
Abstract
Heart failure is one of the common end stages of cardiovascular diseases, the leading cause of death in developed countries. Molecular mechanisms underlying the development of heart failure remain elusive but there is a consistent observation of chronic immune activation and aberrant microRNA (miRNA) expression that is present in failing hearts. This review will focus on the interplay between the immune system and miRNAs as factors that play a role during the development of heart failure. Several studies have shown that heart failure patients can be characterized by a sustained innate immune activation. The role of inflammatory signaling is discussed and TLR4 signaling, IL-1β, TNFα and IL-6 expression appears to coincide with the development of heart failure. Furthermore, we describe the implication of the renin angiotensin aldosteron system in immunity and heart failure. In the past decade microRNAs (miRNAs), small non-coding RNAs that translationally repress protein synthesis by binding to partially complementary sequences of mRNA, have come to light as important regulators of several kinds of cardiovascular diseases including cardiac hypertrophy and heart failure. The involvement of differentially expressed miRNAs in the inflammation that occurs during the development of heart failure is still subject of investigation. Here, we summarize and comment on the first studies in this field and hypothesize on the putative involvement of certain miRNAs in heart failure. MicroRNAs have been shown to be critical regulators of cardiac function and inflammation. Future research will have to point out if dampening the immune response, and the miRNAs associated with it, during the development of heart failure is a therapeutically plausible route to follow.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cardiovascular diseases are the primary cause of human morbidity in developed countries and mortality is quickly increasing [1]. Heart failure is one of the common end stages of cardiovascular diseases with a poor prognosis highlighted by a 5 year mortality of approximately 70% [2]. Understanding the underlying molecular mechanisms that predict and contribute to heart failure is therefore critical.
The adult human heart is an adaptive organ, able to respond to increased demand of circulation or to injury by significant remodeling and hypertrophic growth. Physiological cardiac hypertrophy occurs following exercise and pregnancy and is reversible after removal of the hypertrophic stimulus [3, 4]. Physiological cardiac hypertrophy differs in its structural and molecular profile from pathological cardiac hypertrophy and is characterized by normal or enhanced cardiac function and increased expression of adult isoforms of sarcomeric genes [5].
Pathological cardiac hypertrophy is an important predecessor of heart failure that is observed as a consequence of a plethora of cardiovascular diseases including hypertension, myocardial infarction, endocrine disorders and viral myocarditis. Pathological cardiac hypertrophy is irreversible. It may be initially adaptive in normalizing wall stress and preserving contractile dysfunction, but can proceed to decompensation and heart failure. The progression of hypertrophy from an adaptive form to the pathophysiological state occurs gradually over time and is characterized by cardiac dysfunction, upregulation of fetal gene expression, impaired myocardial vascularization, unfavorable changes in extracellular matrix composition and fibrosis [5–9]. Molecular mechanisms underlying the development of adaptive remodeling into pathological cardiac hypertrophy remain elusive to date, but there is a consistent observation of chronic immune activation and aberrant microRNA (miRNA) expression that is present in pathological hypertrophy [10–12]. Silencing of miRNAs might be therapeutically relevant and efficient in vivo silencing in rodents and nonhuman primates with chemically modified antisense nucleotides has been proven successful in the treatment of cancer and hepatitis C infection [13, 14]. This review will focus on the interplay between the immune system and miRNAs as factors that play a role during the development of heart failure.
Heart failure and the immune system
It has long been known that resident leukocytes are present in the connective tissue of many organs including the heart in a healthy situation, yet little is known about the role of leukocytes in heart failure [15]. In the past decade, the role of the immune system in heart failure has received a considerable amount of attention and it is becoming apparent that it is critical in heart failure development. Devaux et al. investigated the presence of leukocyte subsets in patients with end stage heart failure and found increased amounts of T lymphocytes and macrophages compared to healthy controls, suggesting that these cells may contribute to the structural defects in the myocardium found in heart failure patients [16]. Several studies have shown that heart failure patients can be characterized by a sustained innate immune activation [17]. Proinflammatory molecules, named cytokines are involved in the development of pathological cardiac hypertrophy [12]. Cytokines are a family of low molecular weight, pharmacologically active proteins that function as paracrine or autocrine mediators which are secreted by various cell types including cardiomyocytes and cells of the immune system. While short term activation of proinflammatory cytokines in response to cardiac damage might be cardioprotective, it may occur at the cost of long-term deleterious effects. For example, pretreatment of isolated rat hearts with a low dose of LPS, inducing cytokine release from cardiomyocytes, prior to induction of ischemia/reperfusion damage protected against cardiac dysfunction [18]. However, long term expression of cytokines may produce maladaptive effects [19]. Angiotensin II (AngII), tumor necrosis factor α (TNFα), toll-like receptor 4 (TLR4) and NF-κB are not only markers of heart failure, but actively induce myocardial dysfunction via the c-Jun N-terminal kinase (JNK)/p38 pathway, which promotes apoptosis and cardiac fibrosis [20–22].
TLR4 is a key regulator of inflammation in the myocardium
Recently the TLR4 pathway has emerged as an important player in regulating cardiac hypertrophy via expression levels of TNF-α, IL-1β and IL-6 in the heart after LPS stimulation [23, 24]. TLR4 recognizes pathogen-associated molecular patterns (PAMPs), such as lypopolysacharide (LPS), that are expressed on infectious agents. Evidence has emerged that TLR4 defective mice do not develop LV dysfunction after LPS stimulation whereas wild type mice developed marked defects in LV contraction and relaxation [25]. Furthermore, mice deficient in TLR4 show reduced infarct size and increased LV function after myocardial infarction compared to wild type mice [26, 27]. Interestingly, others have also observed a reduced infarct size, but did not see an improved LV function in TLR4 deficient mice [28].
