
Abstract
The coronavirus disease 19 (COVID-19) pandemic poses a serious global threat to human health and the economy. Based on accumulating evidence, its continuous progression involves not only pulmonary injury but also damage to the cardiovascular system due to intertwined pathophysiological risks. As a point of convergence in the pathophysiologic process between COVID-19 and heart failure (HF), cytokine storm induces the progression of COVID-19 in patients presenting pre-existing or new onset myocardial damage and even HF. Cytokine storm, as a trigger of the progression of HF in patients with COVID-19, has become a novel focus to explore therapies for target populations. In this review, we briefly introduce the basis of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and illuminate the mechanism and links among COVID-19, cytokine storm, and HF. Furthermore, we discuss drugs and therapeutic targets for patients with COVID-19 and HF.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Since the first outbreak in December 2019, coronavirus disease 19 (COVID-19), an emerging infection, has rapidly spread and placed an excessively heavy burden on the medical system worldwide, leading to the loss of life. Cytokine storm represents a vital factor causing the progression of both COVID-19 and heart failure (HF). In patients with COVID-19, serum cytokine levels are obviously elevated, which are beneficial to block viral infection at the early stage [1]. However, persistent and excess cytokine infiltration leads to severe tissue damage and even multi-organ failure, including respiratory and circulatory system failure [2]. In HF, cytokines function as important pathogenic factors and vital biomarkers of the progression of various cardiac diseases that eventually lead to HF. In turn, HF also triggers the release of large amounts of cytokines by inducing systemic inflammation [3]. Consequently, knowledge of the basic pathophysiology and immunological process underlying the clinical manifestations of COVID-19, the mechanism underlying cytokine storm induced by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), and their contributions to the progression of HF by inducing subsequent immune disorders are urgently needed. In this review, we briefly introduce the basic characteristics of SARS-CoV-2 and illuminate the mechanism underlying the associations among SARS-CoV-2, cytokine storm, and HF, as well as the role of hyperinflammation induced by excess cytokine release in COVID-19 and HF pathophysiologic processes. Furthermore, we discuss drugs and therapeutic targets for patients with COVID-19 who have either a pre-existing cardiac disease or new onset myocardial damage, including HF. We hope that this review will become a valuable reference for the prevention and treatment of HF in patients with COVID-19.
The Cytokine Storm in Patients with COVID-19
Virology of COVID-19
SARS-CoV-2, the causative agent of COVID-19, is a spherical, positive single-stranded RNA coronavirus. According to genomic sequencing, SARS-CoV-2 shares 79.5% homology with the sequence of SARS-CoV that caused the outbreak of SARS in 2003 [4]. Compared with the sequences of coronaviruses found in wildlife, SARS-CoV-2 shares 96.2% homology with BatCoV-RaTG13 in bats and approximately 90% homology with coronavirus in pangolins [5, 6]. Consequently, a likely route of viral transmission involves the enlargement of the reproductive scale by SARS-CoV-2 derived from bats infecting one or more intermediate hosts, such as pangolins [7]. In an analysis of the SARS-CoV-2 genome in 103 Chinese patients, SARS-CoV-2 was shown to evolve into type L (~70%) and type S (~30%), and the former type is more invasive and infectious than the ancestral type S [8].
SARS-CoV-2 consists of structural proteins, such as spike (S), membrane (M), envelope (E), and nucleocapsid (N) proteins, as well as hemagglutinin esterase (HE) proteins present in other viruses [9]. Among these proteins, the S protein is a highly glycosylated protein that forms the trimeric spine on the viral surface to recognize receptors in host cells and mediate membrane fusion. It is a critical factor contributing to host cell infection and plays an important role in regulating the process of virus entry into cells [10] (Fig. 1). The S protein forms the S1 and S2 structural domains, both of which maintain the noncovalent binding of the conformation before fusion. After entrance of the virus into host cells, the S protein is cleaved in intracellular vesicles [11, 12]. The S1 domain receptor-binding domain (RBD) interacts with the angiotensin (Ang)-converting enzyme 2 (ACE2) receptor located in homologous host cells, which is beneficial for the adhesion of the virus to the surface of target cells. Under the synergistic effect of transmembrane protease serine 2 (TMPRSS2), ACE2 cleavage, and S protein priming occur, followed by membrane fusion between the virus and host cells mediated by the S2 subunit, enabling SARS-CoV-2 to enter the host cell compartment [13,14,15]. In addition, compared with SARS-CoV, SARS-CoV-2 not only displays better binding affinity and binding site stability, but also contains special furin-like cleavage sites on the border between the S1 and S2 subunits [16, 17]. In pulmonary cells that have lost the ability to express cathepsin L at high levels, the S protein is cleaved at these furin-like S1/S2 cleavage sites before internalization, enhancing the effect of TMPRSS2 and internalization of the virus into cells [18], which may partially explain why COVID-19 has greater transmissibility and/or pathogenicity [19, 20].

Mechanism by which SARS-CoV-2 enters host cells. (1) SARS-CoV-2 adheres to the surface of the target cell after interacting with ACE2 via the S protein. (2) TMPRSS2 binds and then cleaves ACE2. During the process, the S protein is activated to promote membranous fusion between the virus and host cell. (3) The virus enters the host cell. Abbreviations: ACE2, angiotensin-converting enzyme 2; RNA, ribonucleic acid; SARS-CoV-2, severe acute respiratory syndrome coronavirus-2; TMPRSS2, transmembrane serine protease 2
SARS-CoV-2 endocytosis mediated by ACE2 is an important mechanism of viral invasion [13, 14]. The tissue location and expression levels of ACE2 receptors are associated with the symptoms and dysfunction caused by COVID-19. However, specific variants of genes (e.g., ACE2, TMPRSS2, and interleukin-6 (IL-6)) or the differential expression of related genes may explain the individual differences in the sensitivity and severity of COVID-19 by altering the kinetics of the viral interaction and endocytosis [21]. In addition to pulmonary epithelial cells, ACE2 receptors are also expressed at high levels on the surface of cardiomyocytes and vascular endothelial cells [22], which potentially explains the possible complications of SARS-CoV-2 observed in the cardiovascular system. Moreover, patients with hypertension or cardiovascular disease (CVD) exhibit higher ACE2 expression than healthy individuals, which is assumed to increase their susceptibility to SARS-CoV-2 infection and the likelihood of adverse outcomes in these high-risk populations [23]. In this case, single nucleotide polymorphisms (SNPs) in ACE2 and IL-6 genetic polymorphisms may be related to HF in patients with COVID-19 [21].
Mechanism of Cytokine Storm Induced by COVID-19
After SARS-CoV-2 infection, alveolar epithelial cells and alveolar macrophages release pathogen-associated molecular patterns (PAMPs) (e.g., viral RNA) and damage-associated molecular patterns (DAMPs) (e.g., ATP and DNA), both of which activates the secretion of chemokines (e.g., C-C motif chemokine ligand (CCL)-2, CCL-3, and CCL-5) and pro-inflammatory cytokines (e.g., IL-1β, IL-6, tumor necrosis factor (TNF), and interferon (IFN)). The recruitment and accumulation of monocytes and T lymphocytes induced by increasing chemokines and pro-inflammatory cytokines control the infection by executing limited inflammation in the infected focus [2, 24]. While inappropriate external stimuli may introduce limited inflammation into the cytokine storm caused by the release of excess cytokines, the latter is considered the main cause of acute respiratory distress syndrome (ARDS) and multiple organ failure [25]. Activated leukocytes (e.g., macrophages and T/B lymphocytes) can produce cytokines [26]. In terms of inflammatory cells, there are a greater number of macrophages in the bronchoalveolar lavage fluid and/or peripheral blood from patients with severe COVID-19 than from patients with mild COVID-19. The enriched macrophages further promote the secretion of cytokines and chemokines, causing local infiltration of excess cytokines [27]. A decrease in the number of peripheral T lymphocytes has been detected in patients with COVID-19, while convalescent serum samples are effective at treating COVID-19 [2]. T lymphocytes are depleted because they are recruited to the infected focus, indicating the severity of COVID-19, and the clinical effects of serum antibodies suggest that the decrease in the number of B cells also participates in the development of COVID-19 [2, 28]. The number of lymphocytes gradually decreases with the progression of the illness [29]. In addition, older patients with fewer lymphocytes are at a higher risk of an increased length of hospitalization and a worse prognosis [30]. Patients with specific cancers, such as hematopoietic or lymphoid malignancies, immunocompromised patients who are undergoing chemotherapy or radiotherapy; patients with immunodeficiency diseases such as AIDS; or patients with systemic autoimmune diseases receiving immunosuppressive treatment are at higher risk of developing serious complications after being diagnosed with COVID-19. Lymphopenia related to COVID-19 is deemed a criterion indicative of the severity of COVID-19, and its biomarkers are used to evaluate the condition of the SARS-CoV-2 infection [31]. However, the mechanism underlying the exhaustion of T lymphocytes remains unclear. A possible explanation is that infection with a low dose of the virus will activate T- and B-cell responses appropriately; however, high-dose viral exposure may delay viral clearance, because inefficient T- and B-cell immunity will induce the cytokine storm to cause severe COVID-19 further [31].
Regarding cytokines, reports have verified that SARS-CoV causes inflammation by promoting viral replication and the release of pyroptosis products after interfering with IFN responses at the early stage [2, 32]. On the one hand, the rapid replication of SARS-CoV induces inflammatory cell infiltration, including macrophages and monocytes, and then inflammatory cells further secrete cytokines and chemokines to recruit more inflammatory cells, representing a self-amplifying cycle that causes more severe disease manifestations through immune dysregulation. In contrast, lymphopenia or an ineffective lymphocyte response caused by infiltration into the lesion induces a persistently elevated viral load that then stimulates excess cytokine release and hyperinflammation. Although SARS-CoV-2 shares 79% sequence homology with SARS-CoV, whether and how the aforementioned mechanism of eliminating IFN responses occurs in individuals infected with SARS-CoV-2 have not been completely clarified. Importantly, SARS-CoV-2 is more efficient at suppressing the IFN-I response than SARS-CoV [33]. According to published reports, patients with COVID-19 who manifest an obvious inflammatory reaction persistently not only exhibit a decrease in interferon activity [34], but also are insufficient to exert the type I IFN responses to control the infection. In addition, they accompany with elevated pro-inflammatory cytokine levels [35]. Similarly, IL-6 levels in these patients gradually increase over time, and the levels in non-survivors are higher than in survivors [36]. Pyroptosis, a type of programmed cell death, associated with a high level of inflammation. IL-1β is a main cytokine released during pyroptosis, and its enhancement can be observed in patients with COVID-19 [29, 37]. Compared with non-intensive care unit (ICU) patients, the serum levels of cytokines, including TNF-α, are higher in ICU patients [29]. Thus, the rapid and vast replication of the virus inhibits the IFN response and induces the depletion of lymphocytes, further causing the infiltration of inflammatory cells and pro-inflammatory cytokines, which may be one cause of severe tissue injury in patients with COVID-19 (Fig. 2).

