
Abstract
Advanced and recurrent gynecological cancers lack effective treatment and have poor prognosis. Besides, there is urgent need for conservative treatment for fertility protection of young patients. Therefore, continued efforts are needed to further define underlying therapeutic targets and explore novel targeted strategies. Considerable advancements have been made with new insights into molecular mechanisms on cancer progression and breakthroughs in novel treatment strategies. Herein, we review the research that holds unique novelty and potential translational power to alter the current landscape of gynecological cancers and improve effective treatments. We outline the advent of promising therapies with their targeted biomolecules, including hormone receptor-targeted agents, inhibitors targeting epigenetic regulators, antiangiogenic agents, inhibitors of abnormal signaling pathways, poly (ADP-ribose) polymerase (PARP) inhibitors, agents targeting immune-suppressive regulators, and repurposed existing drugs. We particularly highlight clinical evidence and trace the ongoing clinical trials to investigate the translational value. Taken together, we conduct a thorough review on emerging agents for gynecological cancer treatment and further discuss their potential challenges and future opportunities.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Background
Gynecological cancers, particularly endometrial, cervical, and ovarian cancers, have a significant impact on women’s health, with increasing incidence and mortality worldwide. Their symptoms and prognoses, epidemiologic and genetic risk factors, and individual responses to clinical therapy are all diverse. Additionally, the current challenges for management of gynecological cancers are the urgent need for conservative treatments for fertility preserve, especially in endometrial cancer (EC) due to the increasingly younger onset age, and patients in advanced-stage or recurrent condition have limited therapy options [1]; thus, a novel understanding of molecular and cellular biology is required for improving and personalizing drug development.
EC begins in the inner epithelial lining of the uterus (endometrium) [2]. Overweight and unopposed elevated estrogen levels are well-known risk factors for EC. Endometrial cancers are classified into four subtypes based on their molecular characteristics [3]: polymerase-epsilon (POLE) ultramutation, microsatellite instability (MSI) cluster, a copy-number low, and a copy-number high, each of which reveals a unique prognosis [3, 4]. Although the majority of cases are diagnosed after menopause and can be cured by hysterectomy, an increasing number of patients are younger than 40 years old, and most are nulliparous [5]. For patients in advanced stage and those who desire to protect future fertility or preserve their ovaries, there are fewer feasible treatments, which makes EC management challenging [6].
Cervical cancer (CC) is the fourth most common malignancy and the fourth main cause of cancer-related death among women, with an estimated 60,4000 new cases and 34,2000 deaths globally in 2020. The molecular pathogenesis of malignant CC is certainly influenced by exposure to human papillomavirus (HPV), which introduces related viral oncoproteins (E6, E7, and E5) and induces angiogenesis, DNA damage, dysfunction of the immune system, and epigenetic factors. Accordingly, HPV testing and HPV vaccines have been applied to screen and prevent CC. Recurrent and metastatic disease continues to be the leading cause of CC-related mortality, despite the fact that surgery, chemotherapy, and radiation therapy can cure approximately 90% of patients with early-stage cancer [7].
The 5-year survival rate for ovarian cancer (OC) has remained constant at 47% for the past 20 years, making it the deadliest gynecological malignancy. It is important to diagnose OC early, but only 15% of cases are diagnosed at an early stage. The standard treatments for OC include debulking surgery and platinum-based chemotherapy. The majority of patients will relapse within 3 years, despite the high response rate of the first-line treatment [8]. Upon first relapse, up to 25% of patients are platinum-resistant or platinum-refractory, and the response rates of single non-platinum agents (paclitaxel, docetaxel, pegylated liposomal doxorubicin (PLD), gemcitabine, and topotecan) and prognosis are disappointing [9, 10]. Thus, the deeper understanding of the underlying molecular mechanisms that contribute to cancer growth and chemoresistance is crucial to conduct new drugs and promising strategies for OC treatments [11].
In this paper, we summarize the outcomes of preclinical and clinical trials in endometrial, cervical, and ovarian cancers and review the Food and drug Administration (FDA)-approved drugs and the promising agents for gynecological cancer therapy (Fig. 1). We further describe their underlying molecular targets or mechanisms, as well as future directions.

Schematic illustration of pathogenesis and therapeutic targets in gynecological cancers (created by BioRender.com)
2 Methods
We thoroughly reviewed the literatures of promising agents and their clinical efficacies on gynecological cancers. To this end, published reviews, meta-analyses, clinical trials, and other observation studies were searched by PubMed. The information of the associated clinical trials which were completed or ongoing was collected from ClinicalTrials.gov. Based on FDA approval requirements, each approved targeted drug’s indications and references were searched on the website. The following terms were searched: gynecological cancers, ovarian cancer, cervical cancer, endometrial cancer, targeting agents, antiangiogenic agents, poly (ADP-ribose) polymerase (PARP) inhibitors, epigenetic inhibitors, immune checkpoint inhibitors, and each name of the targeted agents (e.g., “bevacizumab,” “pembrolizumab”).
3 Hormone therapeutic strategies
The human endometrium thickens and renews itself to prepare for nourishing an embryo under the dynamic fluctuation of estrogen and progesterone [12,13,14]. However, hormone imbalance, either unopposed estrogen stimulation or insufficient progesterone conditions can result in endometrial pathologies, such as endometrial hyperplasia and endometrial cancer [15]. Obesity and excess exposure to hormone treatments are thought to be the main contributors of hormone imbalance in EC [16,17,18].
To counteract estrogen-induced endometrial proliferation, hormone therapies, including tamoxifen, levonorgestrel intrauterine device (IUD), and progestin (medroxyprogesterone acetate (MPA) and megestrol acetate (MA)), are prescribed for the adjuvant treatment of EC, the reversal of endometrial hyperplasia and the treatment of nulliparous women with low-grade EC [19]. By targeting specific receptors, progestin-mediated responses can impact the functions of numerous genes, such as cyclin D1, Ets-1, and FOXO1, and the activity of MMP proteases, which promote cell cycle arrest and apoptosis in EC cells [20]. The levonorgestrel IUD provides consistent localized progestin exposure, and it showed substantial activity in grade 1 endometrioid endometrial carcinoma in a prospective phase II trial, with minimal adverse effects and modest upfront progesterone resistance [21].
However, with the occurrence of receptor deficiency or drug resistance, the relapse rates of progesterone-treated EC are high [22]. It was reported that MPA treatment did not completely eradicate a carcinomatous lesion, which remained during and after a term pregnancy; therefore, these fertility-preserving options may be temporizing measures [23].