The role of TLR4 signaling in the depression of cardiac function is still controversial. Tavener et al. reported that the primary role of TLR4 mediated cardiac dysfunction can be attributed to the immune system [29], while other reports have shown that TLR4 mediated dysfunction is due to cardiac expression of the protein [30–33].
In humans, reports have demonstrated that TLR4 expression in hearts of advanced heart failure patients is increased [34]. The main pathway through which TLR signaling induces cardiac inflammation ultimately leads to NF-κB activation in immune cells and cardiomyocytes. NF-κB plays a critical role in the expression of genes involved in cell death, inflammatory responses and cell survival [35]. TLR4 signaling that leads to activation of NF-κB involves the adaptor molecule myeloid differentiation factor-88 (MyD88), kinase of IL-1 receptor-associated kinase-1 (IRAK-1), TNF-receptor associated factor 6 (TRAF6), NF-κB-inducing kinase, transforming growth factor (TGF)-β-activated kinase 1 (TAK1) and IκB kinase complexes [36].
Blockade of the downstream pathway of TLR4 by cardiomyocyte specific inhibition of MyD88 reduced cardiac hypertrophy, cardiomyocyte apoptosis and improved cardiac function in rats, further elucidating the importance of TLR4/NF-κB signaling in the development of cardiac dysfunction. Furthermore, a potential role for MyD88/IL-1 signaling in the link between innate sensing and stimulation of the adaptive immune response during heart failure has been demonstrated as MyD88 deficient mice were protected against autoimmune-induced cardiomyocyte death [37].
Taken together, it appears that there is a critical role for TLR4 in cytokine regulation in both humans and other mammals during the development of heart failure. TLR4 expression on hematopoietic cells and cardiac cells is crucial in regulating the hypertrophic response in the heart in response to hypertrophic stimuli. It appears that NF- κB signaling in response to TLR4 stimulation is mediating most of the hypertrophic effects.
NF-κB is the main effector molecule of inflammatory signaling in the heart
NF-κB, the main downstream effector molecule of TLR4 signaling, is a nuclear transcription factor that increases the expression of proinflammatory cytokines like TNF-α, IL-1, IL-6 and IFNγ [38]. Short term NF-κB mediated upregulation of proinflammatory mediators in the heart has beneficial effects during viral infections as it causes increased expression of cell-adhesion molecules and chemokines which attract macrophages and NK cells that can prevent viral replication in host cells, as well as upregulating nitric oxide synthase which is directly toxic to replicating viruses. On the long run however, persistent activation of proinflammatory cytokine expression appears to have detrimental effects. In rats, myocardial infarct size was significantly reduced after chemical NF-κB inhibition, suggesting a detrimental role of NF-κB in challenged hearts [39]. In humans, increased NF-κB activation has also been observed in heart failure patients [40].
These results suggest that NF- κB is a crucial component in the immune response that occurs during the development of cardiac hypertrophy and heart failure. Despite the significant beneficial effects of short term NF- κB upregulation in response to pathological cardiac stimuli, the long term effects of sustained NF- κB exaggerate cardiac damage, supposedly through the upregulation of proinflammatory cytokines.
TNFα and IL-1β act synergistically to depress cardiac function during inflammation
TNFα and IL-1β are proinflammatory cytokines produced by macrophages, T cells and dendritic cells. Upon activation, these cells create a positive feedback loop, catalyzing the activation of nearby T-cells that can in turn help to clear the pathogenic substance that activated the original T cells [41]. Besides being produced in immune cells, TNFα and IL-1β are also synthesized by, among others, cardiomyocytes in response to injury or stress [42, 43]. The expression of these inflammatory mediators in response to cardiac injury led to a series of investigations that elucidated their role in the development of heart failure.
TNFα is able to induce, at physiologically relevant levels, a significant amount of cell death in rat cardiomyocytes in vitro [44]. Moreover, infusion of a pathologically relevant amount of TNFα for 15 days in a rat model of cardiac hypertrophy has shown that TNFα induces a decrease in LV function, cardiac myocyte shortening, and LV dilation [45]. Furthermore, marked LV remodeling in mice overexpressing TNFα in a cardiomyocyte specific manner has been demonstrated [46]. In humans, increased levels of circulating TNFα, together with the soluble TNFα receptors 1 and 2 serve as biomarkers for heart failure. Phase I clinical trials have shown a short term dose dependant improvement of LV structure and function after 3 months of treatment with Etanercept, a soluble TNF antagonist [47]. Results of clinical trials have however been disappointing, as two large independently performed trials have been unable to confirm the beneficial effects of TNF inhibition in the phase I trial [48, 49]. A possible explanation for the inability of TNF antagonism to improve cardiac function is that TNF antagonism may be too specific and a more systemic approach in which multiple proinflammatory cytokines are repressed simultaneously might be a better approach.
The proinflammatory cytokine IL-1β has largely overlapping properties with TNFα. Together with TNFα, IL-1β has been implicated in the pathogenesis of myocardial dysfunction and cardiomyocyte death in ischemia-reperfusion injury and chronic heart failure [43]. Most biological functions of TNFα have also been observed for IL-1β [50, 51]. Moreover, TNFα and IL-1β have overlapping properties and act synergistically as cultured rat cardiomyocytes showed decreased contractile function after administration of these cytokines [52].
The proinflammatory properties of TNFα and IL-1β appear to be crucial in the inflammatory response occurring during the development of heart failure induced through various pathological stimuli. Simultaneous inhibition of multiple proinflammatory cytokines could a therapeutically interesting strategy to improve LV function in HF patients.