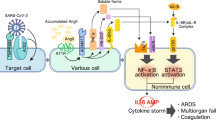
Links among COVID-19, cytokine storms, and HF. After SARS-CoV-2 infection, alveolar epithelial cells and alveolar macrophages release PAMPs and DAMPs, which activate the secretion of CCL-2, CCL-3, and CCL-5 and the pro-inflammatory cytokines IL-1β, IL-6, TNF-α, and IFN to activate and recruit pro-inflammatory cells, including T cells, B cells, and macrophages, to the location of the lesion. Upon exposure to a low dose of SARS-CoV-2, the infection is controlled by limited inflammation, while upon high-dose exposure, T-cell and B-cell exhaustions occur due to the limited inflammation induced by the elevated viral load. Increased IFN levels restrain virus replication, while the SARS-CoV-2 N protein antagonizes signaling through type I IFNs via limiting STAT1 and STAT2 phosphorylation. The persistence of stimulation by SARS-CoV-2 triggers the cytokine storm to secrete large amounts of cytokines that promote the progression of HF. (1) IL-6 induces diastolic dysfunction via positive inotropic effects mediated by the JAK-STAT3 pathway in cardiomyocytes. (2) IL-1β and TNF-α exert a negative inotropic effect by changing intracellular calcium homeostasis in cardiomyocytes to cause systolic dysfunction. (3) IL-1β and TNF-α upregulate AT1R on cardiac fibroblasts and then promote collagen deposition and fibrosis mediated by Ang II. (4) LOX-like 2 expression increases in cardiac fibroblasts via elevated TGF-β induced by TNF-α to promote collagen deposition and cardiac fibrosis. (4) Pro-inflammatory cytokines trigger intravascular oxidative stress in dysfunctional endothelial cells to stimulate the production of PAI-1 and then inhibit fibrinolysis, allowing microvascular thrombi to form. Abbreviations: AT1R, angiotensin type 1 receptors; CCL, chemokine (C-C motif) ligand; COVID-19, coronavirus disease 19; DAMPs, damage-associated molecular patterns; IFN, interferon; IL, interleukin; JAK-STAT3, Janus kinase-signal transducer and activator of transcription pathway; LOX, lysyl oxidase; PAMPs, pathogen-associated molecular patterns; PAI-1, plasminogen activator inhibitor type 1; SARS-CoV-2, severe acute respiratory syndrome coronavirus-2; TGF, transforming growth factor, TNF, tumor necrosis factor
The Role of Cytokine Storm in COVID-19
Limited inflammation eliminates the infection and then restores health after the inflammation is resolved in most individuals. However, patients who are elderly or suffer from co-morbidities generally exhibit a dysfunctional immune response that triggers a cytokine storm, allowing widespread inflammation to occur. Subsequently, localized injuries (e.g., diffuse alveolar damage) and systemic injuries (e.g., multi-organ failure) emerge, particularly in the cardiac, hepatic, and renal systems [2]. The multi-organ failure secondary to cytokine storm results in various clinical manifestations due to tissue damage, such as dry/productive cough associated with lung lesions, palpitations related to heart damage, increased aspartate aminotransferase (AST) levels as a marker of hepatic injury, and hematuria related to kidney damage [1, 38]. The most common cause of death in patients with severe COVID-19 may be ARDS with respiratory failure requiring mechanical ventilation and acute cardiac injury with HF [38]. As expected, myocardial damage and circulatory failure caused by increased levels of cytokines have been observed in patients with COVID-19 [39]. The probable explanation for a higher susceptibility to pathogens and a worse ability to control infection in older adults is that a decrease in the number of CD8+ T cells, which is the most prominent marker of immune aging. The severity of COVID-19 increases with age due to a change in the immune response, which is in keeping with the observation of increased odds of in-hospital death due to COVID-19. Furthermore, a high incidence of co-morbidities, such as cardiac dysfunction, has been observed in the elderly [36, 40]. Additionally, age-associated physiological changes (e.g., endothelial dysfunction) and diseases (e.g., obesity and diabetes) induced by inflammation are the main causes of the high incidence of HF in the elderly [41, 42]. In other words, the sensitivity to COVID-19 and HF in elderly patients with severe COVID-19 may be partially due to inflammation mediated by a decrease in the number of T cells. Furthermore, immunomodulatory therapies targeting cytokine storm reduce mortality and improve outcomes in elderly patients with COVID-19; thus, a reasonable hypothesis is that cytokine storm induced by COVID-19 may be one cause of immune aging in elderly patients. Therefore, the excess inflammation caused by cytokine storm is a major risk factor linking the progression and prognosis of COVID-19, particularly in older patients, with co-morbidities. Moreover, subsequent studies should focus on evaluating the curative effect of immunomodulatory therapies on these special patient populations [1]. Interestingly, the gender difference in the susceptibility to COVID-19 may be attributed to the anti-inflammatory and immunomodulatory actions of estradiol and progesterone (P4). High concentrations of 17β-estradiol (E2) block the migration of neutrophils and monocytes by inhibiting the generation of pro-inflammatory cytokines, as well as stimulating B cells to produce antibodies. P4 promotes the production of anti-inflammatory cytokines and inhibits the generation of pro-inflammatory cytokines [43]. Because E2 and P4 not only blunt innate immune inflammation but also promote B-cell responses, clinical trials investigating these hormones are in progress to evaluate the underlying benefits in male patients with COVID-19 (ClinicalTrials.gov identifiers NCT04359329 and NCT04365127).
HF Induced by Cytokine Storm in Patients with COVID-19
A meta-analysis including eight studies with 46,248 patients with COVID-19 identified hypertension (17 ± 7%, 95% CI 14–22%) and diabetes (8 ± 6%, 95% CI 6–11%) as the most prevalent co-morbidities, followed by cardiovascular diseases (5 ± 4%, 95% CI 4–7%) [1]. Fifty-three patients with COVID-19 and a history of heart diseases and 46 patients without a history of heart diseases were consecutively recruited, and patients with HF accounted for 21% of the total patients and 40% of patients with cardiac diseases [44]. A retrospective case series recruiting 799 moderately to severely ill or critically ill patients with confirmed COVID-19, among which 161 recovered and 113 died, showed that the HF incidence in the patients who died was 49%, a value that is obviously higher than in patients who recovered [45]. HF was deemed one of the most common complications occurring during the exacerbation of COVID-19, and these conditions were more likely to appear in patients with pre-existing cardiac diseases [45]. Thus, the presence of HF in combination with COVID-19, which may occur secondary to a pre-existing cardiac disease or new onset of myocardial damage, is related to an increased mortality risk in patients with COVID-19. Notably, inflammation, which is a factor with dual effects in COVID-19, also interacts with HF to form a chronic vicious cycle by mutually reinforcing the other condition. Therefore, the connections and mechanisms among COVID-19, inflammation and HF are discussed from a pathophysiological perspective in the next section.
The Progression of HF in Patients with COVID-19
HF manifests various clinical indications in the stages of its progression. The patients who remain in stage A and have risk factors for progression to HF but not structural heart disease can be identified by persistent monitoring of B-type natriuretic peptide (BNP) and/or troponin levels. Cardiac structural damage exists in stage B patients who have not yet presented clinical manifestations, while the manifestations of HF, including dyspnea and peripheral edema, will emerge in stage C, both of which represent a transition period from compensated to decompensated HF and eventually lead to dysfunction. Similarly, we are able to identify the HF stages and progression or prognosis by monitoring dynamic changes in the levels of related biomarkers including neurohormones (e.g., renin–Ang–aldosterone system (RAAS) system), myocyte strain-specific molecules (e.g., BNP and pro-BNP), cardiac injury-induced peptides (e.g., troponin), pro-inflammatory mediators, and oxidative stress components. The intricate pathophysiological mechanism involved must be understood to improve the efficiency of therapy for HF induced by SARS-CoV-2. According to current research data, COVID-19 promotes HF progression and maintains patients in a dangerous condition through complex interactions among multiple risk factors.
-
(1)
Direct myocardial injury. SARS-CoV-2 triggers carditis by directly attacking the heart.
-
(2)
Inflammation and thrombosis. The release of cytokines induces endothelial dysfunction and circulatory hypercoagulability to promote plaque instability and thrombosis, respectively, subsequently leading to acute coronary syndrome (ACS).
-
(3)
Hypoxemia. Lung injury induced by viral attack and pulmonary embolism (PTE) induced by thrombosis enhance hypoxia, which increases the pulmonary vascular resistance by inducing pulmonary vasoconstriction.
-
(4)
Downregulation of ACE2. The loss of ACE2 promoted by COVID-19 triggers both vasoconstriction by increasing the expression of Ang and a loss of the protective effects on the heart by decreasing the expression of Ang 1–7.
Together, these mechanisms will promote the progression of HF in patients with COVID-19 [46] (Fig. 3). Meanwhile, researchers have not clearly determined whether cardiac injury is caused by direct infection of cardiomyocytes or is mainly secondary to lung injury and inflammation [47]. The release of cytokines will participate in the aforementioned structural changes associated with HF, such as cardiac remodeling and endothelial dysfunction regulated by TNF-α, the accumulation of interstitial collagen fibers induced by TNF-α and IL-1, and cardiomyocyte hypertrophy mediated by IL-6. Simultaneously, pro-inflammatory cytokines can promote HF damage induced by oxidative stress [48, 49]. Due to the close correlation between cytokine storm and uncertainty about its regulatory mechanism in COVID-19 and HF, we will emphatically discuss the role of cytokine storm in patients with COVID-19 who develop HF.

