(Cl) catalysts as well as bimetallic Ru(N)–Re and Ru(Cl)–Re catalysts at the same composition (3:1) were characterized by XRD technique. The post-reaction characterization indicated that the studied catalysts went through different structural changes during the propane oxidation in O2-rich reaction conditions. As shown in Fig. 13, the used Ru(N) catalyst exhibited the XRD pattern (trace a) with the characteristic diffraction peaks of RuO2 oxide (2Θ = 28.08°, 35.18°, and 54.38°), while no evidence of ruthenium oxide phase was found for the monometallic Ru(Cl) sample (trace c). Nevertheless, oxidized ruthenium species in the Ru(Cl) catalyst (see O2 adsorption data at 250 °C, Table 4) may exist as an amorphous or highly dispersed phase. The XRD pattern of the used Cl-free bimetallic Ru(N)–Re (3:1) catalyst contains also well visible reflections of RuO2 phase (trace b), while in the pattern of the Ru(Cl)–Re (3:1) only very weak reflection at 2Θ of 28.08° is seen (trace d). Therefore, although the both freshly reduced bimetallic Ru–Re (3:1) catalysts contained highly dispersed metallic species (X-ray amorphous, Fig. 2, trace c, Fig. 3, trace b), only in the used Cl-free bimetallic sample oxidized RuOx species were aggregated under the reaction mixture similarly as in the used Ru(N) catalyst. The Re phase was not detected by XRD since oxidized ReOx species exist on the surface of the used catalysts as a highly dispersed monolayer phase [22]. The mean RuO2 crystallite size, calculated from the line broadening of the most intense (110) reflection of RuO2 phase at 2Θ = 28.08° amounts to 10 and 15 nm, for the used Ru(N)–Re (3:1) and Ru(N) catalysts, respectively and it is only slightly higher as compared to the fresh catalysts oxidized at 250 °C for 1 h (Fig. 8). The XRD data evidently show that Cl-containing catalysts (mono- and bi-metallic) under the propane oxidation in O2-rich conditions (up to 280 °C) contained more dispersed ruthenium oxide species than that in the both Cl-free samples. Despite the higher dispersion of the active RuOx phase in the Cl-containing samples, they exhibited lower activity than the Cl-free samples (Fig. 11, Table 5). Therefore, the large differences in the specific activity of the studied samples could not be assigned only to the differences in the number of exposed active sites but mainly to the presence of residual chlorine species especially well visible for the least active bimetallic Ru(Cl)–Re (3:1) catalyst. Probably, chlorine species prefers to occupy the catalyst sites that are directly important for the propane oxidation reaction. Since in the Ru catalysed Deacon process (2 HCl + ½ O2 ↔ H2O + Cl2), a bridge-bonded oxygen in the RuO2 structure, which is the catalytically active surface center, is replaced by Cl ion, forming a stable inactive Cl(br)–Ru site [56] we propose that in our Cl-containing catalysts such structure is also possible.

XRD patterns recorded for the catalysts after three cycles of propane oxidation: 5% Ru(N)/γ-Al2O3 (a), 5% Ru(N)-3%Re/γ-Al2O3 (b), 5% Ru(Cl)/γ-Al2O3 (c), and 5% Ru(Cl)-3%Re/γ-Al2O3 (d)
Concluding, we suppose that the use of chlorinated precursor salts (RuCl3) during the preparation of the bimetallic Ru–Re and monometallic Ru catalysts decrease their activities due to the chemical interaction between residual chlorine and the catalyst surface or to the formation of ruthenium–chlorine species, which are more stable and inactive in propane oxidation reaction. Literature data show that oxidation of saturated hydrocarbons is described by a rate-limiting step which that involves a surface reaction of an alkane–oxygen complex on the catalyst surface. As shown in Table 3, the number of chlorine species on both studied catalysts after catalytic tests was similar (~ 1.8 wt.%), but the overall activity of the bimetallic Ru(Cl)–Re sample was much lower than the monometallic Ru(Cl) catalyst (Fig. 11, Table 5). Probably, during the propane oxidation reaction in an oxygen-rich atmosphere, migration of chlorine species from the ruthenium phase to the interface to form halogenated species with the support, thereby refreshing the metal surface, is easier in the case of Ru(Cl), while in the bimetallic Ru(Cl)–Re sample some of the alumina centers that can adsorb Cl− ions are already occupied by the oxidized rhenium species. Therefore, a higher concentration of ruthenium–chlorine species is expected in the bimetallic Ru(Cl)–Re catalyst, which results in lower activity than in the monometallic Ru(Cl) catalyst. For this reason, in the bimetallic Ru(Cl)–Re catalyst interaction between dispersed ruthenium oxide species and oxidized rhenium species could be weaker, or even absent, as compared to the chlorine-free Ru(N)–Re catalysts, leading also to further reduction activity of the Ru phase under the oxygen-rich and Cl-containing conditions. It should be noted that this is the first reported study which shows that the poisoning effect of chlorine (at the same Cl concentration and Ru content, Table 3) is much greater for the bimetallic Ru(Cl)–Re/γ-Al2O3 catalyst as compared to the monometallic Ru(Cl)/γ-Al2O3 catalyst during the VOCs removal in oxidative conditions. As RuCl3 is often used in the synthesis of the various Ru–Re catalysts (it is available in the market and cheaper than other Ru precursors), the chlorine poisoning effect should be considered in other catalytically important reactions.