IL-6 has synergistic effects with other proinflammatory cytokines and induces STAT3 signaling
As a target of the transcription factor NF-κB, IL-6 is one of the major pro-inflammatory players in the cardiac environment. According to the cytokine hypothesis by Seta et al., heart failure progresses because proinflammatory cytokines exaggerate hemodynamic abnormalities or because they directly induce cardiac cell death [53]. Indeed, increased IL-6 levels have been observed in patients with myocardial dysfunction [54–56]. Moreover, increased plasma levels of IL-6 have been associated with an increased risk of heart failure [57, 58]. IL-6 is secreted by both cardiomyocytes and cardiac fibroblasts in response to cardiac damage and acts synergistically with other cytokines such as IL-1β and TNFα [57, 59].
IL-6 can exert its proinflammatory effect in two distinct ways: via the membrane bound IL-6 receptor (IL-6R), called the classical signaling pathway, and via the soluble IL-6 receptor (sIL-6R), known as trans signaling [60]. Both IL-6R and sIL-6R bind to gp130 to induce down-stream signaling, but only gp130 and sIL-6R are essential for IL-6 signaling in the cardiomyocte to induce hypertrophy via STAT3 and here, gp130 is the rate limiting protein. Interestingly, IL-6 and gp130, but not sIL-6R, correlate with a poor prognosis in HF [53, 60].
IL-6 signaling in the heart is synergistic with other inflammatory cytokines, like IL-1β and TNF-α. Additionally, IL-6 increases STAT3 expression, causing this cytokine to provoke an additional hypertrophic signaling pathway. The dual hypertrophic signaling pathways that IL-6 activates makes it an interesting therapeutical target.
Renin angiotensin aldosteron system
The renin angiotensin aldosteron system (RAAS) is the body’s hormonal system that regulates blood pressure and water balance [61]. Many factors are known to underlie the etiology of heart failure and the RAAS is one of the systems known to be involved. In the RAAS, AngII and aldosterone are important mediators of cardiac hypertrophy [62]. The RAAS was suggested to be involved in the immune reaction that coincides with heart failure by Samsonov et al. who found a relationship between plasma renin activity, angiotensin II, serum levels of angiotensin-converting enzyme, aldosterone and markers of immune activation in congestive heart failure in humans [63].
Besides the well described hemodynamic function of AngII, it also has a well accepted direct role in the development of cardiac remodeling, cardiac hypertrophy and failure. The hypertrophic action of AngII on cardiomyocytes was first suggested by the ability of angiotensin converting enzyme (ACE) inhibitors to reduce the size of hypertrophy in vitro in both neonatal [64] and adult cardiomyocytes [65, 66]. Furthermore, rats treated with ACE inhibitors display a significantly reduced left ventricular (LV) hypertrophic response after hypertrophic stimulation by transverse aortic banding (TAC) [67]. Besides induction of hypertrophic growth in cultured cardiomyocytes, AngII also causes cardiomyocytes to transcribe a fetal phenotype of gene expression in rats including upregulation of factors such as atrial natriuretic factor (ANF), α-skeletal actin (ACTA) and β-myosin heavy chain (β-MHC) [68, 69]. Indirect proof of AngII contributing to inflammation occurring during cardiac damage comes from the study of Kvakan et al., which shows that injection of regulatory T cells (Treg) ameliorated inflammation, reduced fibrosis and cardiac damage after AngII infusion [70]. These combined observations suggest a role for AngII in pathological cardiac hypertrophy that extends beyond its effect on blood pressure. Angiotensin II type 1 receptors (AT1R) are expressed on many cell types including immune cells. AngII is a peptide hormone that, upon binding to AT1R, acts as a proinflammatory agent and induces the expression of NF-κB, resulting in proinflammatory cytokine production and activation of the immune system [71].
Aldosterone levels are elevated in humans with hypertrophic cardiomyopathy [72]. Furthermore, expression of aldosterone is sufficient to cause an upregulation of hypertrophic proteins by phosphorylation of protein kinase D and activation of transforming growth factor-β1 by upregulation of phophoinositide3-kinase in rat cardiomyocytes [72]. It appears that AngII and aldosterone play crucial roles in the activation of the immune system that coincides with heart failure. Upregulation of either RAAS protein is sufficient to initiate a hypertrophic response. It will be interesting to see if there are therapeutically relevant miRNA targets against the AT1R, aldosterone or one of their downstream effector molecules that can attenuate the hypertrophic response and the inflammation that occurs during the development of heart failure.
Heart failure and microRNAs
In the past decade, miRNAs have come to light as important regulators of several kinds of cardiovascular diseases including atherosclerosis, cardiac arrhythmias, hypertrophy and failure [73]. miRNAs are a recently discovered class of highly conserved endogenous small non-coding RNAs, ~22 nucleotides in length, that regulate gene expression by binding to partially complementary sequences of mRNA [74]. Binding of a miRNA to its complementary mRNA causes mRNA degradation or translational repression, effectively silencing the mRNA. Originally it was assumed that miRNAs repressed protein synthesis without much influence on mRNA levels [75, 76]. However, continued research unveiled that lowered mRNA levels account for most of the decreased protein production after miRNA interference [77]. miRNAs alter the translation of 30% to 50% of all genes. Most miRNAs are able to target multiple target mRNAs and most mRNAs are targeted by multiple miRNAs, making research on the function of miRNAs a complex endeavor.