Potential mechanisms of HF deterioration induced by COVID-19. SARS-CoV-2 entry into host cells via ACE2 induces myocardial injury by not only attacking the heart directly to cause myocarditis, but also triggering systemic inflammation and excess immune cell activation to cause a cytokine storm. With the increased release of cytokines, including IL-6 and TNF-α, endothelial cell dysfunction induced by the cytokine storm plays an important role in both plaque instability and circulatory hypercoagulability, promoting thrombosis and ACS. The occurrence of PTE caused by thrombosis worsens the hypoxia induced by lung injury, while pulmonary vasoconstriction under hypoxia increases pulmonary vascular resistance. The loss of ACE2 promoted by COVID-19 triggers both vasoconstriction by increasing the expression of Ang II and a loss of the protective effects on the heart by decreasing the expression of Ang 1–7. In patients with a pre-existing cardiac disease, these mechanisms will work together to promote the progression of HF
Mechanism of HF Induced by Cytokine Storm
Regarding the mechanism, IL-6 induces diastolic dysfunction by exerting positive inotropic effects mediated by the Janus kinase-signal transducer and activator of transcription 3 (JAK-STAT3) pathway [50,51,52] and increases cardiomyocyte stiffness by reducing the phosphorylation of titin [53]. IL-1β and TNF-α exert negative inotropic effects by altering intracellular Ca2+ homeostasis in cardiomyocytes to cause abnormalities in contractile function [54, 55] and may induce cardiomyocyte pyroptosis and apoptosis, respectively [56, 57]. Notably, the levels of these indicators are elevated in patients with COVID-19 [58]. As expected, myocarditis has been reported in some patients infected with SARS-CoV-2 [59, 60]. ACE2 expressed in the heart exerts anti-inflammatory, anti-fibrotic, anti-oxidant, and vasodilatory effects by withstanding the effect of Ang type 1 receptors (AT1R), while the internalization of SARS-CoV-2 decreases cardiac ACE2 expression [61,62,63]. IL-1β and TNF-α upregulate AT1R expression in cardiac fibroblasts and favor collagen deposition and fibrosis mediated by Ang II [64], changes that are ameliorated by IL-1β and TNF-α neutralization [61, 65]. Additionally, the rapid decrease in ACE2 expression will limit the generation of Ang II and Ang (Ang 1–7), weakening the cardiac protection provided by the ACE2/Ang 1–7/Mas receptor axis [66]. Meanwhile, the increased level of Ang II promotes phosphorylation of ADAM-17. ADAM-17 not only increases the release of TNF-α to aggravate systemic inflammation, but also accelerates the detachment and loss of ACE2 from the cytomembrane and then impairs the conversion from Ang II to Ang 1–7. Ang II also upregulates the expression of potent neutrophil chemoattractants, including IL-6, or directly stimulates the activation and proliferation of lymphocytes, resulting in persistent inflammation and cytokine storm [67]. Subsequently, these processes constantly amplify the adverse effects of the positive feedback loop and accelerate the progression of HF [68,69,70]. Failing myocardia express high levels of TNF-α, which has been shown to participate in the development of dilated cardiomyopathy [3]. TNF-α promotes cardiac fibrosis in cardiac fibroblasts. One of the mechanisms is that TNF-α enhances the expression of HF biomarker lysyl oxidase (LOX)-like 2 via elevated transforming growth factor (TGF)-β [71, 72]. Adhesion molecule expression induced by pro-inflammatory cytokines (e.g., IL-1β) in cardiac endothelial cells regulates immune cell adhesion and migration and then accentuates inflammation, while dysfunctional endothelial cells trigger intravascular oxidative stress, which has been observed in patients with SARS-CoV-2 infection [3, 73]. Under oxidative stress, cardiac microvascular endothelial cells cause inhibition of fibrinolysis by stimulating the production of plasminogen activator inhibitor-1 (PAI-1). Subsequently, the persistent existence of microvascular thrombi due to inhibition of fibrinolysis will accelerate the progression of HF [74]. In addition, COVID-19-induced endotheliitis may provide an explanation for the microcirculatory dysfunction in different vascular beds [75]. Briefly, the persistent existence of SARS-CoV-2 stimuli can mediate the cytokine storm and hyperinflammation. Under this state, the progression of HF can be caused through diastolic dysfunction mediated by the Janus kinase-signal transducer and activator of transcription (JAK-STAT) pathway, systolic dysfunction mediated by calcium dyshomeostasis, cardiac fibrosis mediated by elevated TGF-β and AT1R levels, and microvascular thrombi mediated by the inhibition of fibrinolysis (Fig. 2).
The Links among COVID-19, Inflammation, and HF
As mentioned above, the numbers of T/B lymphocytes are decreased in patients with COVID-19, weakening the ability of these cells to confine and clear SARS-CoV-2. Subsequently, persistent viruses stimulate macrophages to activate and release large amounts of cytokines, leading to hyperinflammation and cytokine storm. Systemic inflammation is the basis of acute or chronic HF and elicits the release of large amounts of cytokines and increased cytokine receptor expression, which potentially represent independent predictors of mortality in patients with advanced HF [76, 77]. Meanwhile, the hemodynamic stress of HF and co-morbidities induce a systemic pro-inflammatory state, and inflammation induced by myocardial infection promote the release of pro-inflammatory cytokines as well [41]. A retrospective single-center case series reported significant correlations between plasma troponin T (TnT) levels and the levels of high-sensitivity C-reactive protein (CRP) and N-terminal fragment of pro-BNP (NT-pro-BNP) in patients with COVID-19 [78]. In addition, the levels of TnT and NT-pro-BNP, which are markers of HF and myocardial damage, exhibited a dynamic escalation in patients who died, although a similar clear change was not observed in survivors [78]. Macrophages may also participate in the progression of HF in patients with COVID-19 by releasing pro-inflammatory cytokines. M1 macrophages secrete pro-inflammatory cytokines, such as IL-6, IL-1β, and TNF-α [79]. Interestingly, cardiomyocyte surfaces present some of TNF-α receptors [80]. The overexpression of TNF-α in mice results in ventricular dilatation, interstitial infiltrates, interstitial fibrosis, and ultimately the occurrence of HF [81]. IL-6 and IL-1β levels are simultaneously increased in patients with HF, which suggests that TNF-α is related to the severity of HF [76]. These indicators can be observed in a later stage of COVID-19, which may be related with a high incidence of cardiovascular events in COVID-19 patients, and the causes of death in these patients are mostly attributed to cardiac arrhythmias and HF [82]. Elevated levels of these indicators of HF, myocardial damage, and inflammation prompted us to speculate that hyperinflammation and cytokine storm elicited by SARS-CoV-2 infection may be among the mechanisms underlying the progression of patients with COVID-19 to HF.
Implications for Clinical Applications
The associations among COVID-19, cytokine storm, and HF have been described in the aspect of mechanisms, which can provide underlying therapeutic targets for patients with COVID-19 and pre-existing or new onset HF. In this section, we discuss potential targets derived from the inflammatory mechanism and their roles in COVID-19 and myocardial damage, including HF, allowing recommendations to be proposed for target populations.
Evaluation, Management, and Prognosis of at-Risk Patients
Early identification and timely treatment are beneficial for improving the prognosis of patients with COVID-19 and increasing the utility ratio of strained medical resources. The laboratory index of patients with COVID-19 who develop HF is consistent with acute cardiac injury in retrospective studies [36]; additionally, fatal outcomes in patients with COVID-19, particularly fulminant myocarditis, are consistent with increased levels of inflammatory indicators [39, 83]. Thus, we propose that specific elevated cytokines and appreciably altered inflammatory biomarkers are early warning signals in at-risk patients that may forecast pathogenic conditions and a poor prognosis. The level of CRP in patients with COVID-19 presenting cardiac damage is higher than that in patients without cardiac damage [78]; furthermore, an elevated CRP level is associated with an adverse outcome in patients with HF [84]. The levels of both high-sensitivity troponin I and NT-pro-BNP are increased in patients with COVID-19 presenting with cardiac damage, and troponin has an independent prognostic value for both COVID-19 and HF; however, as a non-independent prognostic marker, NT-pro-BNP is not specific for the diagnosis of concomitant HF in patients with COVID-19 [78, 85]. Additionally, cardiac troponin I levels are substantially increased in patients with severe COVID-19 compared with patients with a mild illness, potentially representing a biomarker to predict the deterioration of the clinical condition of patients with COVID-19 [86]. IL-6, a common characteristic cytokine that participates in cytokine storm, is not only a clinical predictor of mortality in patients with COVID-19 [39], but also an underlying target for decreasing atherosclerosis-related cardiovascular morbidity and mortality [87, 88]. Serum IL-1β and TNF-α levels are obviously elevated in patients with severe COVID-19 [29, 58]. Regarding TNF-α, the proposition that anti-TNF therapy should be initiated at the early and middle stages in patients with COVID-19 to prevent progression has been refuted [89, 90]. Importantly, TNF-α and soluble TNF receptor (TNFR) levels in the circulation are independent predictors of mortality in patients with HF [77]. In addition, IL-1β and its receptor are therapeutic targets in MI and HF [91]; additionally, IL-1β inhibitors improve hyperinflammation and the clinical outcomes of patients with COVID-19 [92, 93]. Because of these increased indexes in patients with COVID-19 presenting with HF, we speculate reasonably that the degree of change and levels of cytokines and related indicators may be related with evaluations of the severity and therapeutic effect, stratified management, and determination of the prognosis of such patients. In addition, the clinical management of chronic conditions should include specific considerations of the underlying cause of a higher risk of death in patients with COVID-19 who have chronic diseases, such as cardiovascular disorders. A novel management protocol that considers cardiovascular diseases and diabetes has been developed, as described below.
-
(1)
Telephonic triage by screening the risk factors, symptoms, signs of COVID-19, and body mass index (BMI) should be conducted, and then some recommendations, including self-isolation and close monitoring, should be provided to patients with a BMI of 30.
-
(2)
A telemedicine consultation should be provided to all patients to assess the underlying factors influencing diabetes and cardiovascular diseases.
-
(3)
Patients who require an immediate medical assessment must visit an emergency department, while others should receive a teleconsultation.
-
(4)
Patients who present at the emergency department must undergo diagnostic testing for COVID-19.
-
(5)
Related measures must be implemented to protect healthcare workers when patients who require surgery are COVID-19 positive.
-
(6)
Patients should ensure that they maintain a proper quarantine after discharge and follow public health guidelines [94].
Corticosteroids
The use of corticosteroids to treat COVID-19 remains highly controversial. On the one hand, corticosteroids lead to a delay in viral clearance by impairing innate immunity [95, 96]. On the other hand, corticosteroids reverse the tissue damage caused by cytokine storm due to their powerful anti-inflammatory effects [97,98,99]. According to many clinical studies, corticosteroid treatment is not beneficial to improving tissue injury caused by SARS-CoV-2, which can induce the increasing severity of viral infection, increased mortality rate, and prolonged hospital stays. Given the unsatisfactory clinical outcomes and side effects, corticosteroids are not recommended by the current WHO guidelines (released Jan 28, 2020) for the clinical management of severe COVID-19 [100]. However, the powerful anti-inflammatory effects of corticosteroids on cytokine storms should not be neglected in patients with COVID-19. The mortality of patients with influenza A (H1N1) pdm09 viral pneumonia presenting with partial pressure of oxygen (PaO2)/fraction of inspired oxygen (FiO2) values less than 300 mmHg was reduced by corticosteroid treatment at a low-to-moderate dose (25–150 mg/day methylprednisolone or equivalent) [101]. Because the point-in-time of the peak of the viral load is similar for both SARS-CoV-2 and influenza virus, corticosteroid treatment is worth considering in patients with COVID-19. Notably, methylprednisolone reduced the mortality of patients with COVID-19 presenting ARDS more than 15% and shortened the duration of mechanical ventilation [102]. An early short course of methylprednisolone improved the clinical outcomes and reduced the escalation of care in patients with moderate to severe COVID-19 [97, 102]. Similarly, dexamethasone reduced the mortality of patients with COVID-19 on oxygen therapy or invasive mechanical ventilation, and water-sodium retention was less likely to occur than with the application of methylprednisolone, partially relieving the concern about the use of corticosteroids [96, 103].