5 Conclusions
The following conclusions can be drawn based on the results reported in this study:
-
An increase of the heating temperature in hydrogen from 500 to 800 °C of the bimetallic Ru–Re/γ-Al2O3 catalysts leads to some lowering of the metal dispersion. Depending on the amount of Re added to the Ru/γ-Al2O3 catalyst, three types of nanoparticles were identified, monometallic Ru NPs and Re NPs and as a major fraction Ru–Re bimetallic NPs. The size of Ru–Re NPs is below 2 nm, independent of the Ru/Re atomic ratio and the chemical nature of the Ru precursors evidencing very high resistance of Ru–Re NPs against sintering. Since Ru–Re catalysts are active for a variety of industrially important reactions, the unique resistance of Ru–Re NPs to sintering may recommend them for the application and focus the interest of them, stimulating more research of catalytic systems containing Ru and Re.
-
In the oxygen atmosphere, Ru–Re bimetallic NPs were unstable and at a moderate temperature of 250 °C segregation of metals is visible. This process leads to RuO2 agglomeration, which was lower in the Ru–Re catalysts containing a higher amount of Re and in the presence of residual Cl species, and to the transformation of the Re phase into a highly dispersed ReOx species.
-
The Ru precursor significantly affected the catalytic activity of the bimetallic Ru–Re catalysts, with Ru(NO)(NO3)3 resulting in better performance in terms of propane oxidation rates with comparison to Ru(Cl)3. Residual chlorine ions prevent the effective interaction between ruthenium and rhenium species, which is clearly seen in the bimetallic Ru(N)–Re catalysts based on the chlorine-free precursors. Thus, the existence of a synergetic effect between Ru and Re species was observed only on the Cl-free bimetallic catalysts.
-
Finally, the reported data reveal the need to rely on the simultaneous use of complementary macroscopic (XRD) and microscopic methods (TEM, HAADF-STEM) to accurately reveal the complex structure of the bimetallic Ru–Re catalysts, consisting of the mixture of different type of particles whose size range and compositions change as a function of Ru/Re atomic ratio and the atmosphere (reductive or oxidative).
References
Argyle MD, Bartholomew CH (2015) Catalysts 5:145. https://doi.org/10.3390/catal5010145
Liu L, Corma A (2018) Chem Rev 118:4981. https://doi.org/10.1021/acs.chemrev.7b00776
DeLaRiva AT, Hansen TW, Challa SR, Datye AK (2013) J Catal 308:291. https://doi.org/10.1016/j.jcat.2013.08.018
Hansen TW, DeLaRiva AT, Challa SR, Datye AC (2013) Acc Chem Res 46:1720. https://doi.org/10.1021/ar3002427
Dietze EM, Abild-Pedersen F, Plessow FN (2018) J Phys Chem C 122:26563. https://doi.org/10.1021/acs.jpcc.8b09303
Cao A, Lu R, Veser G (2010) Phys Chem Chem Phys 12:13499. https://doi.org/10.1039/C0CP00729C
Alayoglu S, Eichhorn B (2008) J Am Chem Soc 130:17479. https://doi.org/10.1021/ja8061425
Chinchilla LE, Olmos C, Kurttepeli M, Bals S, Van Tendeloo G, Villa A, Prati L, Blanco G, Calvino JJ, Chen X, Hungría AB (2016) Part Part Syst Charact 33:419. https://doi.org/10.1002/ppsc.201600057
Cao A, Veser G (2010) Nat Mater 9:75. https://doi.org/10.1038/nmat2584