In the heart, miRNA expression is crucial during development as cardiac specific Dicer deletion, a crucial protein in miRNA synthesis, leads to premature death in mice [78]. In the adult heart miRNA expression remains important to maintain proper pump function. Inducible cardiac specific knockout of the Dicer or DCGR8 gene, both required for the synthesis of miRNA, in adult mice caused a marked cardiomyocyte hypertrophy, fibrosis and induction of fetal gene expression [78, 79]. Several microarray studies have revealed signature pattern expression of specific miRNAs that are consistently aberrantly expressed in heart failure patients. miR-1, -29, -30, -133 and -150 are found to be downregulated in heart failure patients, whereas miR-21, -23a, -125, -146, -195, -199 and -214 are upregulated [9, 80–83]. Interestingly, most of the miRNAs that are constitutively downregulated during HF, miR-1, -29, -30 and -133, are also downregulated in cardiomyocyte specific Dicer knockouts, suggesting a cardiomyocyte origin of these miRNAs. Likewise, most of the upregulated miRNAs that are upregulated during HF, miR-21,-23a,-125, and -146, are also upregulated in cardiomyocyte specific Dicer knockouts. Interestingly, these cardiomyocyte miRNA-deficient mice developed heart failure with supposedly consequent influx of non-myocyte cells like leukocytes and fibroblasts as contributors to the increased presence of these miRNAs. Therefore, the cardiomyocyte specific Dicer knockout model is highly relevant for the selection of non-myocyte miRNAs with a role in HF.
The involvement of myocytic miRNAs in HF has been subject of other reviews [80, 84]. The involvement of differentially expressed miRNAs in the inflammation that occurs during the development of heart failure is still subject of investigation. Here, we summarize and comment on the first studies in this field. The following sections will discuss the potential roles of miR-155, miR-146a/b, -223,-21 and the miR17 ~ 92 cluster.
miR-155 has a proinflammatory role in the immune system and might be implicated in heart failure
miR-155 has an important role in the mammalian immune system and is abundantly, though not exclusively, expressed in T-cells, B-cells and monocytes [85, 86]. miR-155 −/− mice are immunodeficient and show a bias towards an anti-inflammatory TH2 cytokine production [87]. Recently, a first in vivo study showed a pro-inflammatory role for miR-155, where miR155 deficient mice were protected against experimental autoimmune encephalomyelitis following defective T-cell responses [88]. It thus appears that miR-155 provokes a proinflammatory response in immune cells.
In a cardiovascular setting, in human endothelial and vascular smooth muscle cells, miR-155 is co-transcribed with, and represses the expression of the angiotensin II type 1 receptor (AT1R) [89]. Moreover, a polymorphism in the human AT1R is associated with cardiovascular disease, possibly mediated through enhanced AT1R expression. Indeed, the presence of this polymorphism interrupts base-pairing of miR-155 with the AT1R binding site for miR-155, derepressing AT1R expression and increasing AngII signaling [89]. The AT-1R binding site for miR-155 is not conserved among species. Still, in the absence of cardiomyocyte derived miRNAs, miR-155 expression increases during HF in mice, suggesting a role for non-cardiomyocyte derived miR-155 production in heart failure [78]. Presumably, this increased expression is due to immune activation during HF.
These results reveal a crucial role for miR-155 in the immune system and unveil a possible role for miR-155 during HF. Further research on this miRNA could illustrate interesting molecular interactions and a possible therapeutic relevance of miR-155 treatment in HF.
miR-146a/b is expressed in monocytes and cardiomyocytes and prevents pathological signaling
miR-146a/b is abundantly expressed in the heart and is upregulated upon activation of NF-κB, a transcription factor known to be involved in the development of heart failure [10, 39]. Indeed, miR-146 was found to be upregulated in juvenile and adult mice with a cardiomyocyte specific deficiency for the Dicer protein, suggesting a non-cardiomyocyte origin of miR-146 [78]. Originally, miR-146a/b was described in immune cells by Taganov et al. who observed an NF-κB dependent upregulation of miR-146 after LPS stimulation of monocytes [86]. The increased expression of miR-146a/b in monocytes leads to the repression of IRAK and TRAF6 proteins which are key adaptor molecules downstream of Toll like and cytokine receptor signaling. By inhibiting the expression of IRAK and TRAF6, the expression of miR-146a/b creates an anti-inflammatory feedback loop, inhibiting NF-κB induced proinflammatory cytokine production [86]. In the heart, in doxorubicin induced model of heart failure miR-146a/b was found to be increased upon induction of cardiotoxicity and inhibited ErbB-2 and -4 expression [90]. ErbB-2 and -4 are receptors for the neuregulin-1 (NRG-1) protein, which is essential for normal cardiac development and induces hypertrophic growth in ventricular rat cardiomyocytes [91].
MiR-146a/b appears to be a key player in both immune cell signaling and cardiomyocyte responses with its expression being triggered by the activation of both cell types. In both monocytes and cardiomyocytes, miR-146a/b expression is dependent on NF-κB and in both cell types miR-146a/b creates a negative feedback loop preventing pathological signaling by inhibiting expression of pathological molecules such as NF-κB and TNFα. It seems likely that there is a relationship between the inflammatory and cardiac properties of miR-146a/b, but no publications have currently confirmed this statement to our knowledge.
miR-223 cardioprotective and anti-inflammatory properties
miR-223 regulates glucose metabolism in the heart by increasing Glut4 expression in cardiomyocytes [92]. In diabetes type II patients, Glut4 expression is decreased and an increase of miR-223 is observed. Interestingly, it is also known that pathological cardiac events such as MI and cardiac ischemia increase the expression level of miR-223 in mice [93]. Furthermore, it is known that MI and cardiac ischemia cause an upregulation of glucose uptake of the heart in humans, raising the possibility that miR-223 induced glucose uptake via increased Glut4 expression provides a cardioprotective mechanism during pathological cardiac events.
In the immune system, miR-223 has a myeloid specific expression pattern and its expression is dependent on CCAAT/enhancer-binding protein (C/EBP) activity, a known regulator of granulopoiesis [94]. miR-223 knockout mice have an expanded granulocytic compartment [95]. Furthermore, the same study showed an increased production, differentiation and activation of granulocytes in miR-223 deficient mice, resulting in inflammation and exaggerated tissue destruction. Therefore, miR-223 was suggested to act as a regulator of granulocyte production and the inflammatory response.