Several hormone therapeutic strategies have been investigated in recent years. The aromatase enzyme is responsible for converting androgen to estrogen and presents in 33–81% of ovarian tumor tissues [24]. Accordingly, aromatase inhibitors (letrozole and anastrozole) have received growing attention as therapeutic strategies against gynecological cancers [25, 26]. Letrozole exerts great antitumor effects and is well tolerated in patients with recurrent low-grade or borderline OC [27]. Letrozole combined with ribociclib (a cyclin kinase inhibitor) is safe and effective in patients with estrogen receptor (ER)-positive OC and EC, particularly in those with low-grade serous OC or EC, which represents a promising treatment option (NCT02657928) [28]. Recently, in a phase II study (NCT03675893), abemaciclib, a CDK4/6 inhibitor, in combination with letrozole exerted promising and durable antitumor efficacy in ER-positive EC patients with recurrent condition [29]. Anastrozole exhibits promising efficacy when combined with mTOR inhibitor vistusertib in hormone receptor-positive recurrent or metastatic EC patients (NCT02730923) [30].
Gonadotropin-releasing hormone (GnRH) signal transduction and intracellular actions are also involved for gynecological cancer therapy. GnRH is the central neuropeptide released from neurons in the hypothalamus and induces the synthesis and secretion of luteinizing hormone (LH) and follicle-stimulating hormone (FSH) from the anterior pituitary gland [31]. GnRH agonists (GnRH-as) are recommended in the treatment for OC and EC because of the critical role it plays in cell proliferation and metastasis. In EC, GnRH-as cause a reduction in cell proliferation by inhibiting epidermal growth factor receptor (EGF-R) signal transduction via extracellular signal-regulated kinase 1/2 (ERK1/2) or phosphoinositide 3-kinase (PI3K)/AKT pathway. GnRH also modulates apoptosis by activating nuclear factor-kappa B (NF-κB) or Fas ligand, and GnRH-as can activate the c-Jun N-terminal kinase (JNK)/activator protein-1 (AP-1) pathway, resulting in an increase of G0/1-phase cells and decreased DNA synthesis [32]. Furthermore, several studies have reported that patients treated with GnRH-as showed a significantly higher rate of implantation and clinical pregnancy than those treated with GnRH antagonists (GnRH-ants), while the mechanisms were unclear [33, 34].
Recently, androgen has been reported to be a new factor regulating squamous differentiation involved in the early progression of cervical intraepithelial neoplasia (CIN), suggesting a novel nonsurgical hormone-induced differentiation therapy could be used against CIN1 and CIN2 [35].
4 Targeting epigenetic modifiers in gynecological cancers
Epigenetics is defined as heritable gene expression changes that do not alter the underlying DNA sequence. The epigenetic modifications mainly include histone methylation and acetylation, and DNA methylation [36]. In recent decades, the disturbances in epigenome resulting from epigenetic dysregulation have been proved to play a crucial role in the development and metastasis of a variety of cancers. Therefore, reprogramming the cancer-associated epigenome landscape is one of the most attractive targetable therapies in both reversing and treatments for a variety of malignancies [37]. Multiple inhibitors targeting epigenetic modifiers have been applied for cancer treatment for decades, predominantly in hematological disorders. At present, there are several candidate drugs targeting epigenetic-modified enzymes, typically DNA methyltransferase inhibitors (DNMTis) and histone deacetylase inhibitors (HDACis), entering clinical research, potentially providing new treatment approaches for patients with gynecologic cancers (Fig. 2) [38].

The conceptual diagram illustrating the functions and inhibitors of key epigenetic modifiers in gynecological cancers. DNA writers, such as DNMTs (DNA methyltransferases) add methyl groups (Me) and can be inhibited by 5-AZA (5-azacitidine), RG108, and decitabine. Histone writers, HMTs (histone methyltransferases) and HDMs (histone demethylases), add Me or acetyl groups (Ac) and can be inhibited by GSK343, GSK126, SP-2577. Histone erasers, HDACs (histone deacetylases), remove Ac and can be inhibited by approved drugs, including entinostat, VPA (valproic acid), apicidin, SAHA (suberoylanilide hydroxamic acid), belinostat and vorinostat. Histone readers such as the BET proteins can recognize acetylated lysine residues on histone tails, and they can be inhibited by R06870810 (created with BioRender.com)
4.1 DNA methylation events in gynecological cancers
DNA methyltransferases (DNMTs), such as DNMT1, DNMT3a, and DNMT3b, mediate DNA methylation in CpG islands [39], and aberrant DNA methylation is associated with endometrial tumorigenesis, induced by the change of gene transcription, including DNA repair factors, tumor suppressors and steroid receptors [40]. Interestingly, both type I and II EC exhibit aberrant DNA methylation profiles. In type I cancer cells, DNMT1 and DNMT3b were preferably upregulated, but downregulated in type II cancer cells [40]. In a retrospective study, elevated methylation of hMLH1 and O6-MGMT, genes relating to DNA mismatch repair (MMR), were reported in atypical endometrial hyperplasia and continuously increased in tumor tissues, which seems to be an early event in EC carcinogenesis [41]. In addition, gene methylation, especially in ADCYAP1 and HAND2, can be identified prior to the diagnosis of EC [42]. Hypermethylated PCDHGB7 has been identified as a new cancer marker and is applicable in early screening for EC and CC [43]. Thus, abnormal DNA methylation could serve as a promising indicator for the detection of EC and CC.
In cervical carcinogenesis, alterations in DNA methylation pattern both affect the expression of persistent oncogenic HPV and disrupt cell cycle control, through which the epithelial host cells acquire immortal and malignant phenotype and further progress to invasive stage [44]. The fragile histidine triad, which acts as a negative cell growth regulator, is significantly downregulated in cervical neoplasia due to gene promoter hypermethylation [45, 46]. Transcription inhibition of the pro-apoptotic factor death-associated protein kinase (DAPK1) has also been reported in most cervical cancers [47, 48]. Similarly, cyclin A1 (CCNA1) promoter hypermethylation, probably induced by the infection of HPV, is common in CC and the decreased gene expression is specific to the invasive phenotype [49, 50], suggesting a potential role of CCNA1 for early diagnosis of invasive CC. Hypermethylated PCDHGB7 was also found in CC, which can be applied for early cancer screening [51].