Given the urgency of the clinical demands and the uncertainty of the current evidence, the following expert consensus statement regarding corticosteroid treatment of COVID-19 has been proposed: (1) the benefits and harms should be carefully weighed before using corticosteroids; (2) corticosteroids should be used prudently in critically ill patients with novel coronavirus (2019-nCoV) pneumonia; (3) the further use of corticosteroids should be applied with caution in patients with hypoxemia due to underlying diseases or patients who regularly use corticosteroids for chronic diseases; and (4) the dosage should be low to moderate (≤ 0.5–1 mg/kg per day methylprednisolone or equivalent) and the duration should be short (≤ 7 days) [104, 105]. Current guidelines indicate the use of corticosteroids in severe hospitalized COVID-19 patients with ARDS. Furthermore, shorter and relatively lower-dose corticosteroid therapy could be considered [106]. Ciclesonide and mometasone are inhaled corticosteroids (ICSs) that exert an attenuated effect on the replication of SARS-CoV-2 in vitro and are recommended for application in the early stage of COVID-19 [107]. Furthermore, when a patient experiences a cytokine storm, immunosuppression by steroids may be considered. The early administration of a short course of corticosteroids improves the clinical outcome and immune hyper-reactivity in patients with severe COVID-19 [108].
For patients with severe viral myocarditis, the early intravenous administration of adequate corticosteroids is suggested, as this approach exerts an early anti-inflammatory effect and preserves the dying myocardium [109]. Myocarditis may progress to HF with irreversible dilatation if treatment is delayed or not administered [110]. A Cochrane meta-analysis of 719 patients with viral myocarditis revealed that corticosteroids were beneficial treatments for improving cardiac function and that the patients in this group had a higher left ventricular ejection fraction (LVEF) than patients in the control group at the 1- to 3-month follow-up [111]. According to a previous case report, myocarditis induced by the immune checkpoint inhibitor nivolumab was successfully improved by corticosteroid treatment [112]. Similarly, one report has it that corticosteroids followed by tocilizumab and the aldose reductase inhibitor AT-001 may help the recovery of patients with suspected myocarditis in a patient with severe ARDS and suspected myocarditis with ensuing cardiogenic shock caused by COVID-19 [110]. Patients with fulminant myocarditis caused by SARS-CoV-2 have been treated with methylprednisolone combined with i.v. immunoglobulins [110].
Due to drug interactions at the mechanistic level, glucocorticoids not only activate multiple anti-inflammatory genes, but also inhibit the expression of pro-inflammatory cytokines such as TNF-α, IL-1, and IL-6 by activating glucocorticoid receptors. In terms of mechanism, activated glucocorticoid receptors can inhibit the production of pro-inflammatory cytokines by activating relevant transcription factors (e.g., nuclear factor-kappa B (NF-kB) and activator protein-1). Interestingly, activated glucocorticoid receptors will be depleted due to the excess production of activator protein-1 in patients with chronic inflammatory disorders treated with glucocorticoids, that is, the anti-inflammatory effect of glucocorticoids can be disrupted by overly activating relevant transcription factors [113]. Moreover, glucocorticoids inhibit IFN-γ in a concentration-dependent manner, which will reduce the benefit of IFN responses [114]. Given the benefit of IFN responses and the effect of overly activated transcription factors, it is speculated that the use of inhibitors targeting transcription factors may be more appropriate than glucocorticoid therapy in patients with chronic inflammatory disorders. However, evidence is still lacking to support this, and further studies are required.
In conclusion, corticosteroid therapy appears to be a potential COVID-19 treatment that promotes the recovery of myocardial injury, but additional clinical and fundamental experiments aiming to further define whether and how the use of corticosteroids is beneficial to patients with COVID-19 presenting myocardial injury are lacking.
Interleukin Inhibitors
IL-6 is a pro-inflammatory cytokine that initiates the immune response via the classical cis or trans signaling pathway. The classical cis signaling pathway only exists in immunogenic cells, in which the IL-6 and membrane-bound receptor for IL-6 (mIL-6R) complex activates the JAK-STAT pathway or mitogen-activated protein kinase (MAPK)-NF-kB-IL-6 pathway after binding to the signal transducer glycoprotein 130 (gp130), subsequently triggering the responses of innate and acquired immune system. Meanwhile, IL-6 activates the trans signaling pathway by interacting with soluble receptor for IL-6 (sIL-6R) to induce an extensive pro-inflammatory response in all cells, specifically various endothelial cells [87, 115,116,117]. Tocilizumab is an mIL-6R and sIL-6R inhibitor that reverses the increases in serum CRP and IL-6 levels in patients with COVID-19. Tocilizumab was approved for treatment in patients with serious COVID-19 presenting elevated IL-6 levels by the Chinese National Medical Products Administration (NMPA) in March 2020, and this treatment has been proposed as a candidate therapy for severe COVID-19 [118, 119]. A retrospective analysis showed that the abnormally increased CRP levels decreased after administering tocilizumab based on conventional treatment. Except that, tocilizumab treatment decreased oxygen intake in 75% of seriously ill patients with COVID-19 and promoted lung lesion opacity absorption in 90.5% of the patients [120]. A meta-analysis showed a 12% decrease in the mortality of patients treated with tocilizumab compared with the control group [121]. Similarly, the latest data from a cohort of 1229 hospitalized patients with COVID-19 suggested that tocilizumab contributed to reducing the death risk, ICU admission rate, or mortality in patients whose baseline CRP levels were greater than 150 mg/L, but had no effect on patients with CRP levels less than 150 mg/L [119]. Regarding safety, the use of tocilizumab as a first-line treatment to inhibit cytokine storm does not cause serious adverse events and improves tachycardia and fever associated with cytokine storm [122,123,124]. Thus, IL-6R inhibition contributes to the development of novel treatments for COVID-19. In addition, tocilizumab may decrease the burden on the heart during COVID-19 treatment. However, recent research discoveries revealed that both IL-6 and IL-6R blockade may reduce the benefit in patients with COVID-19. The binary complex formed by IL-6 and IL-6R transforms macrophages from the M1 phenotype that promotes inflammation into the anti-inflammatory M2 phenotype [125]. sgp130Fc is a fusion protein of gp130 with the Fc portion of a human immunoglobulin antibody that specifically binds sIL-6R to inhibit intracellular signal transduction and protect tissue from excess inflammatory damage [115, 126, 127]. Therefore, selective blockade of sIL-6R may be a better treatment for patients with COVID-19. Otherwise, JAK-STAT signaling inhibitors, including baricitinib and ruxolitinib, exert potent effects on preventing the progression of a cytokine storm. Baricitinib inhibits endocytosis to limit viral infection at the early stage and decreases IL-6 levels. Consequently, baricitinib can be used at the late stage of COVID-19. Similarly, ruxolitinib reduces the secretion of pro-inflammatory cytokines, such as TNF-α and IL-6, in inflammatory human macrophages. Based on early clinical evidence, ruxolitinib improved the condition of 4 hospitalized patients with COVID-19 who required supplemental oxygen. Additionally, emerging data from a randomized trial analyzing 43 patients with COVID-19 showed a shorter time to an improvement in clinical symptoms and a better survival rate in the therapeutic group treated with ruxolitinib [128]. These IL-6-related signaling targets, including sIL-6R and the JAK-STAT signaling pathway, may represent new approaches to limit hyperinflammation and cytokine storms in patients with COVID-19. Because multiple cytokines exert their effects through these signaling pathways, the inhibitors targeting these signaling pathways may exert broader effects than an inhibitor of a single cytokine.
Interestingly, in a large MI case control study, circulating sIL-6R levels were obviously increased and associated with MI risk and cardiovascular mortality in patients with st - segment elevation myocardialinfarction (STEMI) [129]. A recent study involving a Swedish cohort of 60-year-old men and women revealed a monotone association between an elevated binary/ternary complex ratio and cardiovascular events, leading to the conclusion that IL-6 trans signaling and IL-6:sIL-6R:sgp130 are associated with cardiovascular events [129]. A separate study of patients with stable coronary disease investigating the relationship between circulating levels of sgp130 and the severity of coronary atherosclerosis showed a negative correlation between the serum sgp130 concentration and the risk of cardiovascular events [130]. Consequently, the progression of cardiovascular events may be prevented and treated by inhibitors of IL-6 trans signaling. In conclusion, we reasonably postulate that strategies targeting IL-6R trans signaling may be beneficial for the prevention and treatment of patients with severe COVID-19 and pre-existing cardiac disease or new onset myocardial damage, even HF. Nevertheless, we must consider the situation in which patients with severe COVID-19 who were treated with tocilizumab experienced an increased incidence of secondary infections, and tocilizumab was useless in patients with a hypoinflammatory status or other special conditions [131]. Therefore, further studies are urgently needed to resolve the uncertainties of anti-IL-6 therapy.
IL-1 beta (IL-1β), which is a main activator of IL-6 expression, has attracted our attention in the exploration of COVID-19 therapies [132]. Anakinra, an inhibitor of IL-1β, has been used to treat hyperinflammation at a high dose. A retrospective cohort study of patients with COVID-19 presenting with hyperinflammation (ferritin > 1000 ng/mL and/or d-dimers > 1.5 μg/mL, plus IL-6 < 40 mg/mL) and ARDS (PaO2/FiO2 < 300) showed that the mortality of patients treated with anakinra was similar to patients administered with tocilizumab. In terms of the curative effect, 55.6% patients administered with anakinra showed a favorable outcome, similar to the tocilizumab-treated matched cohort [133]. In addition, anakinra reduced the return of patients with corticosteroid-dependent and colchicine-resistant recurrent pericarditis [134]. A randomized, placebo-controlled trial showed that canakinumab, a monoclonal antibody for IL-1β, decreased hospitalization for HF in patients with ongoing subclinical inflammation and a history of MI [135]. Thus, anti-inflammatory therapy using an inhibitor of IL-1β may provide a mechanistically distinct approach to the treatment of HF. Namely, the inhibition of IL-1β deserves consideration for patients with COVID-19 presenting with myocardial injury due to its anti-inflammatory activity. Regrettably, adequate clinical data are not available to evaluate the effect of anakinra on patients with COVID-19, but related clinical trials are in progress (CT04330638, NCT04341584, NCT04339712, and NCT04324021) [58].