In conclusion, miR-223 appears to have cardio-protective and anti-inflammatory properties by enhancing glucose metabolism and inhibiting granulocyte activation. It will be interesting to see if future reports that investigate the effect of miR-223 on the immune system in the development of heart failure can confirm the anti-pathological properties that have so far been reported.
miR-21 expression in fibroblasts might contribute to prohypertrophic signaling in the heart
miR-21 expression is induced in several types of cancers and is, together with miR-155, also important in monocyte differentiation [96–99]. A cardiac role for miR-21 has been attributed to its function in fibroblasts by Thum et al. [100]. Here, it was identified as a critical regulator of the extracellular signal-regulated kinase-mitogen-activated protein kinase (ERK-MAPK) signaling pathway by inhibition of the protein sprouty homologue 1 (SPRY1) [100, 101]. In vivo silencing of miR-21 protected against pressure overload-induced fibrosis and attenuated myocardial dysfunction. Recently however, Patrick et al. revealed that miR-21 is not required for cardiac hypertrophy, fibrosis, or loss of contractile function. Thus the role of miR-21 in heart failure has not yet been unambiguously determined but the observation that it has distinguished roles in both the myocardium and the immune system makes it an interesting target for future investigation.
Members of the miR-17 ~ 92 cluster are upregulated during heart failure
The miR-17 ~ 92 cluster is a group of miRNAs that are transcribed as a polycystronic cluster. Constitutive expression of miR-17 ~ 92 causes inhibition of BIM and p21 proteins, causing lymphoproliferative disorders and autoimmunity [102]. The cluster regulates hematopoiesis and immune function and has a role in cardiac development [103, 104]. Targeted inhibition of miR-17 ~ 92 causes postnatal lethality in mice due to cardiac and pulmonary defects [104]. MiR-17 ~ 92 has been reported to target connective tissue growth factor (CTGF) which, in the heart, is associated with adverse structural remodeling during heart failure [105–107].
Again, a miRNA with functions in the heart as well as the immune system emerges. Future research may unravel their interconnection during heart failure.
Conclusion
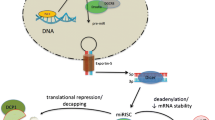
The last decade has seen a considerable amount of research on miRNAs in heart failure and on inflammation in heart failure. Both topics are to date not yet fully unraveled and the interplay of miRNAs and inflammation has hardly been touched in the field of cardiovascular research. Inflammation is believed to be important in heart failure by the vast majority of researchers today as animal models that inhibit the inflammatory response show a marked decrease in cardiac dysfunction after induction of a pathological stimulus. miRNAs have been shown to be critical regulators of cardiac function as alteration of miRNA expression has a drastic influence on cardiac performance (Fig. 1). Future research will have to point out if dampening the immune response, and the miRNAs that are associated with it, during the development of heart failure is a therapeutically plausible route to follow.

A central role for miRNAs and cytokines in heart failure. Upon activation of stress signals in cardiomyocytes, NF-κB expression is increased which results in the production of the proinflammatory cytokines TNFα, IL-1β and IL-6. These cytokines have a plethora of roles in the development of heart failure, including induction of apoptosis in myocytes, activation and migration of immune cells and activation of fibroblasts. In these events, specific miRNAs play an important role as depicted in the individual cell types
References
Van Herck PL, Vrints CJ, Carlier SG. Coronary circulation and interventional cardiology. Ann Biomed Eng. 2005;33:1735–42.
Levi F, Lucchini F, Negri E, La Vecchia C. Trends in mortality from cardiovascular and cerebrovascular diseases in Europe and other areas of the world. Heart. 2002;88:119–24.
Fagard RH. Impact of different sports and training on cardiac structure and function. Cardiol Clin. 1997;15:397–412.
Pluim BM, Zwinderman AH, van der Laarse A, van der Wall EE. The athlete’s heart. A meta-analysis of cardiac structure and function. Circulation. 2000;101:336–44.
Iemitsu M, Miyauchi T, Maeda S, et al. Physiological and pathological cardiac hypertrophy induce different molecular phenotypes in the rat. Am J Physiol Regul Integr Comp Physiol. 2001;281:R2029–36.
De Boer RA, Pinto YM, Van Veldhuisen DJ. The imbalance between oxygen demand and supply as a potential mechanism in the pathophysiology of heart failure: the role of microvascular growth and abnormalities. Microcirculation. 2003;10:113–26.
Hilfiker-Kleiner D, Limbourg A, Drexler H. STAT3-mediated activation of myocardial capillary growth. Trends Cardiovasc Med. 2005;15:152–7.
Olson EN. A decade of discoveries in cardiac biology. Nat Med. 2004;10:467–74.
Thum T, Galuppo P, Wolf C, et al. MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation. 2007;116:258–67.
Cheng Y, Ji R, Yue J, et al. MicroRNAs are aberrantly expressed in hypertrophic heart: do they play a role in cardiac hypertrophy? Am J Pathol. 2007;170:1831–40.
Niebauer J, Volk HD, Kemp M, et al. Endotoxin and immune activation in chronic heart failure: a prospective cohort study. Lancet. 1999;353:1838–42.
Torre-Amione G. Immune activation in chronic heart failure. Am J Cardiol. 2005;95:3C–8. discussion 38C-40C.
Lanford RE, Hildebrandt-Eriksen ES, Petri A, et al. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science. 2010;327:198–201.
Petri A, Lindow M, Kauppinen S. MicroRNA silencing in primates: towards development of novel therapeutics. Cancer Res. 2009;69:393–5.
Spencer SC, Fabre JW. Characterization of the tissue macrophage and the interstitial dendritic cell as distinct leukocytes normally resident in the connective tissue of rat heart. J Exp Med. 1990;171:1841–51.