A variety of genes were identified to be hypermethylated in the evolution of CC [52,53,54,55,56,57,58,59,60]. Ras association domain family 1 isoform A (RASSF1A) is a key regulator of apoptosis [61], and its gene promoter is uniquely hypermethylated in HPV-negative CC cell lines, but not in HPV-positive or primary cervical tumors [62]. In addition, RASSF1A promoter methylation was found in squamous cell carcinomas (10%), adenocarcinomas (20%), and adenosquamous carcinomas (45%), indicating that silenced RASSF1A may be involved in cervical adenocarcinoma progression [63, 64].
As for OC, aberrant DNA methylation is a contributing factor for tumor development and metastasis, chemotherapy resistance, and the survival of cancer stem cells [36]. It is possible to pinpoint specific methylated loci linked to poor progression-free survival (PFS) by comparing the level of aberrant methylation and the number of hypermethylated loci in OC, both of which are directly correlated with the progression and recurrence of ovarian tumors [65, 66]. Based on platinum-resistant DNA methylation signature, epigenetic therapies may reverse the transcriptional suppression that results in chemoresistance and restore sensitivity to platinum-based chemotherapeutics [67]. Homozygous methylation of BRCA1 was viewed as a vigorous indicator of reaction to PARP inhibitors (PARPis) in an Ariel 2 clinical test of rucaparib. Furthermore, patterns of blood DNA methylation have been recently connected to prognosis of OC patients. Taken together, methylation markers in OC might be helpful for evaluation of therapeutic effects and recognition of chemoresistance-related pathways [68,69,70,71].
4.2 Histone modification events in gynecological cancers
Histone methylation is regulated by histone methyltransferases (HMTs) and histone demethylases (HDMs). Methylation can occur both on the lysine residues (mono-, di-, or tri-methylation) and arginine residues (mono- or di-methylation) of histones. The dysregulation of these complicated methylation degrees was reported to be involved in gynecological cancers; thus, the modulators of histone-modifying enzymes have been in the forefront in gynecological cancer research because abnormal histone modification alters gene expression and may have adverse clinical effects [72].
Histone acetylation is regulated by the dynamic balance of histone acetyltransferases (HATs) and histone deacetylases (HDACs), which determines chromosome accessible or inaccessible to transcription factors and thus influences gene expression [73]. Two principal superfamilies of HATs have been recognized: the GNAT and MYST families, by which acetyl groups from lysine residues were added at the histone N-terminal tails [73]. The four classes of HDACs—class I (HDAC1, 2, 3, and 8), class II (HDAC4, 5, 6, 7, 9, and 10), class III (SIRT1, 2, 3, 4, 5, 6, and 7), and class IV (HDAC11)—are responsible for removing acetyl groups [74]. In addition, acetylation/deacetylation of histones influences the gene expression of nonhistone proteins, which are also involved in tumorigenesis, tumor progression, and metastasis [75, 76]. Thus, HDACis may be promising therapeutic targets for gynecological cancers.
HATs in EC are rarely reported, while numerous studies have shown that histone deacetylation causes tumor suppressor gene silencing and contributes to malignant transformation. Histone acetyltransferase MOF is known to regulate ER activity and maintain ER stability, which suppresses EC progression. The levels of HDACs (HDAC1, HDAC2, and HDAC3) are elevated in EC compared to normal endometrium, which have been reported to be related to a poor prognosis. On the contrary, higher SIRT1 level in EC is positively correlated with a better prognosis [77, 78].
For CC, elevated expressions of HDAC1 and HDAC2 were observed in both dysplasia and invasive carcinomas of cervical tissues. Overexpression of HDAC8 was reported in HeLa cells. Histone acetylation mainly affects crucial signaling pathways associated with CC progression by modulating the expression of key genes via acetylation/deacetylation. Loss of MGMT is linked to a decreased level of acetylated histones, which affects DNA repair in CC [79]. In HeLa cells, DICKKOPF-1 (DKK-1) is transcriptionally suppressed by histone deacetylation and inhibits the Wnt signaling pathway, which is crucial for HPV-infected cervical cells to undergo clonal proliferation at the early stage of malignancy [80]. It has been reported that the HPV E7 oncoprotein prevents HDACs from interacting with HIF-1 and results in HIF-1-dependent transcription [81].
The histone modification is essential for the tumorigenesis and progression in OC. Class I HDACs are elevated during ovarian carcinogenesis and are independent prognostic factors for malignant ovarian tumors. Compared with normal ovarian tissues, SIRT1 is significantly elevated in malignant ovarian tumors [82], and it was described to induce chemoresistance and correlate with poor prognosis in OC [83, 84].
4.3 Interaction between DNA methylation and histone modification in gynecological cancers
Gynecological malignancies exhibit interaction between DNA methylation and the histone modification. In CC cell lines, aberrant histone modification and DNA methylation are responsible for silenced protein osteoprotegerin (OPG) [85], and retinoic acid receptor beta2 (RAR beta2) is epigenetically silenced either by DNA hypermethylation or repressive histone modification [86]. Liu et al. have demonstrated that HDAC1 and DNMT3a are linked to the suppression of octamer-binding transcription factor 4 (Oct4) in CC cells [87]. In EC, DICER1 regulates tumor invasion via histone acetylation and methylation [88]. These studies propose the possibility of combination treatment with DNMTis and HDACis for gynecological cancers.
4.4 Emerging epigenetic targets for gynecological cancer treatment
4.4.1 Inhibitors targeting DNMTs, HMTs and HDMs, and HDACs in endometrial cancer
Epigenetic dysregulations, especially aberrant DNA methylation and histone modification, have been reported in the development and progression of EC, and several inhibitors targeting epigenetic regulators are considered to be effective against EC by a few preclinical studies [89]. Two emerging DNMTis have been investigated in EC cell lines. 5-azacytidine (5-AZA) suppressed EC cell proliferation through downregulating of cyclin D1 and β-catenin, while RG108 induced apoptosis of EC cells by demethylating the MMR gene hMLH1 [90].
Inhibitors targeting HMTs and HDMs mainly include DOT1L inhibitors, EZH2 inhibitors, and LSD1 inhibitors, among which EZH2 inhibitors and LSD1 inhibitors exert therapeutic potential in EC. Studies demonstrated that EZH2 selective inhibitors significantly inhibited cell growth, of which GSK343 upregulated miR-361 and decreased Twist expression, and GSK126 induced apoptosis [91, 92]. A phase I clinical trial (NCT04611139) investigated the synergetic effect between LSD inhibitor (SP-2577) and pembrolizumab, an anti-programmed death 1 (PD-1) antibody on EC patients in 2020, however, it was withdrawn maybe due to severe toxicities.