Luo W, Sankar M, Beale AM, He Q, Kiely ChJ, Bruijnincx PCA, Weckhuysen BM (2015) Nat Commun 6:6540. https://doi.org/10.1038/ncomms7540
Beamson G, Papworth AJ, Philipps Ch, Smith AM, Whyman R (2011) J Catal 278:228. https://doi.org/10.1016/j.jcat.2010.12.009
Ma L, He D (2010) Catal Today 149:148. https://doi.org/10.1016/j.cattod.2009.03.015
Li KT, Yen RH (2018) Nanomaterials 8:153. https://doi.org/10.3390/nano8030153
Qu EH, Luo JJ, Di X, Li C, Liang CH (2020) J Nanosci Nanotech 20:1144. https://doi.org/10.1166/jnn.2020.16965
Zhou L, Lin W, Liu K, Wang Z, Liu Q, Cheng H, Zhang Ch, Arai M, Zhao F (2020) Catal Sci Technol 10:222. https://doi.org/10.1039/C9CY01909J
Raróg-Pilecka W, Miśkiewicz E, Szmigiel D, Kowalczyk Z (2005) J Catal 231:11. https://doi.org/10.1016/j.jcat.2004.12.005
Hayashi F, Iwamoto M (2011) Micropor Mesopor Mater 146:184. https://doi.org/10.1016/j.micromeso.2011.03.035
Ayvalı T, Fazzini PF, Lecante P, Mayoral A, Philippot K, Chaudret B (2017) Dalton Trans 46:15070. https://doi.org/10.1039/C7DT02287E
Ma L, Sun K, Luo M, Yan L, Jiang Z, Lu AH, Ding YJ (2018) J Phys Chem C 122:23011. https://doi.org/10.1021/acs.jpcc.8b06748
Mom RV, Ivashenko O, Frenken JWM, Groot IMN, Siastad AO (2018) J Phys Chem C 122:8967. https://doi.org/10.1021/acs.jpcc.8b00993
Okal J (2009) Mater Res Bull 44:318. https://doi.org/10.1016/j.materresbull.2008.05.018
Okal J, Kępiński L, Krajczyk L, Drozd M (1999) J Catal 188:140. https://doi.org/10.1006/jcat.1999.2634
Okal J, Tylus W, Kępiński L (2004) J Catal 225:498. https://doi.org/10.1016/j.jcat.2004.05.004
Baranowska K, Okal J, Miniajluk N (2014) Catal Lett 144:447. https://doi.org/10.1007/s10562-013-1169-1
Massalski TB (1990) Binary alloy phase diagrams. ASM International, Materials Park
Baranowska K, Okal J (2015) Appl Catal A 499:158. https://doi.org/10.1016/j.apcata.2015.04.023
Baranowska K, Okal J (2016) Catal Lett 146:72. https://doi.org/10.1007/s10562-015-1619-z
Okal J, Zawadzki M, Baranowska K (2016) Appl Catal B 194:22. https://doi.org/10.1016/j.apcatb.2016.04.038
Okal J, Zawadzki M, Kępiński L, Krajczyk L, Tylus W (2007) Appl Catal A 319:202. https://doi.org/10.1016/j.apcata.2006.12.005
Vuurman MA, Stufkens DJ, Oskam A, Wachs IE (1992) J Mol Catal 76:263. https://doi.org/10.1016/0304-5102(92)80164-C
Yusenko KV, Korolkov IV, Martynova SA, Gromilov SA (2009) Z Kristallogr Suppl 30:269. https://doi.org/10.1524/zksu.2009.0039
Mondal Ch, Barman ChK, Sarkar S, Barman SR, Alam A, Pathaki B (2020) Phys Rev B 101:155108. https://doi.org/10.1103/PhysRevB.101.155108
Chadzynski GW, Kubicka H (1990) Thermochim. Acta 158:353. https://doi.org/10.1016/0040-6031(90)80083-B
Almohalla M, Gallegos-Suarez E, Arcoya A, Alvarez-Rodriguez J, Rodriguez-Ramos I, Guerrero-Ruiz A (2017) Appl Catal A 535:61. https://doi.org/10.1016/j.apcata.2017.02.007
Mieth JA, Schwarz JA (1989) J Catal 118:218. https://doi.org/10.1016/0021-9517(89)90312-6
Narita T, Miura H, Ohira M, Hondou H, Sugiyama K, Matsuda T, Gonzalez RD (1987) Appl Catal 32:185. https://doi.org/10.1016/S0166-9834(00)80624-7
Bare SR, Kelly SD, Vila FD, Boldingh E, Karapetrova E, Kas J, Mickelson GE, Modica FS, Yang N, Rehr JJ (2011) J Phys Chem C 115:5740. https://doi.org/10.1021/jp1105218