Devaux B, Scholz D, Hirche A, Klovekorn WP, Schaper J. Upregulation of cell adhesion molecules and the presence of low grade inflammation in human chronic heart failure. Eur Heart J. 1997;18:470–9.
Testa M, Yeh M, Lee P, et al. Circulating levels of cytokines and their endogenous modulators in patients with mild to severe congestive heart failure due to coronary artery disease or hypertension. J Am Coll Cardiol. 1996;28:964–71.
Chao W. Toll-like receptor signaling: a critical modulator of cell survival and ischemic injury in the heart. Am J Physiol Heart Circ Physiol. 2009;296:H1–12.
Mann DL, Topkara VK, Evans S, Barger PM. Innate immunity in the adult mammalian heart: for whom the cell tolls. Trans Am Clin Climatol Assoc. 2010;121:34–50. discussion −1.
Dorn 2nd GW, Brown JH. Gq signaling in cardiac adaptation and maladaptation. Trends Cardiovasc Med. 1999;9:26–34.
Dorn 2nd GW, Force T. Protein kinase cascades in the regulation of cardiac hypertrophy. J Clin Invest. 2005;115:527–37.
Liang Q, Molkentin JD. Redefining the roles of p38 and JNK signaling in cardiac hypertrophy: dichotomy between cultured myocytes and animal models. J Mol Cell Cardiol. 2003;35:1385–94.
Baumgarten G, Knuefermann P, Nozaki N, Sivasubramanian N, Mann DL, Vallejo JG. In vivo expression of proinflammatory mediators in the adult heart after endotoxin administration: the role of toll-like receptor-4. J Infect Dis. 2001;183:1617–24.
Frantz S, Kobzik L, Kim YD, et al. Toll4 (TLR4) expression in cardiac myocytes in normal and failing myocardium. J Clin Invest. 1999;104:271–80.
Nemoto S, Vallejo JG, Knuefermann P, et al. Escherichia coli LPS-induced LV dysfunction: role of toll-like receptor-4 in the adult heart. Am J Physiol Heart Circ Physiol. 2002;282:H2316–23.
Chong AJ, Shimamoto A, Hampton CR, et al. Toll-like receptor 4 mediates ischemia/reperfusion injury of the heart. J Thorac Cardiovasc Surg. 2004;128:170–9.
Oyama J, Blais Jr C, Liu X, et al. Reduced myocardial ischemia-reperfusion injury in toll-like receptor 4-deficient mice. Circulation. 2004;109:784–9.
Kim SC, Ghanem A, Stapel H, et al. Toll-like receptor 4 deficiency: smaller infarcts, but no gain in function. BMC Physiol. 2007;7:5.
Tavener SA, Long EM, Robbins SM, McRae KM, Van Remmen H, Kubes P. Immune cell Toll-like receptor 4 is required for cardiac myocyte impairment during endotoxemia. Circ Res. 2004;95:700–7.
Fallach R, Shainberg A, Avlas O, et al. Cardiomyocyte Toll-like receptor 4 is involved in heart dysfunction following septic shock or myocardial ischemia. J Mol Cell Cardiol. 2010;48:1236–44.
Baumgarten G, Knuefermann P, Schuhmacher G, et al. Toll-like receptor 4, nitric oxide, and myocardial depression in endotoxemia. Shock. 2006;25:43–9.
Binck BW, Tsen MF, Islas M, et al. Bone marrow-derived cells contribute to contractile dysfunction in endotoxic shock. Am J Physiol Heart Circ Physiol. 2005;288:H577–83.
Boyd JH, Mathur S, Wang Y, Bateman RM, Walley KR. Toll-like receptor stimulation in cardiomyoctes decreases contractility and initiates an NF-kappaB dependent inflammatory response. Cardiovasc Res. 2006;72:384–93.
Birks EJ, Felkin LE, Banner NR, Khaghani A, Barton PJ, Yacoub MH. Increased toll-like receptor 4 in the myocardium of patients requiring left ventricular assist devices. J Heart Lung Transplant. 2004;23:228–35.
Baeuerle PA, Baltimore D. NF-kappa B: ten years after. Cell. 1996;87:13–20.
Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406:782–7.
Ha T, Hua F, Li Y, et al. Blockade of MyD88 attenuates cardiac hypertrophy and decreases cardiac myocyte apoptosis in pressure overload-induced cardiac hypertrophy in vivo. Am J Physiol Heart Circ Physiol. 2006;290:H985–94.
Muller JM, Ziegler-Heitbrock HW, Baeuerle PA. Nuclear factor kappa B, a mediator of lipopolysaccharide effects. Immunobiology. 1993;187:233–56.
Morishita R, Sugimoto T, Aoki M, et al. In vivo transfection of cis element “decoy” against nuclear factor-kappaB binding site prevents myocardial infarction. Nat Med. 1997;3:894–9.
Frantz S, Fraccarollo D, Wagner H, et al. Sustained activation of nuclear factor kappa B and activator protein 1 in chronic heart failure. Cardiovasc Res. 2003;57:749–56.
Gallucci S, Matzinger P. Danger signals: SOS to the immune system. Curr Opin Immunol. 2001;13:114–9.
Azzawi M, Hasleton P. Tumour necrosis factor alpha and the cardiovascular system: its role in cardiac allograft rejection and heart disease. Cardiovasc Res. 1999;43:850–9.
Cain BS, Meldrum DR, Dinarello CA, et al. Tumor necrosis factor-alpha and interleukin-1beta synergistically depress human myocardial function. Crit Care Med. 1999;27:1309–18.
Krown KA, Page MT, Nguyen C, et al. Tumor necrosis factor alpha-induced apoptosis in cardiac myocytes. Involvement of the sphingolipid signaling cascade in cardiac cell death. J Clin Invest. 1996;98:2854–65.