Although more research has been done on histone acetylation than on histone methylation in EC, only one phase I clinical trial was initiated for EC treatment (NCT03018249) evaluating the therapeutic effect of combination with HDACi entinostat (MS-275) and hormonal therapy (MPA). Base on the knowledge that the expression level of progesterone receptor (PR) in the endometrium is positively related to MPA responsiveness, and epigenetically silenced PR resulted from histone modification could promote MPA resistance, the trial enrolled 22 patients in the MPA group and 20 in the entinostat/MPA group. Despite the fact that entinostat had no detectable impact on PR expression in this short period, the significantly decreased expression level of Ki-67 in the combination group compared to MPA group suggests entionstat exerts a synergetic effect with MPA in EC patients and this novel finding provides premise for progressing the combination strategy between entionstat and MPA towards a treatment trial [93].
4.4.2 Inhibitors targeting DNMTs and HDACs in cervical cancer
Hydralazine is a DNMTi that was well tolerated in clinical trial and plays a role in demethylating and reactivating several tumor suppressors in patients with CC [94]. Several HDACis have been reported to exert powerful anticancer effects in CC. Valproic acid (VPA), a potent HDACi, promotes apoptotic cell death by downregulation of Akt1, and it also hyperacetylates p53, which then increases p53 activity by preventing it from degradation by oncogenic HPV protein [95, 96]. In CC cells, the HDAC inhibitor Apicidin could preferentially downregulate DNM1 expression and induce repressive histone modifications by recruiting corepressor complex [97]. In HeLa cells, suberoylanilide hydroxamic acid (SAHA) synergistically causes apoptosis when treated with the proteasome inhibitor bortezomib through activation of caspase-3 and raising the bax/bcl-2 expression ratio [98].
4.4.3 Clinical trials of inhibitors targeting epigenetic modulators in ovarian cancer
DNMTis such as 5-AZA and decitabine, have been validated to be effective in regaining platinum sensitivity in platinum-resistant OC [99]. Decitabine performed better than 5-AZA to improve the responsiveness with a 35% objective response rate (ORR), a 10.2-month PFS, and the significant demethylation of tumor suppressor genes MLH1, RASSF1A, HOXA10, and HOXA11 [100].
Belinostat, an HDACi, was once given to patients with platinum-resistant OC with no therapeutic results because of the termination due to serious side effects including neutropenia, thrombocytopenia, and vomiting [101]. Similarly, despite the partial response seen in clinical trial, vorinostat, a pan-HDACi, also caused severe hematological toxicities when treated with carboplatin or gemcitabine, resulting in the study’s cancelation [102].
Accordingly, clinical trials of epigenetic therapies in single-agent regimen have proved unsatisfactory for ovarian cancer treatment, so the focus of preclinical research has been shifted to the combination of various epigenetic medications. To improve anticancer therapy and overcome drug resistance for OC, epigenetic strategies have been initially combined with standard treatments in a few clinical trials.
The synergy efficiency of DNMTi and HDACi combination can be explained by the augmentation activity of HDACis in regulating chromatin accessibility and the recovery of abnormally silenced genes induced by DNMTis. In a xenograft model, the combination of decitabine and belinostat induced more platinum responsiveness in OC than either drug used alone [103].
It is generally acknowledged that combination therapy reduces drug toxicity in part because a low administered concentration has equivalent antitumor effects. However, in platinum-resistant epithelial ovarian cancer (EOC), the combination of 5-AZA, VPA, and carboplatin was extremely toxic, which resulted in the early termination of the study because 80% of participants experienced grade 3 or higher adverse effects, such as vomiting, neutropenia, and fatigue [104]. The severe toxicity profiles of 5-AZA and VPA may be because their targets span the epigenome, which emphasizes the necessity of more specific epigenetic modulators. Although the above clinical outcomes are less satisfactory, combined therapy is still a desirable treatment option, as many patients showed substantial and long-lasting clinical responses in non-small cell lung cancer when DNMTis and HDACis were combined [103].
Immunotherapy combined with DNMTis and/or HDACis enables the immune system to attack tumor cells unrestrainedly. In a syngeneic OC mouse model, combining decitabine and anti-CTLA-4 therapy dramatically inhibited tumor progression and extended survival when compared to either drug used alone [105]. Decitabine could stimulate the production of chemokines and attract CD8 and natural killer (NK) cells to the tumor microenvironment, which prolonged the cytotoxic lymphocyte response and then increased mouse survival. Epigenetic treatments combined with immune checkpoint inhibition have been tested in clinical trials based on the preclinical findings. Patients on a DNMTi combined with a HDACi mounted a potent and long-lasting response after receiving immune checkpoint therapy in non-small cell lung cancer, which supported the utility of triple combination therapy in solid tumors [106].
The combination therapy of entinostat (HDAC1/3 inhibitor) and avelumab, an antibody targeting programmed death ligand 1 (PD-L1), is now being tested in patients with chemo-resistant EOC, and the preliminary results are encouraging (NCT02915523). According to Odunsi et al., DNA methylation inhibits the expression of the cancer testis antigen NY-ESO-1. Combining a vaccination against this antigen with decitabine and doxorubicin induced a partial clinical response in six out of ten patients with recurrent EOC [107].
Other epigenetic modulators besides DNMTs and HDACs that are being studied in clinical trials include the histone lysine methyltransferase EZH2 and BET proteins which comprise BRD2, BRD3, BRD4, and BRDT and contain bromodomains that identify acetylated lysine residues on histone tails [108, 109]. BET inhibition suppresses the expression of MYC, an oncogene whose expression is favorably controlled by BRD4. BRD4 is typically overexpressed in OC and is linked to a poor prognosis. Cell cycle arrest is brought on by BET inhibition, which also prevents tumor growth [108]. BET inhibitors increase the DNA damage caused by PARP inhibition in cancer cells, mainly through lowering homologous recombination [110]. A phase Ib clinical trial combining RO6870810 with atezolizumab is also being carried out to evaluate the BET inhibitor and anti-PD-L1 therapy in advanced OC patients (NCT03292172).