Amada Y, Shinimi Y, Koso S, Kubota T, Nakanawa Y, Tomishige K (2011) Appl Catal B 105:117. https://doi.org/10.1016/j.apcatb.2011.04.001
Koso S, Wanatabe H, Okamura K, Nakagawa Y, Tomishige K (2012) Appl Catal B 111–112:27. https://doi.org/10.1016/j.apcatb.2011.09.015
Ebashi T, Ishida Y, Nakagawa Y, Ito S, Kubota T, Tomishige K (2010) J Phys Chem C 114:6518. https://doi.org/10.1021/jp911908c
Mazzieri V, Figoli N, Pascual FC, L’argentiere P (2005) Catal Lett 102:79. https://doi.org/10.1007/s10562-005-5206-6
Prestvik R, Moljord K, Grande K, Holmen A (1998) J Catal 174:119. https://doi.org/10.1006/jcat.1998.1956
Gracia FJ, Miller JT, Kropf AJ, Wolf EE (2002) J Catal 209:341. https://doi.org/10.1006/jcat.2002.3601
Oran U, Uner D (2004) Appl Catal B 54:183. https://doi.org/10.1016/j.apcatb.2004.06.011
Roth D, Gelin P, Primet M, Tena E (2000) Appl Catal A 203:37. https://doi.org/10.1016/S0926-860X(00)00465-8
Okal J, Zawadzki M (2009) Appl Catal B 89:22. https://doi.org/10.1016/j.apcatb.2008.11.024
Okal J, Zawadzki M, Kraszkiewicz P, Adamska K (2018) Appl Catal A 549:161. https://doi.org/10.1016/j.apcata.2017.09.036
Debecker DP, Farin B, Gaigneaux EM, Sanchez C, Sassoye C (2014) Appl Catal A 481:11. https://doi.org/10.1016/j.apcata.2014.04.043
Ledwa KA, Kępiński L, Ptak M, Szukiewicz R (2020) Appl Catal B 274:119090. https://doi.org/10.1016/j.apcatb.2020.119090
Ledwa KA, Pawlyta M, Kępiński L (2018) Appl Catal B 230:135. https://doi.org/10.1016/j.apcatb.2018.02.037
Kurnatowska M, Miśta W, Mazur P, Kępiński L (2014) Appl Catal B 148:123. https://doi.org/10.1016/j.apcatb.2013.10.047
Novio F, Monahan D, Coppel Y, Antorrena G, Lecante P, Philippot K, Chaudret B (2014) Chem Eur J 20:1287. https://doi.org/10.1002/chem.201303935
Hu Z, Wang Z, Guo Y, Wang L, Guo Y, Zhang J, Zhan W (2018) Environ Sci Technol 52:9531. https://doi.org/10.1021/acs.est.8b03448
Liao WM, Liu YR, Zhao PP, Cen BH, Tang C, Jia AP, Lu JQ, Luo MF (2020) Appl Catal A 590:117337. https://doi.org/10.1016/j.apcata.2019.117337
Taylor MN, Zhou W, Garcia T, Solsona B, Carley AF, Kiely ChJ, Taylor SH (2012) J Catal 285:103. https://doi.org/10.1016/j.jcat.2011.09.019
Seitsonen AP, Over H (2010) J Phys Chem C 114:22624. https://doi.org/10.1021/jp108603a
Acknowledgements
The authors wish to thank: Mrs. Ewa Bukowska for the XRD measurements, and Dr. Damian Szymański for the EDS experiments.
Funding
The STEM-HAADF study received funding from the European Union Seventh Framework Programme under Grant Agreement 312483—ESTEEM2 (Integrated Infrastructure Initiative—13). This work was carried out within the Reintegration programme of the Foundation for Polish Science co-financed by the European Union under the European Regional Development Fund, project no. POIR.04.04.00-00-5F33/18-00.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Okal, J., Adamska, K. Thermal Stability of Ru–Re NPs in H2 and O2 Atmosphere and Their Activity in VOCs Oxidation: Effect of Ru Precursor. Catal Lett 152, 55–74 (2022). https://doi.org/10.1007/s10562-021-03607-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-021-03607-7