Because IL-6 trans signaling and IL-1β play vital roles in both COVID-19 and HF, their inhibitors may be effective and novel treatments to prevent the progression of HF in patients with COVID-19.
Interferons
IFNs are natural broad-spectrum antivirals, and type I IFNs (IFN-α/β) and type III IFNs (IFN-λ) have been applied in the clinic [136, 137]. The immunopathology of SARS-CoV-infected mice was improved by the early administration of type I IFNs [27]. Compared with SARS-CoV, SARS-CoV-2 does not induce an obvious IFN response in infected human lung tissues [138]. The SARS-CoV-2 N protein may explain this phenomenon because it antagonizes type I IFN signaling by limiting STAT1 and STAT2 phosphorylation [139] (Fig. 2). Thus, the interference in the IFN response is more evident in patients with COVID-19. As a result, an uncontrolled viral load leads to damage to various tissues in these patients with impaired IFN response. A multicenter, prospective phase 2 trial showed that a triple combination of IFN beta-1b (IFN β-1b), lopinavir–ritonavir, and ribavirin dominated by IFN β-1b is superior to the control treatment in alleviating symptoms among patients with mild to moderate COVID-19. Moreover, the SARS-CoV-2 load and hospital stays were decreased under such treatment as well [140]. Furthermore, regarding adverse effects, no differences were observed between the two groups. Thus, IFN β-1b is safe for clinical applications [140]. In addition, IFN β-1b improves virus-induced lung fibrosis in mice, and this in vivo experiment provides a theoretical basis for the treatment of patients with acute respiratory distress syndrome caused by SARS-CoV-2 [141]. Consequently, IFN β-1b may be a rational and effective treatment for COVID-19 by launching or improving the antiviral response. Given the peak of the viral load at the time of presentation, the early application of IFNs has been advised to improve injury and prevent the serious complications associated with COVID-19.
In addition to the effect of IFN β on COVID-19, IFN β also plays an important role in HF [142,143,144]. On the one hand, cardiotropic viruses interfere with the physical function of cardiomyocytes [145, 146]. In contrast, the negative inotropic cytokines induced by the virus may induce hemodynamic deterioration [147]. However, the viral genomes disappear after IFN-β administration, and a decrease in the number of T lymphocytes and an obvious improvement in LV dysfunction have been reported [148]. Thus, IFN β-1b exerts positive effects on virus clearance and the reversion of LV function in patients with myocardial viral persistence. Meanwhile, the partial virus-induced myocardial dysfunction is reversible. Early diagnosis and therapy with IFN-β are important for myocardial dysfunction caused by the virus, even HF. Regarding safety, IFN-β does not exert cardiac-specific adverse effects. Moreover, its clinical benefit may persist due to its antiviral effect [148].
Because type I IFNs induce a systemic inflammatory response to the virus, some scientists have focused on IFN-λ [149, 150]. In contrast to IFN-β, IFN-λ exerts a stronger anti-inflammatory effect on protecting tissues, but lacks the forceful pro-inflammatory effect of type I IFNs [151, 152]. If the infection escapes the control of IFN-λ, the systemic pro-inflammatory response caused by type I IFNs will occur, resulting in further tissue damage due to hyperinflammation [151]. Pegylated or recombinant forms of IFN-λ have been used to inhibit viral replication and block cytokine storms in patients with COVID-19 [152, 153]. IFNs promote ACE2 expression, while the effect of IFNs on blocking SARS-CoV-2 replication may counterbalance the replication-promoting benefits mediated by increased ACE2 expression [150]. Thus, IFN-λ has an anti-virus activity and decreases further damage caused by inflammation. IFN-λ may be inhibited in patients with COVID-19 and then lead to serious complications, including myocardial injury.
In conclusion, IFN-β and IFN-λ may represent effective approaches for the treatment of patients with mild to moderate COVID-19 to prevent or reverse cardiac complications. However, the exact mechanism of IFNs in COVID-19 treatments remains unclear, and the time points associated with the action or treatment of different IFN types still require further exploration.
RAAS Inhibitors
At present, controversy exists regarding whether patients with COVID-19 should be treated with RAAS inhibitors. On the one hand, ACE2 resists inflammation and tissue damage in the inflammatory environment and may exert protective effects on severe pulmonary complications. RAAS inhibitors also restore reduced ACE2 levels and prevent or even reverse the phenotype of HF. On the other hand, the use of RAAS inhibitors will increase the expression of ACE2, which is important for SARS-CoV-2 to enter host cells. The long-term use of RAAS inhibitors increases the levels of Ang 1–7 [154], which will theoretically increase the sensitivity of host cells to entry and transmission and lead to a higher risk of infection in these patients. Due to the lack of sufficient medical evidence, a clear conclusion regarding the risks and benefits of RAAS inhibitors has not been reached, but the answer will become increasingly clear with ongoing clinical trials.
Recently, a large case control study including 6272 patients with COVID-19 and 30,759 patients as controls did not observe a correlation between the use of RAAS inhibitors and the risk of COVID-19 in a multivariate analysis (angiotensin receptor blockers (ARB), adjusted OR = 0.95 [95% CI 0.86–1.05]; Ang-converting-enzyme inhibitor (ACEI), adjusted OR = 0.96 [95% CI 0.87–1.07]) [155]. Another trial involving 1,128 patients with COVID-19 presenting hypertension showed that the continued use of RAAS inhibitors reduced the risk of death in these patients [156].
In summary, according to the available data, the application of RAAS inhibitors, including ACEIs and ARBs, should be maintained or started in patients with COVID-19 because the benefits of RAAS inhibitors may outweigh the risks. In clinically unstable patients, particularly patients at risk of HF or myocardial infarction, treatment with RAAS inhibitors should not be stopped or switched to treatment with alternative drugs. Because the sudden withdrawal of RAAS inhibitors may lead to an increased early risk of COVID-19-related myocardial injury and accelerate the deterioration of cardiac function, potentially even leading to the development of HF in days to weeks. In brief, withdrawal of RAAS inhibitors would result in an increased risk of cardiovascular death in patients with COVID-19 [103, 157]. Many clinical trials have been launched to investigate the therapeutic value of ACEIs and ARBs (NCT04312009 and NCT04311177). Based on the premise that supplementation of ACE2 will decrease the susceptibility to SARS-CoV-2 and protect the heart while also inhibiting inflammation, a clinical trial of a recombinant form of human ACE2 (rhACE2) Apn01 is ongoing in Europe [158]. Because Ang 1–7 may cause a hyperinflammatory condition, a non-randomized interventional clinical trial designed to evaluate the therapeutic efficacy of Ang 1–7 is ongoing (NCT04375124) [159].
Conclusions
COVID-19 is a great public health emergency that may cause serious heart damage and result in a worse prognosis and urgently needs to be solved. The cytokine storm caused by SARS-CoV-2 induces systemic hyperinflammation, deteriorating pre-existing cardiac damage or eliciting new onset myocardial injury in patients with COVID-19, which has become a focus for exploring underlying therapeutic targets in COVID-19. In addition, the mechanism underlying the inhibition of cytokine storm may have common targets to inhibit both the progression of COVID-19 and HF, enabling the development of inhibitors of related targets for the treatment of patients with COVID-19 presenting with pre-existing or new onset cardiac disease, even HF. In this review, we sought to reveal the association between COVID-19 and HF in terms of the pathophysiological mechanism, which centers on the cytokine storm. We provided insights into cytokine storm as a trigger of HF progression in patients with COVID-19 and discussed potential anti-inflammatory drugs worthy of further exploration as treatments for COVID-19.
As early warning signals in at-risk patients, clinicians should pay more attention and continuously monitor cytokines and related laboratory indexes. Furthermore, studies are needed to evaluate whether the detection of serum levels of specific cytokines at the early stage contributes to the choice of treatment plans or exerts a curative effect. Hence, large-scale studies are needed to confirm our analysis. We hope that the large number of ongoing prospective, randomized trials will provide more high-quality data and clinical evidence, which will be important for further determining management strategies and optimizing the prognosis of patients with COVID-19.
Data Availability
Not applicable
References
Bhaskar S, Sinha A, Banach M, Mittoo S, Weissert R, Kass JS, et al. Cytokine Storm in COVID-19-Immunopathological Mechanisms, Clinical Considerations, and Therapeutic Approaches: The REPROGRAM Consortium Position Paper. Front Immunol. 2020;11:1648.
Tay MZ, Poh CM, Renia L, MacAry PA, Ng LFP. The trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol. 2020;20(6):363–74.
Van Linthout S, Tschope C. Inflammation - cause or consequence of heart failure or both? Curr Heart Fail Rep. 2017;14(4):251–65.
Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382(8):727–33.
Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–3.
Lam TT, Shum MH, Zhu HC, Tong YG, Ni XB, Liao YS, et al. Identifying SARS-CoV-2 related coronaviruses in Malayan pangolins. Nature. 2020;583:282–5.
Graham RL, Baric RS. SARS-CoV-2: combating coronavirus emergence. Immunity. 2020;52(5):734–6.
Tang X, Wu C, Li X, Song Y, Yao X, Wu X, et al. On the origin and continuing evolution of SARS-CoV-2. Natl Sci Rev. 2020;7:1012–23.
Fehr AR, Perlman S. Coronaviruses: an overview of their replication and pathogenesis. Methods Mol Biol. 2015;1282:1–23.
Hofmann H, Hattermann K, Marzi A, Gramberg T, Geier M, Krumbiegel M, et al. S protein of severe acute respiratory syndrome-associated coronavirus mediates entry into hepatoma cell lines and is targeted by neutralizing antibodies in infected patients. J Virol. 2004;78(12):6134–42.
Xiao X, Chakraborti S, Dimitrov AS, Gramatikoff K, Dimitrov DS. The SARS-CoV S glycoprotein: expression and functional characterization. Biochem Biophys Res Commun. 2003;312(4):1159–64.
Bosch BJ, Bartelink W, Rottier PJ. Cathepsin L functionally cleaves the severe acute respiratory syndrome coronavirus class I fusion protein upstream of rather than adjacent to the fusion peptide. J Virol. 2008;82(17):8887–90.
Heurich A, Hofmann-Winkler H, Gierer S, Liepold T, Jahn O, Pohlmann S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J Virol. 2014;88(2):1293–307.
Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271–80 e8.