4.5 Other promising molecules involved in epigenetic regulation
4.5.1 MicroRNA-related epigenetic mechanisms in endometrial cancer
microRNAs (miRNAs) have drawn substantial interest in EC, from diagnostics and pathophysiology to therapeutics. miRNA-related epigenetic mechanisms can be summarized in three patterns: (1) miRNAs have the ability to directly bind to and silence target genes, serving as oncomiRNAs or tumor suppressors (such as miR-182 and miR-230); (2) CpG-rich regions in miRNA loci can be hypo- or hypermethylated, which leads to increased or decreased expression of these miRNAs, respectively (such as miR-34b and miR-129–2); and (3) miRNAs can increase/decrease the methylation of target genes (such as miR-30d and miR-191) [111]. In addition to their regulation of malignant cell phenotypes, miRNAs are also involved in chemosensitivity and angiogenesis in endometrioid EC [112, 113]. Through exosomal delivery, miRNAs (miRNA-21, miR-26a/b-5p) also mediate cellular communication between cancer cells and stromal or immune cells, which may be a potential mechanism underlying the construction of the immune microenvironment in EC progression [114, 115]. miR-26a-5p have also been demonstrated to contribute to lymph node metastasis, suggesting a specific target for EC therapies [116]. Due to their extensive implications in cancer progression, miRNA analysis has been explored as a promising factor in the management of patients with EC[117].
4.5.2 m6A modification in endometrial cancer
As the most common epigenetic modification of messenger RNA (mRNA), N6-methyladenosine (m6A) affects the pathogenesis of many diseases, including a wide range of cancers [118]. m6A modifications also intrinsically regulate tumor immunogenicity and modulate immune cells implicated in anti-tumor responses. Its dysregulation promotes cancer occurrence and development by driving aberrant transcription and translation programs, and immune cell responses in tumor cell are also affected by m6A alterations in the tumor microenvironment [36]. He Chuan et al. demonstrated that reduced m6A mRNA methylation is an oncogenic factor in EC [119]. Endometrial cancer cells proliferate and become more tumorigenic as a result of a mutation in METTL14 (R298P), a crucial part of the methyltransferase complex, which activates the AKT pathway [119]. Demethylation of m6A modifications by FTO and ALKBH5 promotes EC metastasis and invasion through the activation of Wnt and IGF1R signaling pathways, respectively [120, 121]. YTHDF2 is an m6A reader protein that promotes IRS1 mRNA degradation and inhibits cell proliferation and invasion by weakening IRS1/AKT signaling in EC [122]. These results suggest a protective role of m6A against EC progression. In liver cancer cells, YTHDF2 is positively correlated with the stem cell phenotype and cancer metastasis [123]. Insulin-like growth factor 2 mRNA-binding protein 1 (IGF2BP1), another “reader” of m6A sites in the 3′UTR of mRNA, enhances the mRNA stability of PEG10 and SOX2, thereby promoting cell cycle progression and tumor progression in EC [124, 125]. Thus, m6A modification may be a "double-edged sword" in endometrial tumorigenesis, and therapies targeting m6A should be thoroughly investigated before application.
5 Angiogenesis and antiangiogenic agents
Angiogenesis is crucial for the development of tumors and the evolution of gynecological cancers in the female reproductive system. The understanding of vascularization in tumor growth has facilitated antiangiogenic therapy for gynecological cancers. Unfortunately, in a small subset of patients, antiangiogenic drugs have shown minimal clinical success, and despite initial benefit, many patients eventually acquire resistance to these drugs. Due to this inadequate efficacy, it is urgently necessary to identify new methods for controlling tumor vascularization and enhancing patient survival in ongoing preclinical investigations and clinical trials.
5.1 The role of angiogenesis in gynecological cancers
5.1.1 Angiogenesis in endometrial cancer
As successful implantation and pregnancy depend on angiogenesis, the human endometrium shows stronger angiogenic potential than other female reproductive tract tissues. Due to an increase of proangiogenic and downregulation of antiangiogenic molecules, angiogenesis control is lost in CC. Furthermore, increased microvessel density (MVD) is positively correlated with aggressive phenotypes of tumor. The association between angiogenesis and estrogen signaling is a distinctive feature of EC, in addition to alterations of traditional angiogenic biomarkers in other tumors, including vascular endothelial growth factor (VEGF) and its receptors, hypoxia-inducible factor-1α (HIF-1α), fibroblast growth factor (FGF) and other angiogenic factors [126]. Previous studies have showed that estrogen promotes angiogenesis by inducing production of VEGF-A and the AKT, NF-κB or HIF-1 signaling are responsible for elevated VEGF levels. Through platelet-activating factor (PAF)-driven NF-κB activation and phospholipase A2 (PLA2) production, estrogen also promotes endometrial angiogenesis [126,127,128]. However, there is no evidence supporting the usage of estrogen signaling as a prognostic or predictive biomarker for EC.
5.1.2 Angiogenesis in cervical cancer
Angiogenesis is relevant in both premalignant cervical lesions and invasive CC. High vascularity is correlated with the poor prognosis of CC, and MVD is directly associated with VEGF expression [129]. HPV oncoproteins E6 and E7 have significant impacts on the angiogenesis of CC. E7 stimulates HIF-1 and inactivates the tumor suppressor protein retinoblastoma, and E6 promotes the degradation of p53, which are responsible for angiogenesis activation through the upregulation of VEGF [130, 131]. Furthermore, VEGF genetic polymorphisms influence cancer susceptibility and survival in early stage of CC through regulation of tumor angiogenesis [132,133,134,135].
5.1.3 Angiogenesis in ovarian cancer
EOCs are typically strongly vascularized, and the peritoneal vasculature is thick towards carcinomatosis, while the artery circulation is poor, which accelerate the development of edema and inflammation [136]. As the major subset of stromal cells in a number of human malignancies, cancer-associated fibroblasts (CAFs) can contribute to vascular stabilization in EOCs [137]. CAFs promote angiogenesis through activation of the tumor-derived proangiogenic growth factors, platelet-derived growth factor (PDGF), VEGF, FGF, and transforming growth factor-beta (TGF-β), and by secreting stromal cell-derived factor-1 (SDF-1), which draws endothelial progenitor cells to the tumor stroma [138].
Ovarian cancer stem cells (CSCs) also play a crucial role in the process of angiogenesis in malignancies [139, 140]. During hypoxia, the CSCs-derived angiogenesis serves as an alternative to sprouting angiogenesis, which arises from neighboring normal blood vessels and the interactions between ovarian CSCs. Besides, angiogenesis via VEGF, Wnt, Notch, and Sonic hedgehog signaling results in vascular cooperation, leading to metastasis of the tumor cells [141,142,143]. The anti-VEGF antibody bevacizumab significantly increased disease-free survival in both primary and recurrent OCs in clinical trials; hence drugs targeting CSCs that can enhance chemotherapy response and prevent recurrence will be the way forward [140].