Bozkurt B, Kribbs SB, Clubb Jr FJ, et al. Pathophysiologically relevant concentrations of tumor necrosis factor-alpha promote progressive left ventricular dysfunction and remodeling in rats. Circulation. 1998;97:1382–91.
Sivasubramanian N, Coker ML, Kurrelmeyer KM, et al. Left ventricular remodeling in transgenic mice with cardiac restricted overexpression of tumor necrosis factor. Circulation. 2001;104:826–31.
Bozkurt B, Torre-Amione G, Warren MS, et al. Results of targeted anti-tumor necrosis factor therapy with etanercept (ENBREL) in patients with advanced heart failure. Circulation. 2001;103:1044–7.
Anker SD, Coats AJ. How to RECOVER from RENAISSANCE? The significance of the results of RECOVER, RENAISSANCE, RENEWAL and ATTACH. Int J Cardiol. 2002;86:123–30.
Mann DL, McMurray JJ, Packer M, et al. Targeted anticytokine therapy in patients with chronic heart failure: results of the Randomized Etanercept Worldwide Evaluation (RENEWAL). Circulation. 2004;109:1594–602.
Dinarello CA, Wolff SM. The role of interleukin-1 in disease. N Engl J Med. 1993;328:106–13.
Last-Barney K, Homon CA, Faanes RB, Merluzzi VJ. Synergistic and overlapping activities of tumor necrosis factor-alpha and IL-1. J Immunol. 1988;141:527–30.
Kumar A, Thota V, Dee L, Olson J, Uretz E, Parrillo JE. Tumor necrosis factor alpha and interleukin 1beta are responsible for in vitro myocardial cell depression induced by human septic shock serum. J Exp Med. 1996;183:949–58.
Seta Y, Shan K, Bozkurt B, Oral H, Mann DL. Basic mechanisms in heart failure: the cytokine hypothesis. J Card Fail. 1996;2:243–9.
Prabhu SD. Cytokine-induced modulation of cardiac function. Circ Res. 2004;95:1140–53.
Shan K, Kurrelmeyer K, Seta Y, et al. The role of cytokines in disease progression in heart failure. Curr Opin Cardiol. 1997;12:218–23.
Terrell AM, Crisostomo PR, Wairiuko GM, Wang M, Morrell ED, Meldrum DR. Jak/STAT/SOCS signaling circuits and associated cytokine-mediated inflammation and hypertrophy in the heart. Shock. 2006;26:226–34.
Gwechenberger M, Mendoza LH, Youker KA, et al. Cardiac myocytes produce interleukin-6 in culture and in viable border zone of reperfused infarctions. Circulation. 1999;99:546–51.
Vasan RS, Benjamin EJ, Larson MG, et al. Plasma natriuretic peptides for community screening for left ventricular hypertrophy and systolic dysfunction: the Framingham heart study. JAMA. 2002;288:1252–9.
Long CS. The role of interleukin-1 in the failing heart. Heart Fail Rev. 2001;6:81–94.
Szabo-Fresnais N, Lefebvre F, Germain A, Fischmeister R, Pomerance M. A new regulation of IL-6 production in adult cardiomyocytes by beta-adrenergic and IL-1 beta receptors and induction of cellular hypertrophy by IL-6 trans-signalling. Cell Signal. 2010;22:1143–52.
Duprez DA. Role of the renin-angiotensin-aldosterone system in vascular remodeling and inflammation: a clinical review. J Hypertens. 2006;24:983–91.
Weber KT, Brilla CG. Pathological hypertrophy and cardiac interstitium. Fibrosis and renin-angiotensin-aldosterone system. Circulation. 1991;83:1849–65.
Samsonov M, Lopatin J, Tilz GP, et al. The activated immune system and the renin-angiotensin-aldosterone system in congestive heart failure. J Intern Med. 1998;243:93–8.
Baker KM, Booz GW, Dostal DE. Cardiac actions of angiotensin II: role of an intracardiac renin-angiotensin system. Annu Rev Physiol. 1992;54:227–41.
Kim S, Iwao H. Molecular and cellular mechanisms of angiotensin II-mediated cardiovascular and renal diseases. Pharmacol Rev. 2000;52:11–34.
Wada H, Zile MR, Ivester CT, Cooper GT, McDermott PJ. Comparative effects of contraction and angiotensin II on growth of adult feline cardiocytes in primary culture. Am J Physiol. 1996;271:H29–37.
Baker KM, Chernin MI, Wixson SK, Aceto JF. Renin-angiotensin system involvement in pressure-overload cardiac hypertrophy in rats. Am J Physiol. 1990;259:H324–32.
Izumo S, Lompre AM, Matsuoka R, et al. Myosin heavy chain messenger RNA and protein isoform transitions during cardiac hypertrophy. Interaction between hemodynamic and thyroid hormone-induced signals. J Clin Invest. 1987;79:970–7.
Izumo S, Nadal-Ginard B, Mahdavi V. Protooncogene induction and reprogramming of cardiac gene expression produced by pressure overload. Proc Natl Acad Sci USA. 1988;85:339–43.
Kvakan H, Kleinewietfeld M, Qadri F, et al. Regulatory T cells ameliorate angiotensin II-induced cardiac damage. Circulation. 2009;119:2904–12.
Phillips MI, Kagiyama S. Angiotensin II as a pro-inflammatory mediator. Curr Opin Investig Drugs. 2002;3:569–77.
Tsybouleva N, Zhang L, Chen S, et al. Aldosterone, through novel signaling proteins, is a fundamental molecular bridge between the genetic defect and the cardiac phenotype of hypertrophic cardiomyopathy. Circulation. 2004;109:1284–91.
Lu M, Zhang Q, Deng M, et al. An analysis of human microRNA and disease associations. PLoS ONE. 2008;3:e3420.