Glowacka I, Bertram S, Muller MA, Allen P, Soilleux E, Pfefferle S, et al. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J Virol. 2011;85(9):4122–34.
Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181(2):281–92 e6.
Coutard B, Valle C, de Lamballerie X, Canard B, Seidah NG, Decroly E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir Res. 2020;176:104742.
Hoffmann M, Kleine-Weber H, Pohlmann S. A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human Lung cells. Mol Cell. 2020;78:779–784.e5.
Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72314 cases from the Chinese Center for Disease Control and Prevention. JAMA. 2020.
Mahase E. Coronavirus covid-19 has killed more people than SARS and MERS combined, despite lower case fatality rate. BMJ. 2020;368:m641.
Ovsyannikova IG, Haralambieva IH, Crooke SN, Poland GA, Kennedy RB. The role of host genetics in the immune response to SARS-CoV-2 and COVID-19 susceptibility and severity. Immunol Rev. 2020;296(1):205–19.
Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203(2):631–7.
Zheng YY, Ma YT, Zhang JY, Xie X. COVID-19 and the cardiovascular system. Nat Rev Cardiol. 2020;17(5):259–60.
Ye Q, Wang B, Mao J. The pathogenesis and treatment of the ‘cytokine storm’ in COVID-19. J Inf Secur. 2020;80(6):607–13.
Chousterman BG, Swirski FK, Weber GF. Cytokine storm and sepsis disease pathogenesis. Semin Immunopathol. 2017;39(5):517–28.
Behrens EM, Koretzky GA. Review: cytokine storm syndrome: looking toward the precision medicine era. Arthritis Rheumatol. 2017;69(6):1135–43.
Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, et al. Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe. 2016;19(2):181–93.
Zheng HY, Zhang M, Yang CX, Zhang N, Wang XC, Yang XP, et al. Elevated exhaustion levels and reduced functional diversity of T cells in peripheral blood may predict severe progression in COVID-19 patients. Cell Mol Immunol. 2020;17(5):541–3.
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506.
Bermejo-Martin JF, Almansa R, Menendez R, Mendez R, Kelvin DJ, Torres A. Lymphopenic community acquired pneumonia as signature of severe COVID-19 infection. J Inf Secur. 2020;80(5):e23–e4.
Azkur AK, Akdis M, Azkur D, Sokolowska M, van de Veen W, Bruggen MC, et al. Immune response to SARS-CoV-2 and mechanisms of immunopathological changes in COVID-19. Allergy. 2020;75(7):1564–81.
Channappanavar R, Fehr AR, Zheng J, Wohlford-Lenane C, Abrahante JE, Mack M, et al. IFN-I response timing relative to virus replication determines MERS coronavirus infection outcomes. J Clin Invest. 2019;129(9):3625–39.
Xia H, Cao Z, Xie X, Zhang X, Chen JY, Wang H, et al. Evasion of type I interferon by SARS-CoV-2. Cell Rep. 2020;108234.
Merad M, Martin JC. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat Rev Immunol. 2020;20(6):355–62.
Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. 2020;369:718–24.
Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395(10229):1054–62.
Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun. 2005;73(4):1907–16.
Oussalah A, Gleye S, Urmes IC, Laugel E, Barbe F, Orlowski S, et al. The spectrum of biochemical alterations associated with organ dysfunction and inflammatory status and their association with disease outcomes in severe COVID-19: a longitudinal cohort and time-series design study. EClinicalMedicine. 2020;100554.
Ruan Q, Yang K, Wang W, Jiang L, Song J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 2020;46(5):846–8.
Goronzy JJ, Weyand CM. Mechanisms underlying T cell ageing. Nat Rev Immunol. 2019;19(9):573–83.
Murphy SP, Kakkar R, McCarthy CP, Januzzi JL Jr. Inflammation in heart failure: JACC state-of-the-art review. J Am Coll Cardiol. 2020;75(11):1324–40.
Dharmarajan K, Rich MW. Epidemiology, Pathophysiology, and prognosis of heart failure in older adults. Heart Fail Clin. 2017;13(3):417–26.
Mauvais-Jarvis F, Klein SL, Levin ER. Estradiol, Progesterone, Immunomodulation, and COVID-19 Outcomes. Endocrinology. 2020;161(9).
Inciardi RM, Adamo M, Lupi L, Cani DS, Di Pasquale M, Tomasoni D, et al. Characteristics and outcomes of patients hospitalized for COVID-19 and cardiac disease in northern Italy. Eur Heart J. 2020;41(19):1821–9.
Chen T, Wu D, Chen H, Yan W, Yang D, Chen G, et al. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: retrospective study. BMJ. 2020;368:m1091.
Wei ZY, Geng YJ, Huang J, Qian HY. Pathogenesis and management of myocardial injury in coronavirus disease 2019. Eur J Heart Fail. 2020.
Bojkova D, Wagner JUG, Shumliakivska M, Aslan GS, Saleem U, Hansen A, et al. SARS-CoV-2 infects and induces cytotoxic effects in human cardiomyocytes. Cardiovasc Res. 2020;116:2207–15.
Tanai E, Frantz S. Pathophysiology of heart failure. Compr Physiol. 2015;6(1):187–214.
van der Meer P, Gaggin HK, Dec GW. ACC/AHA versus ESC guidelines on heart failure: JACC guideline comparison. J Am Coll Cardiol. 2019;73(21):2756–68.
Hilfiker-Kleiner D, Shukla P, Klein G, Schaefer A, Stapel B, Hoch M, et al. Continuous glycoprotein-130-mediated signal transducer and activator of transcription-3 activation promotes inflammation, left ventricular rupture, and adverse outcome in subacute myocardial infarction. Circulation. 2010;122(2):145–55.
Fischer P, Hilfiker-Kleiner D. Role of gp130-mediated signalling pathways in the heart and its impact on potential therapeutic aspects. Br J Pharmacol. 2008;153(Suppl 1):S414–27.
Shirazi LF, Bissett J, Romeo F, Mehta JL. Role of inflammation in heart failure. Curr Atheroscler Rep. 2017;19(6):27.
Savvatis K, Muller I, Frohlich M, Pappritz K, Zietsch C, Hamdani N, et al. Interleukin-6 receptor inhibition modulates the immune reaction and restores titin phosphorylation in experimental myocarditis. Basic Res Cardiol. 2014;109(6):449.
Thaik CM, Calderone A, Takahashi N, Colucci WS. Interleukin-1 beta modulates the growth and phenotype of neonatal rat cardiac myocytes. J Clin Invest. 1995;96(2):1093–9.
Yokoyama T, Vaca L, Rossen RD, Durante W, Hazarika P, Mann DL. Cellular basis for the negative inotropic effects of tumor necrosis factor-alpha in the adult mammalian heart. J Clin Invest. 1993;92(5):2303–12.
Takahashi M. NLRP3 inflammasome as a novel player in myocardial infarction. Int Heart J. 2014;55(2):101–5.
Condorelli G, Morisco C, Latronico MV, Claudio PP, Dent P, Tsichlis P, et al. TNF-alpha signal transduction in rat neonatal cardiac myocytes: definition of pathways generating from the TNF-alpha receptor. FASEB J. 2002;16(13):1732–7.
Jamilloux Y, Henry T, Belot A, Viel S, Fauter M, El Jammal T, et al. Should we stimulate or suppress immune responses in COVID-19? Cytokine and anti-cytokine interventions. Autoimmun Rev. 2020;19(7):102567.
Zeng JH, Liu YX, Yuan J, Wang FX, Wu WB, Li JX, et al. First case of COVID-19 complicated with fulminant myocarditis: a case report and insights. Infection. 2020;48:773–7.
Inciardi RM, Lupi L, Zaccone G, Italia L, Raffo M, Tomasoni D, et al. Cardiac involvement in a patient with coronavirus disease 2019 (COVID-19). JAMA Cardiol. 2020.
Peng J, Gurantz D, Tran V, Cowling RT, Greenberg BH. Tumor necrosis factor-alpha-induced AT1 receptor upregulation enhances angiotensin II-mediated cardiac fibroblast responses that favor fibrosis. Circ Res. 2002;91(12):1119–26.
Tomasoni D, Italia L, Adamo M, Inciardi RM, Lombardi CM, Solomon SD et al. COVID-19 and heart failure: from infection to inflammation and angiotensin II stimulation. Searching for evidence from a new disease. Eur J Heart Fail. 2020.
Xu D, Ma M, Xu Y, Su Y, Ong SB, Hu X et al. Single-cell transcriptome analysis indicates new potential regulation mechanism of ACE2 and NPs signaling among heart failure patients infected with SARS-CoV-2. medRxiv. 2020.
Gurantz D, Cowling RT, Varki N, Frikovsky E, Moore CD, Greenberg BH. IL-1beta and TNF-alpha upregulate angiotensin II type 1 (AT1) receptors on cardiac fibroblasts and are associated with increased AT1 density in the post-MI heart. J Mol Cell Cardiol. 2005;38(3):505–15.
Wang Y, Li Y, Wu Y, Jia L, Wang J, Xie B, et al. 5TNF-alpha and IL-1beta neutralization ameliorates angiotensin II-induced cardiac damage in male mice. Endocrinology. 2014;155(7):2677–87.
Das SK. The Pathophysiology, Diagnosis and Treatment of Corona Virus Disease 2019 (COVID-19). Indian J Clin Biochem. 2020:1–12.
Chang Y, Wei W. Angiotensin II in inflammation, immunity and rheumatoid arthritis. Clin Exp Immunol. 2015;179(2):137–45.
Haga S, Yamamoto N, Nakai-Murakami C, Osawa Y, Tokunaga K, Sata T, et al. Modulation of TNF-alpha-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-alpha production and facilitates viral entry. Proc Natl Acad Sci U S A. 2008;105(22):7809–14.
Patel VB, Clarke N, Wang Z, Fan D, Parajuli N, Basu R, et al. Angiotensin II induced proteolytic cleavage of myocardial ACE2 is mediated by TACE/ADAM-17: a positive feedback mechanism in the RAS. J Mol Cell Cardiol. 2014;66:167–76.
Xu J, Sriramula S, Xia H, Moreno-Walton L, Culicchia F, Domenig O, et al. Clinical relevance and role of neuronal AT1 receptors in ADAM17-mediated ACE2 shedding in neurogenic hypertension. Circ Res. 2017;121(1):43–55.
Voloshenyuk TG, Hart AD, Khoutorova E, Gardner JD. TNF-alpha increases cardiac fibroblast lysyl oxidase expression through TGF-beta and PI3Kinase signaling pathways. Biochem Biophys Res Commun. 2011;413(2):370–5.