5.2 Clinical research of anti-angiogenic therapy
Bevacizumab monotherapy was investigated in a phase II clinical trial for patients with persistent or recurrent EOCs. The results showed a 21% clinical response rate, 4.7-month median PFS, and 17-month overall survival (OS), with favorable tolerability [144].
In addition, combination therapy of bevacizumab and chemotherapy was performed by numerous clinical trials. Patients who were treated with bevacizumab and chemotherapy (topotecan/paclitaxel or cisplatin/paclitaxel) had prolonged OS (17 versus 13.3 months), a longer PFS (8.2 versus 5.9 months), and a higher response rate (48% versus 36%) than those treated with chemotherapy alone, according to the Gynecologic Oncology Group (GOG)-240 phase III trial [145]. Accordingly, FDA approved bevacizumab plus chemotherapy as a combination therapy for metastatic and recurrent CC in 2014 [145, 146].
The addition of bevacizumab to carboplatin and paclitaxel chemotherapy followed by extended bevacizumab therapy (GOG-218 trial) in early diagnosed advanced EOC patients significantly prolonged the median PFS by 3.8 months compared to the chemotherapy plus placebo group. Meanwhile, this combination did not reduce patients’ quality of life [147]. Based on these findings, bevacizumab combined with platinum and paclitaxel has been approved by the European Medicine Agency (EMA) as a first-line chemotherapy regimen for OC.
The International Collaborative Ovarian Neoplasm (ICON) 7 trial showed a lower dose and a shorter maintenance period of bevacizumab in patients with OC. Compared with standard therapy (carboplatin and paclitaxel) group, patients assigned carboplatin-paclitaxel-bevacizumab regimen showed a better PFS, especially for those at high risk for progression (14.4 months vs. 18.1 months; P = 0.002). Additionally, patients with a poor prognosis had prolonged OS [148, 149]. This might be explained by the greater demand for angiogenesis in patients with high-risk progression, and the above combination strategy also prolonged the median PFS and OS in platinum-sensitive recurrent OC [150]. The combination of bevacizumab plus gemcitabine and carboplatin followed by bevacizumab maintenance therapy also led to significant improvement in PFS, ORR, and duration of response (DOR) in patients with platinum-sensitive recurrent EOC [151]. Based on these findings, both combination regimens were approved for platinum-sensitive recurrent OC. However, compared with carboplatin-paclitaxel, the treatment for advanced and recurrent EC with carboplatin-paclitaxel-bevacizumab did not significantly improve PFS in a randomized phase II trial (the MITO END-2 experiment) [152, 153].
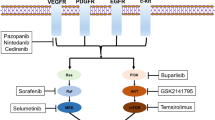
Pazopanib and lapatinib are tyrosine kinase inhibitors (TKIs) that target VEGFR and PDGFR or EGFR and HER/neu, respectively. In a phase II clinical trial, pazopanib or lapatinib monotherapy or pazopanib-lapatinib combined regimen was given to patients with primary stage IVB or recurrent CC. When compared with lapatinib group, patients received pazopanib had significantly longer PFS (17.1 vs. 18.1 weeks; P = 0.013) and OS (39.1 vs. 50.7 weeks; P = 0.045). However, the combination therapy was low efficiency and more toxic than monotherapy [154]. As for the usage of pazopanib monotherapy in EC treatment, a case report showed that a 57-year-old patient with recurrent metastatic EC responded favorably to pazopanib monotherapy [155]. A phase III trial (OVAR-16) testing pazopanib maintenance therapy for OC patients whose disease did not progress during first-line chemotherapy showed a significant improvement in the median PFS, while there was no benefit in OS [156]. A phase II trial (MITO 11) enrolled patients with platinum-resistant or platinum-refractory advanced OC and found that PFS was greatly longer in the pazopanib plus paclitaxel group than that in paclitaxel group (6.35 months vs. 3.49 months; P = 0.0002), and that the adverse events, such as neutropenia, fatigue, leucopenia, and hypertension, were more common in the combination group [157].
Nintedanib is an oral triple angiokinase inhibitor of VEGFR, PDGFR, and EGFR. In newly diagnosed advanced OC patients, the phase 3 trial AGO-OVAR 12 investigated the combination therapy of nintedanib with the standard treatment (carboplatin and paclitaxel). The results showed that patients who received carboplatin-paclitaxel-nintedanib therapy had longer PFS than patients from the carboplatin-paclitaxel-placebo group [158].
Cediranib is an oral antiangiogenic VEGFR1/2/3 inhibitor. According to a phase 3 trial (ICON6), when combined with chemotherapy and continued as maintenance therapy, cediranib greatly increased PFS in recurrent platinum-sensitive OC patients, albeit with added toxic effects, which was similar with the PFS benefit of bevacizumab observed in the GOG-0213 and OCEANS trials [159]. Considering that TKIs are commonly multitarget inhibitors, the balance between increased toxicity and clinical benefits needs to be further investigated in future trials.
A single-arm clinical trial (AMG 386; IND#111,071) of 32 patients with persistent/recurrent EC assessed anti-angiopoietin therapy with trebananib, a peptibody that selectively neutralizes angiopoietin 1/2. The OS and PFS were 2.0 and 6.6 months, respectively. Eight patients exhibited stable progression, five had 6-month event-free survival, and one patient displayed a partial response. Unfortunately, trebananib monotherapy has no obvious efficiency [160].
6 Therapeutic agents targeting abnormal signaling pathways
Tumor-intrinsic signaling pathways that drive cancer initiation and progression provide intriguing targets for antitumor therapies. Due in part to the upregulated expression of drug transporters and reprogramming of the resistant microenvironment, CSCs are responsible for metastasis, therapy resistance and cancer relapse [161, 162]. In EC, stemness and cell fate are tightly modulated by abnormal signaling pathways, such as Wnt/β-Catenin, Notch, and PI3K/AKT/mTOR signaling [163, 164]. Dysregulations of components in these cascades have also been identified in several OC subtypes [165], and the stemness of ovarian CSCs is directly regulated by PI3K/PTEN/AKT pathway, which causes enrichment and phenotype maintenance of CSCs, as well as promotes multidrug resistance [166]. Currently, in gynecological malignancies, several molecules have been targeted for promising therapeutic strategies, including HER2, PI3K/mTOR, and other signaling targets.