Lee Y, Kim M, Han J, et al. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004;23:4051–60.
Olsen PH, Ambros V. The lin-4 regulatory RNA controls developmental timing in Caenorhabditis elegans by blocking LIN-14 protein synthesis after the initiation of translation. Dev Biol. 1999;216:671–80.
Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993;75:855–62.
Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–40.
da Costa Martins PA, Bourajjaj M, Gladka M, et al. Conditional dicer gene deletion in the postnatal myocardium provokes spontaneous cardiac remodeling. Circulation. 2008;118:1567–76.
Rao PK, Toyama Y, Chiang HR, et al. Loss of cardiac microRNA-mediated regulation leads to dilated cardiomyopathy and heart failure. Circ Res. 2009;105:585–94.
Ikeda S, Kong SW, Lu J, et al. Altered microRNA expression in human heart disease. Physiol Genomics. 2007;31:367–73.
Tatsuguchi M, Seok HY, Callis TE, et al. Expression of microRNAs is dynamically regulated during cardiomyocyte hypertrophy. J Mol Cell Cardiol. 2007;42:1137–41.
van Rooij E, Sutherland LB, Liu N, et al. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci USA. 2006;103:18255–60.
Wang X, Zhang X, Ren XP, et al. MicroRNA-494 targeting both proapoptotic and antiapoptotic proteins protects against ischemia/reperfusion-induced cardiac injury. Circulation. 2010;122:1308–18.
Schroen B, Heymans S. MicroRNAs and beyond: the heart reveals its treasures. Hypertension. 2009;54:1189–94.
Haasch D, Chen YW, Reilly RM, et al. T cell activation induces a noncoding RNA transcript sensitive to inhibition by immunosuppressant drugs and encoded by the proto-oncogene, BIC. Cell Immunol. 2002;217:78–86.
Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci USA. 2006;103:12481–6.
Rodriguez A, Vigorito E, Clare S, et al. Requirement of bic/microRNA-155 for normal immune function. Science. 2007;316:608–11.
Tili E, Michaille JJ, Cimino A, et al. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J Immunol. 2007;179:5082–9.
Martin MM, Buckenberger JA, Jiang J, et al. The human angiotensin II type 1 receptor +1166 A/C polymorphism attenuates microrna-155 binding. J Biol Chem. 2007;282:24262–9.
Arola OJ, Saraste A, Pulkki K, Kallajoki M, Parvinen M, Voipio-Pulkki LM. Acute doxorubicin cardiotoxicity involves cardiomyocyte apoptosis. Cancer Res. 2000;60:1789–92.
Zhao YY, Feron O, Dessy C, Han X, Marchionni MA, Kelly RA. Neuregulin signaling in the heart. Dynamic targeting of erbB4 to caveolar microdomains in cardiac myocytes. Circ Res. 1999;84:1380–7.
Lu H, Buchan RJ, Cook SA. MicroRNA-223 regulates Glut4 expression and cardiomyocyte glucose metabolism. Cardiovasc Res. 2010;86:410–20.
van Rooij E, Sutherland LB, Thatcher JE, et al. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci USA. 2008;105:13027–32.
Fazi F, Racanicchi S, Zardo G, et al. Epigenetic silencing of the myelopoiesis regulator microRNA-223 by the AML1/ETO oncoprotein. Cancer Cell. 2007;12:457–66.
Johnnidis JB, Harris MH, Wheeler RT, et al. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature. 2008;451:1125–9.
Hashimi ST, Fulcher JA, Chang MH, Gov L, Wang S, Lee B. MicroRNA profiling identifies miR-34a and miR-21 and their target genes JAG1 and WNT1 in the coordinate regulation of dendritic cell differentiation. Blood. 2009;114:404–14.
Jazbutyte V, Thum T. MicroRNA-21: from cancer to cardiovascular disease. Curr Drug Targets. 2010;11:926–35.
Schmeier S, MacPherson CR, Essack M, et al. Deciphering the transcriptional circuitry of microRNA genes expressed during human monocytic differentiation. BMC Genomics. 2009;10:595.
Selcuklu SD, Donoghue MT, Spillane C. miR-21 as a key regulator of oncogenic processes. Biochem Soc Trans. 2009;37:918–25.
Thum T, Gross C, Fiedler J, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980–4.
Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci USA. 1995;92:7686–9.
Xiao C, Srinivasan L, Calado DP, et al. Lymphoproliferative disease and autoimmunity in mice with increased miR-17–92 expression in lymphocytes. Nat Immunol. 2008;9:405–14.
Baltimore D, Boldin MP, O’Connell RM, Rao DS, Taganov KD. MicroRNAs: new regulators of immune cell development and function. Nat Immunol. 2008;9:839–45.
Ventura A, Young AG, Winslow MM, et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell. 2008;132:875–86.
Ernst A, Campos B, Meier J, et al. De-repression of CTGF via the miR-17–92 cluster upon differentiation of human glioblastoma spheroid cultures. Oncogene. 2010;29:3411–22.
Izumiya Y, Kim S, Izumi Y, et al. Apoptosis signal-regulating kinase 1 plays a pivotal role in angiotensin II-induced cardiac hypertrophy and remodeling. Circ Res. 2003;93:874–83.
Schellings MW, Vanhoutte D, van Almen GC, et al. Syndecan-1 amplifies angiotensin II-induced cardiac fibrosis. Hypertension. 2010;55:249–56.
Acknowledgements
We acknowledge M.F. Corsten for critical reading of the manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
van de Vrie, M., Heymans, S. & Schroen, B. MicroRNA Involvement in Immune Activation During Heart Failure. Cardiovasc Drugs Ther 25, 161–170 (2011). https://doi.org/10.1007/s10557-011-6291-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10557-011-6291-y