Yang J, Savvatis K, Kang JS, Fan P, Zhong H, Schwartz K, et al. Targeting LOXL2 for cardiac interstitial fibrosis and heart failure treatment. Nat Commun. 2016;7:13710.
Panfoli I. Potential role of endothelial cell surface ectopic redox complexes in COVID-19 disease pathogenesis. Clin Med (Lond). 2020.
Okada H, Woodcock-Mitchell J, Mitchell J, Sakamoto T, Marutsuka K, Sobel BE, et al. Induction of plasminogen activator inhibitor type 1 and type 1 collagen expression in rat cardiac microvascular endothelial cells by interleukin-1 and its dependence on oxygen-centered free radicals. Circulation. 1998;97(21):2175–82.
Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395(10234):1417–8.
Torre-Amione G, Kapadia S, Benedict C, Oral H, Young JB, Mann DL. Proinflammatory cytokine levels in patients with depressed left ventricular ejection fraction: a report from the studies of left ventricular dysfunction (SOLVD). J Am Coll Cardiol. 1996;27(5):1201–6.
Deswal A, Petersen NJ, Feldman AM, Young JB, White BG, Mann DL. Cytokines and cytokine receptors in advanced heart failure: an analysis of the cytokine database from the Vesnarinone trial (VEST). Circulation. 2001;103(16):2055–9.
Shi S, Qin M, Shen B, Cai Y, Liu T, Yang F, et al. Association of cardiac injury with mortality in hospitalized patients with COVID-19 in Wuhan, China. JAMA Cardiol. 2020.
Frantz S, Nahrendorf M. Cardiac macrophages and their role in ischaemic heart disease. Cardiovasc Res. 2014;102(2):240–8.
Torre-Amione G, Kapadia S, Lee J, Bies RD, Lebovitz R, Mann DL. Expression and functional significance of tumor necrosis factor receptors in human myocardium. Circulation. 1995;92(6):1487–93.
Kubota T, McTiernan CF, Frye CS, Slawson SE, Lemster BH, Koretsky AP, et al. Dilated cardiomyopathy in transgenic mice with cardiac-specific overexpression of tumor necrosis factor-alpha. Circ Res. 1997;81(4):627–35.
Mehra MR, Ruschitzka F. COVID-19 illness and heart failure: a missing link? JACC Heart Fail. 2020;8(6):512–4.
Lal S, Hayward CS, De Pasquale C, Kaye D, Javorsky G, Bergin P, et al. COVID-19 and acute heart failure: screening the critically ill - a position statement of the Cardiac Society of Australia and New Zealand (CSANZ). Heart Lung Circ. 2020;29:e94–8.
Radenovic S, Loncar G, Busjahn A, Apostolovic S, Zdravkovic M, Karlicic V, et al. Systemic inflammation and functional capacity in elderly heart failure patients. Clin Res Cardiol. 2018;107(4):362–7.
Zhang Y, Coats AJS, Zheng Z, Adamo M, Ambrosio G, Anker SD, et al. Management of heart failure patients with COVID-19: a joint position paper of the Chinese heart failure Association & National Heart Failure Committee and the heart failure Association of the European Society of cardiology. Eur J Heart Fail. 2020;22:941–56.
Lippi G, Lavie CJ, Sanchis-Gomar F. Cardiac troponin I in patients with coronavirus disease 2019 (COVID-19): evidence from a meta-analysis. Prog Cardiovasc Dis. 2020;63(3):390–1.
Ziegler L, Gajulapuri A, Frumento P, Bonomi A, Wallen H, de Faire U, et al. Interleukin 6 trans-signalling and risk of future cardiovascular events. Cardiovasc Res. 2019;115(1):213–21.
Ferrante G, Condorelli G. Interleukin-6 trans-signalling and risk of future cardiovascular events: a new avenue for atheroprotection? Cardiovasc Res. 2019;115(1):8–9.
Feldmann M, Maini RN, Woody JN, Holgate ST, Winter G, Rowland M, et al. Trials of anti-tumour necrosis factor therapy for COVID-19 are urgently needed. Lancet. 2020;395(10234):1407–9.
Chen XY, Yan BX, Man XY. TNFalpha inhibitor may be effective for severe COVID-19: learning from toxic epidermal necrolysis. Ther Adv Respir Dis. 2020;14:1753466620926800.
Segiet OA, Piecuch A, Mielanczyk L, Michalski M, Nowalany-Kozielska E. Role of interleukins in heart failure with reduced ejection fraction. Anatol J Cardiol. 2019;22(6):287–99.
Grom AA, Horne A, De Benedetti F. Macrophage activation syndrome in the era of biologic therapy. Nat Rev Rheumatol. 2016;12(5):259–68.
Cavalli G, De Luca G, Campochiaro C, Della-Torre E, Ripa M, Canetti D, et al. Interleukin-1 blockade with high-dose anakinra in patients with COVID-19, acute respiratory distress syndrome, and hyperinflammation: a retrospective cohort study. Lancet Rheumatol. 2020;2(6):e325–e31.
Bhaskar S, Rastogi A, Chattu VK, Adisesh A, Thomas P, Alvarado N, et al. Key strategies for clinical management and improvement of healthcare Services for Cardiovascular Disease and Diabetes Patients in the coronavirus (COVID-19) settings: recommendations from the REPROGRAM consortium. Front Cardiovasc Med. 2020;7:112.
Arabi YM, Mandourah Y, Al-Hameed F, Sindi AA, Almekhlafi GA, Hussein MA, et al. Corticosteroid therapy for critically ill patients with Middle East respiratory syndrome. Am J Respir Crit Care Med. 2018;197(6):757–67.
Singh AK, Majumdar S, Singh R, Misra A. Role of corticosteroid in the management of COVID-19: a systemic review and a clinician’s perspective. Diabetes Metab Syndr. 2020;14(5):971–8.
Fadel R, Morrison AR, Vahia A, Smith ZR, Chaudhry Z, Bhargava P, et al. Early short course corticosteroids in hospitalized patients with COVID-19. Clin Infect Dis. 2020.
Chen Q, Zheng Z, Zhang C, Zhang X, Wu H, Wang J, et al. Clinical characteristics of 145 patients with corona virus disease 2019 (COVID-19) in Taizhou, Zhejiang, China. Infection. 2020;48:543–51.
Gasparini L, Ongini E, Wenk G. Non-steroidal anti-inflammatory drugs (NSAIDs) in Alzheimer’s disease: old and new mechanisms of action. J Neurochem. 2004;91(3):521–36.
Russell CD, Millar JE, Baillie JK. Clinical evidence does not support corticosteroid treatment for 2019-nCoV lung injury. Lancet. 2020;395(10223):473–5.
Li H, Yang SG, Gu L, Zhang Y, Yan XX, Liang ZA, et al. Effect of low-to-moderate-dose corticosteroids on mortality of hospitalized adolescents and adults with influenza a(H1N1)pdm09 viral pneumonia. Influenza Other Respir Viruses. 2017;11(4):345–54.
Wu C, Chen X, Cai Y, Xia J, Zhou X, Xu S, et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med. 2020;180:934–43.
Rizk JG, Kalantar-Zadeh K, Mehra MR, Lavie CJ, Rizk Y, Forthal DN. Pharmaco-Immunomodulatory therapy in COVID-19. Drugs. 2020;80(13):1267–92.
Zhao JP, Hu Y, Du RH, Chen ZS, Jin Y, Zhou M, et al. Expert consensus on the use of corticosteroid in patients with 2019-nCoV pneumonia. Zhonghua Jie He He Hu Xi Za Zhi. 2020;43:E007.
Shang L, Zhao J, Hu Y, Du R, Cao B. On the use of corticosteroids for 2019-nCoV pneumonia. Lancet. 2020;395(10225):683–4.
Alhazzani W, Moller MH, Arabi YM, Loeb M, Gong MN, Fan E, et al. Surviving sepsis campaign: guidelines on the Management of Critically ill Adults with coronavirus disease 2019 (COVID-19). Crit Care Med. 2020;48(6):e440–e69.
Lipworth B, Kuo CR, Chan R. Emerging pharmacotherapy for COVID-19. J R Coll Physicians Edinb. 2020;50(2):133–7.
Kolilekas L, Loverdos K, Giannakaki S, Vlassi L, Levounets A, Zervas E, et al. Can steroids reverse the severe COVID-19 induced “cytokine storm”? J Med Virol. 2020;92:2866–9.
Hu H, Ma F, Wei X, Fang Y. Coronavirus fulminant myocarditis saved with glucocorticoid and human immunoglobulin. Eur Heart J. 2020.
Coyle J, Igbinomwanhia E, Sanchez-Nadales A, Danciu S, Chu C, Shah N. A recovered case of COVID-19 myocarditis and ARDS treated with corticosteroids, Tocilizumab, and experimental AT-001. JACC Case Rep. 2020;2:1331–6.
Chen HS, Wang W, Wu SN, Liu JP. Corticosteroids for viral myocarditis. Cochrane Database Syst Rev. 2013;10:CD004471.
Matsuo K, Ishiguro T, Najama T, Shimizu Y, Kobayashi Y, Mutou M. Nivolumab-induced myocarditis successfully treated with corticosteroid therapy: a case report and review of the literature. Intern Med. 2019;58(16):2367–72.
Barnes PJ. Anti-inflammatory actions of glucocorticoids: molecular mechanisms. Clin Sci (Lond). 1998;94(6):557–72.
Rutten S, Schrodl W, Abraham G. Modulation of TNF-alpha, IL-1Ra and IFN-gamma in equine whole blood culture by glucocorticoids. Vet Immunol Immunopathol. 2019;210:1–5.
Saha A, Sharma AR, Bhattacharya M, Sharma G, Lee SS, Chakraborty C. Tocilizumab: a therapeutic option for the treatment of cytokine storm syndrome in COVID-19. Arch Med Res. 2020;51(6):595–7.
Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta. 2011;1813(5):878–88.
Wolf J, Rose-John S, Garbers C. Interleukin-6 and its receptors: a highly regulated and dynamic system. Cytokine. 2014;70(1):11–20.
Crisafulli S, Isgro V, La Corte L, Atzeni F, Trifiro G. Potential role of anti-interleukin (IL)-6 drugs in the treatment of COVID-19: rationale, clinical evidence and risks. BioDrugs. 2020;34:415–22.
Martinez-Sanz J, Muriel A, Ron R, Herrera S, Perez-Molina JA, Moreno S, et al. Effects of Tocilizumab on mortality in hospitalized patients with COVID-19: a multicenter cohort study. Clin Microbiol Infect. 2020.
Xu X, Han M, Li T, Sun W, Wang D, Fu B, et al. Effective treatment of severe COVID-19 patients with tocilizumab. Proc Natl Acad Sci U S A. 2020;117(20):10970–5.