6.1 HER2-targeted inhibitors
Targeting HER2 has been established as a therapeutic strategy for large subsets of breast cancer patients. Accordingly, trastuzumab, pertuzumab, lapatinib, neratinib and trastuzumab emtansine (T-DM1) have been approved for the treatment of HER2-positive breast cancers [167].
High gene amplification and/or protein expression of HER2 is also involved in EC, especially in serous carcinomas, which provides an opportunity for targeted therapy [168, 169]. Although no HER2-targeting agents have been approved for gynecological cancer treatment, a number of translational studies and clinical trials are in progress, which may alter the paradigm of treating gynecologic cancers that express HER2 [170].
Pertuzumab is a monoclonal antibody against HER-mediated signaling and tolerated in relapsed OC patients, with response rate (RR) of 4.3% [171]. For advanced, platinum-resistant EOC patients, the combination of pertuzumab and gemcitabine demonstrated a higher RR and a trend towards improved PFS[172]. However, pertuzumab did not substantially favor carboplatin-based chemotherapy with a comparable PFS in platinum-sensitive OC patients [173]. The randomized phase III clinical research demonstrated that the combination of pertuzumab with chemotherapy did not greatly increase PFS [174].
In a clinical trial evaluating the feasibility of trastuzumab in OC, the overall RR of 7.3% suggested a limited clinical value for single-agent monoclonal antibody therapy, which may be due to low objective response or low incidence of HER2 overexpression [175]. Single-agent trastuzumab therapy also demonstrated few activities against EC even with HER2 amplification or overexpression [176]. However, trastuzumab increased PFS and OS when combined with carboplatin/paclitaxel in patients with HER2/Neu-positive advanced or recurrent uterine-serous-carcinoma (USC) [177], and trastuzumab appears to be safe and exhibit a manageable toxicity profile in patients with HER2/Neu-positive USC, both in combination with chemotherapy and as a single-agent maintenance [178]. The combination therapy of trastuzumab and carboplatin-paclitaxel significantly prolonged PFS without unexpected safety signals in USC patients with HER2/neu overexpression [179]. Adavosertib is a specific and effective inhibitor of the WEE1 kinase. The monotherapy of adavosertib demonstrated durable and promising anti-tumor efficacy in USC (29.4% ORR) [180]. In addition to the direct cytotoxic effect in gynecological cancers, adavosertib also reversed trastuzumab resistance in HER2-positive cancers, which supports further clinical development [181, 182].
Currently, the therapeutics targeting HER2 face many challenges in CC [183]. Firstly, a truncated form of HER2, p95HER2, was reported to be highly expressed in high-grade EC, which led to trastuzumab resistance due to the lack of N-terminal trastuzumab binding. Secondly, abnormal activation of the PI3K pathway and Notch signaling are associated with the resistance of HER2 therapies [183, 184]. On the contrary, HER3 was increased upon PI3K pathway inhibition via mTOR, AKT, or direct PI3K blockage. Thirdly, the expression of EGFR (HER1, ERBB1) was also related to decreased sensitivity of trastuzumab [183].
6.2 PI3K/AKT/mTOR signaling and targeted inhibitors
The PI3K/AKT/mTOR pathway is involved in the pathogenesis of gynecological cancers and in crosstalk with the estrogen receptor signals and RAS/RAF/MEK pathways [185]. Mechanisms underlying abnormal PI3K/AKT/mTOR pathway activation include amplification or mutation of PI3K/AKT, activation of growth factor receptors, loss function of tumor suppressor PTEN, and exposure to carcinogens [186]. Through a diverse set of downstream targets, the PI3K/AKT/mTOR pathway responds to several endogenous or exogenous stimuli to determine EC cell fate and AKT can be catalytically activated by the MLLT11-TRIL complex, which promotes cancer progression [187]. In EC, PI3K/AKT/mTOR inhibition is responsible for metformin-induced cell proliferation, while its activation promotes progestin resistance [188, 189]. In EOC, abnormal activation of AKT is closely linked with poor PFS and OS [190]. The stemness of OC is directly regulated by PI3K/PTEN/AKT pathway, which mediates stem cell enrichment and phenotype maintenance, as well as multidrug resistance [166], resulting in aberrant cell proliferation and epithelial-mesenchymal transition (EMT) [191].
Many clinical trials have been conducted to investigate the efficacy of mTOR inhibitors in the treatment of cancer, especially everolimus and LY3023414. Everolimus is an oral rapamycin analog, and its monotherapy showed an encouraging clinical benefit in patients with recurrent EC [192]. Although in recurrent ovarian, peritoneal, and fallopian tube cancers, everolimus did not distinctly improve the response when combined with bevacizumab [193], it is well tolerated at fully approved doses and is effective in patients with ER- and/or PR-positive ovarian or endometrial cancers when combined with aromatase inhibitor (anatrozole) [194]. Thus, everolimus-based combination therapies exhibited a promising effect and need to be further explored in future clinical trials. LY3023414 is a dual PI3K/mTOR inhibitor and exhibits a manageable safety profile and modest single-agent effect for advanced EC patients [195].
Gedatolisib (PF-05212384) demonstrated moderate activity with acceptable tolerability in patients with recurrent EC, as evaluated in a phase II study (NCT01420081) [196]. Ridaforolimus showed encouraging activity in advanced EC but was associated with significant toxicity [197], while in recurrent or metastatic EC, it had modest activity and was reasonably tolerated [198].
Temsirolimus exhibits encouraging antitumor effect in EC, and the responsiveness of chemotherapy-naive patients is more favorable than chemotherapy-treated patients [199], however, in both CC and persistent/recurrent EOC it shows modest activity as a single agent [200, 201]. The PI3K inhibitor buparlisib combined with the MEK1/2 inhibitor trametinib shows promising antitumor activity for KRAS-mutant OC [202].
6.3 Notch signaling and targeted inhibitors
Notch signaling is evolutionarily conserved and essential for adult tissue homeostasis and the progression of multiple cancer types. The pathway is comprised of five canonical Notch ligands (DLL1, DLL3, DLL4, Jagged1, and Jagged2) and four Notch receptor paralogs (Notch1/2/3/4) [203]. Upon ligand binding, the Notch receptor is cleaved in the extracellular domain and then trans-endocytosed by the ligand-expressing cell. Following the second cleavage caused by γ-secretase activity, the intracellular fragment of Notch (ICN) is released and allowed to nuclear translocation where it associates with the CBF1/Su(H)/Lag-1 (CSL) transcription factor complex, resulting in subsequent activation of the target genes (MYC, p21, and HES family members) [203, 204].