Malgie J, Schoones JW, Pijls BG. Decreased mortality in COVID-19 patients treated with Tocilizumab: a rapid systematic review and meta-analysis of observational studies. Clin Infect Dis. 2020.
Fitzgerald JC, Weiss SL, Maude SL, Barrett DM, Lacey SF, Melenhorst JJ, et al. Cytokine release syndrome after chimeric antigen receptor T cell therapy for acute lymphoblastic leukemia. Crit Care Med. 2017;45(2):e124–e31.
Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. 2016;127(26):3321–30.
Masia M, Fernandez-Gonzalez M, Padilla S, Ortega P, Garcia JA, Agullo V, et al. Impact of interleukin-6 blockade with tocilizumab on SARS-CoV-2 viral kinetics and antibody responses in patients with COVID-19: a prospective cohort study. EBioMedicine. 2020;60:102999.
Mauer J, Chaurasia B, Goldau J, Vogt MC, Ruud J, Nguyen KD, et al. Signaling by IL-6 promotes alternative activation of macrophages to limit endotoxemia and obesity-associated resistance to insulin. Nat Immunol. 2014;15(5):423–30.
Rabe B, Chalaris A, May U, Waetzig GH, Seegert D, Williams AS, et al. Transgenic blockade of interleukin 6 transsignaling abrogates inflammation. Blood. 2008;111(3):1021–8.
Rose-John S, Waetzig GH, Scheller J, Grotzinger J, Seegert D. The IL-6/sIL-6R complex as a novel target for therapeutic approaches. Expert Opin Ther Targets. 2007;11(5):613–24.
Yeleswaram S, Smith P, Burn T, Covington M, Juvekar A, Li Y, et al. Inhibition of cytokine signaling by ruxolitinib and implications for COVID-19 treatment. Clin Immunol. 2020;218:108517.
Ritschel VN, Seljeflot I, Arnesen H, Halvorsen S, Eritsland J, Fagerland MW et al. Circulating levels of IL-6 receptor and gp130 and long-term clinical outcomes in ST-elevation myocardial infarction. J Am Heart Assoc. 2016;5(6).
Korotaeva AA, Samoilova EV, Chepurnova DA, Zhitareva IV, Shuvalova YA, Prokazova NV. Soluble glycoprotein 130 is inversely related to severity of coronary atherosclerosis. Biomarkers. 2018;23(6):527–32.
Hedrick TL, Murray BP, Hagan RS, Mock JR. COVID-19: clean up on IL-6. Am J Respir Cell Mol Biol. 2020;63(4):541–3.
Hunter CA, Jones SA. IL-6 as a keystone cytokine in health and disease. Nat Immunol. 2015;16(5):448–57.
Iglesias-Julian E, Lopez-Veloso M, de-la-Torre-Ferrera N, Barraza-Vengoechea JC, Delgado-Lopez PD, Colazo-Burlato M, et al. High dose subcutaneous Anakinra to treat acute respiratory distress syndrome secondary to cytokine storm syndrome among severely ill COVID-19 patients. J Autoimmun. 2020;102537.
Brucato A, Imazio M, Gattorno M, Lazaros G, Maestroni S, Carraro M, et al. Effect of Anakinra on recurrent pericarditis among patients with colchicine resistance and corticosteroid dependence: the AIRTRIP randomized clinical trial. JAMA. 2016;316(18):1906–12.
Everett BM, Cornel JH, Lainscak M, Anker SD, Abbate A, Thuren T, et al. Anti-inflammatory therapy with Canakinumab for the prevention of hospitalization for heart failure. Circulation. 2019;139(10):1289–99.
Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, et al. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol. 2003;4(1):69–77.
Sheppard P, Kindsvogel W, Xu W, Henderson K, Schlutsmeyer S, Whitmore TE, et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat Immunol. 2003;4(1):63–8.
Chu H, Chan JF, Wang Y, Yuen TT, Chai Y, Hou Y, et al. Comparative replication and immune activation profiles of SARS-CoV-2 and SARS-CoV in human lungs: an ex vivo study with implications for the pathogenesis of COVID-19. Clin Infect Dis. 2020;71:1400–9.
Mu J, Fang Y, Yang Q, Shu T, Wang A, Huang M, et al. SARS-CoV-2 N protein antagonizes type I interferon signaling by suppressing phosphorylation and nuclear translocation of STAT1 and STAT2. Cell Discov. 2020;6:65.
Hung IF, Lung KC, Tso EY, Liu R, Chung TW, Chu MY, et al. Triple combination of interferon beta-1b, lopinavir-ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: an open-label, randomised, phase 2 trial. Lancet (London, England). 2020;395(10238):1695–704.
Luckhardt TR, Coomes SM, Trujillo G, Stoolman JS, Vannella KM, Bhan U, et al. TLR9-induced interferon beta is associated with protection from gammaherpesvirus-induced exacerbation of lung fibrosis. Fibrogenesis Tissue Repair. 2011;4:18.
Lutton CW, Gauntt CJ. Ameliorating effect of IFN-beta and anti-IFN-beta on coxsackievirus B3-induced myocarditis in mice. J Interf Res. 1985;5(1):137–46.
Bartlett EJ, Lenzo JC, Sivamoorthy S, Mansfield JP, Cull VS, James CM. Type I IFN-beta gene therapy suppresses cardiac CD8+ T-cell infiltration during autoimmune myocarditis. Immunol Cell Biol. 2004;82(2):119–26.
Deonarain R, Cerullo D, Fuse K, Liu PP, Fish EN. Protective role for interferon-beta in coxsackievirus B3 infection. Circulation. 2004;110(23):3540–3.
Badorff C, Lee GH, Lamphear BJ, Martone ME, Campbell KP, Rhoads RE, et al. Enteroviral protease 2A cleaves dystrophin: evidence of cytoskeletal disruption in an acquired cardiomyopathy. Nat Med. 1999;5(3):320–6.
Dorner A, Pauschinger M, Schwimmbeck PL, Kuhl U, Schultheiss HP. The shift in the myocardial adenine nucleotide translocator isoform expression pattern is associated with an enteroviral infection in the absence of an active T-cell dependent immune response in human inflammatory heart disease. J Am Coll Cardiol. 2000;35(7):1778–84.
Bozkurt B, Kribbs SB, Clubb FJ Jr, Michael LH, Didenko VV, Hornsby PJ, et al. Pathophysiologically relevant concentrations of tumor necrosis factor-alpha promote progressive left ventricular dysfunction and remodeling in rats. Circulation. 1998;97(14):1382–91.
Kuhl U, Pauschinger M, Schwimmbeck PL, Seeberg B, Lober C, Noutsias M, et al. Interferon-beta treatment eliminates cardiotropic viruses and improves left ventricular function in patients with myocardial persistence of viral genomes and left ventricular dysfunction. Circulation. 2003;107(22):2793–8.
Broggi A, Granucci F, Zanoni I. Type III interferons: Balancing tissue tolerance and resistance to pathogen invasion. J Exp Med. 2020;217(1).
Busnadiego I, Fernbach S, Pohl MO, Karakus U, Huber M, Trkola A et al. Antiviral Activity of Type I, II, and III Interferons Counterbalances ACE2 Inducibility and Restricts SARS-CoV-2. mBio. 2020;11(5).
Galani IE, Triantafyllia V, Eleminiadou EE, Koltsida O, Stavropoulos A, Manioudaki M, et al. Interferon-lambda mediates non-redundant front-line antiviral protection against influenza virus infection without compromising host fitness. Immunity. 2017;46(5):875–90 e6.
Davidson S, McCabe TM, Crotta S, Gad HH, Hessel EM, Beinke S, et al. IFNlambda is a potent anti-influenza therapeutic without the inflammatory side effects of IFNalpha treatment. EMBO Mol Med. 2016;8(9):1099–112.
Andreakos E, Tsiodras S. COVID-19: lambda interferon against viral load and hyperinflammation. EMBO Mol Med. 2020;12(6):e12465.
Luque M, Martin P, Martell N, Fernandez C, Brosnihan KB, Ferrario CM. Effects of captopril related to increased levels of prostacyclin and angiotensin-(1-7) in essential hypertension. J Hypertens. 1996;14(6):799–805.
Mancia G, Rea F, Ludergnani M, Apolone G, Corrao G. Renin-angiotensin-aldosterone system blockers and the risk of Covid-19. N Engl J Med. 2020;382(25):2431–40.
Zhang P, Zhu L, Cai J, Lei F, Qin JJ, Xie J, et al. Association of Inpatient use of angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers with mortality among patients with hypertension hospitalized with COVID-19. Circ Res. 2020;126(12):1671–81.
Curfman G. Renin-angiotensin-aldosterone inhibitors and susceptibility to and severity of COVID-19. JAMA. 2020;324(2):177–8.
Guzik TJ, Mohiddin SA, Dimarco A, Patel V, Savvatis K, Marelli-Berg FM, et al. COVID-19 and the cardiovascular system: implications for risk assessment, diagnosis, and treatment options. Cardiovasc Res. 2020;116(10):1666–87.
Datta PK, Liu F, Fischer T, Rappaport J, Qin X. SARS-CoV-2 pandemic and research gaps: understanding SARS-CoV-2 interaction with the ACE2 receptor and implications for therapy. Theranostics. 2020;10(16):7448–64.
Funding
This work was supported by grants from the National Natural Science Foundation of China (grant nos. 91739113 and 81971715 to JWT and 81827806 to BY), the National Key R&D Program of China (grant no. 2016YFC1301100 to BY), the Fok Ying-Tong Education Foundation for Young Teachers (grant no. 171032 to JWT), and the Foundation of Guangxi Key Laboratory of Diabetic Systems Medicine (grant no. GKLCDSM-20200101-01 to JWT).
Author information
Authors and Affiliations
Contributions
Xiang Peng and Yani Wang designed and wrote the review, supervised the process, and critically reviewed the complete manuscript. Xiangwen Xi and Ying Jia performed the literature search and prepared the figures. Jiangtian Tian, Bo Yu, and Jinwei Tian revised the text and critically discussed the completed manuscript. All authors read and approved the final manuscript. Xiang Peng and Yani Wang contributed equally to this work and should be regarded as joint first authors.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflicts of interest.
Ethical Approval
Not applicable
Consent to Participate
Not applicable
Consent for Publication
Not applicable
Code Availability
Not applicable
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Peng, X., Wang, Y., Xi, X. et al. Promising Therapy for Heart Failure in Patients with Severe COVID-19: Calming the Cytokine Storm. Cardiovasc Drugs Ther 35, 231–247 (2021). https://doi.org/10.1007/s10557-020-07120-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10557-020-07120-8