As one of the most active pathways in cancer cells, Notch signaling plays a crucial role in the linkage between angiogenesis and CSC self-renewal [205]. In CD133+ EC cells, blocking the Notch pathway enhanced the suppressive effects of EGFR inhibitors on cancer cell proliferation [206]. Thus, targeting Notch signal transduction is a promising pharmacological intervention to eradicate endometrial CSCs. While numerous Notch inhibitors have been developed and investigated for cancer therapies, only RO4929097 has been explored in clinical research. It is a gamma-secretase inhibitor with minimal single-agent activity for patients with platinum-resistant OC [207].
6.4 JAK/STAT signaling pathway and targeted inhibitors
Abnormally constitutive activation of the JAK/STAT pathway is associated with tumor progression and a poor prognosis in OC [208]. JAK/STAT pathway mediates tumor progression through the induction of numerous proteins and cytokines attributed to cell proliferation, stemness, and evasion from antitumor immunity [209]. By inducing the synthesis of immunological checkpoints (e.g., PD-1, PD-L1, PD-L2, and CTLA-4), STAT is a major contributor to the resistance of radiation therapy and chemotherapy and the failure of targeted immunotherapies [210].
Ruxolitinib, a JAK1/2 inhibitor, was demonstrated to be active in various peripheral T cell lymphoma subtypes [211]. Though the JAK1 inhibitor itacitinib showed manageable safety and clinical activity when combined with chemotherapy in patients with advanced solid tumors, it was terminated for negative phase III results in patients with previously treated advanced pancreatic cancer [212]. JAK2 inhibitor TG101348 is well tolerated and provides durable therapeutic benefit for myelofibrosis patients [213]. However, these inhibitors have not been evaluated in gynecological cancers.
6.5 Wnt/β-catenin pathway
A large number of genes related to tumor formation and progression have been identified to be transcriptionally activated by Wnt/β-Catenin signaling [214]. In CD133/CD44+ endometrial CSCs, the activation of Wnt signaling was complicated by the upregulation of stemness-associated genes (such as SOX2, OCT4, and NANOG), which can be decreased by targeting SMOC-2 [215].
Aberrant activation of Wnt/β-Catenin signaling, especially mutation of CTNNB1, AXIN, and APC, was observed in endometrial and mucinous EOC subtypes [216]. The Wnt pathway is essential for the occurrence, progression, metastasis, angiogenesis, and chemoresistance of OC due to its crucial roles in CSC self-renewal, EMT, invasion capabilities, and tumor immunity suppression [217].
7 Poly (adenosine diphosphate-ribose) polymerase inhibitors
7.1 The DNA damage response in gynecological cancers
The DNA damage response (DDR) induces programmed cell death in the presence of severe DNA damage in reproducing cells by stopping cell cycle progression, which makes it easier to repair DNA lesions to stop mutagenesis and maintain genomic stability [218].
Localized DDR activation is triggered by HPV replication centers in CC [219, 220], and there is evidence that HPV E7 oncoprotein and HPV genomic integration may induce the upregulation of DDR proteins [221, 222]. Compared to their HPV-negative counterparts, HPV-positive cancer cells are more radiosensitive; however, resistance to DNA-damaging therapy still exists as shown by the poor prognosis of HPV-positive advanced CC [223, 224]. Therapeutic regulation of the DDR is an appealing technique to improve treatment response. Further research is required to understand how HPV affects the DDR and how it affects the in vivo sensitivity of HPV-associated malignancies to DNA-damaging agents.
Many aspects of OC biology are affected by abnormal DDR and DNA repair deficiency, including tumor initiation, progression from low-grade towards high-grade phenotype, responsiveness to chemotherapy, and development of chemoresistance. Numerous single nucleotide polymorphisms in genes linked to the DNA repair and DDR are expected to enhance the risk of OC [225].
PUMA, a new agent with therapeutic potential for OC, produces DNA breaks and activates kinases involved in DNA damage. Therefore, it was proposed to enhance the DDR in OC cell lines, increasing the rate of apoptosis [226, 227]. Upregulation of SEI1 (also known as TRIPBr1 and SERTAD1) causes increased DNA strand breaks, and DDR proteins were significantly downregulated and the number of micronuclei was greatly decreased upon SEI1 knockdown. Accordingly, SEI1 controls genomic stability by altering the DDR when cancer cells progress towards malignant phenotype [228].
Platinum–DNA damage tolerance and cisplatin sensitivity were found to be highly correlated [229]. OC patients had higher levels of DNA damage and a failure in DNA repair than did healthy individuals. Additionally, platinum-sensitive individuals had higher DNA damage levels than platinum-resistant patients [230].
A lack of DNA MMR proteins, such as MLH1, MSH2, MSH6, and/or PMS2, is seen in 30–40% of endometrioid tumors. This may be caused by germline mutations like in Lynch syndrome, or MLH1 promoter hypermethylation [231]. A possible role for therapies that target DNA repair processes and take advantage of an already underdeveloped repair pathway is suggested by the loss of MMR proteins, which are important in the repair of DNA single-strand breaks (SSBs). Additionally, MSI is triggered by mismatch repair-deficient (dMMR) and results in a phenotype that would be more vulnerable to checkpoint inhibition. There are significant indicators for prospective targeted therapy, such as PARP inhibition, in this molecular subtyping.
7.2 PARP inhibitors in gynecological cancers
The detection and repair of SSBs depend on PARP enzymes, and PARPis inactivate these enzymes which allows the persistence of spontaneous SSBs and inhibits the release of PARP from DNA (“PARP trapping”). Both processes result in double-strand breaks (DSBs), collapsed replication forks, and persistent SSBs. DSBs can be repaired in two ways: through homologous recombination repair (HRR) or nonhomologous end-joining (NHEJ). NHEJ is an error-prone process that results in genetic instability, whereas homologous recombination repairs DNA with high fidelity [232]. As a result of the functional termination of two DNA repair processes in cells with HRR deficits (HRD), PARPis cause “synthetic lethality,” which results in a dependence on NHEJ and ultimately cell death brought on by the accumulation of genetic damage [233, 234]. Therefore, malignancies with HRD respond particularly well to PARPis. HRR depends on BRCA proteins, and germline BRCA (gBRCA) mutations and somatic BRCA (sBRCA) mutations can cause malignant transformation and make cancers vulnerable to PARPis [235]. The HRR pathway also experiences additional genetic and epigenetic alterations, providing other potential targets for PARP inhibition. In BRCA1-deficient OC, inhibition of PARP could induce PD-L1 expression via JAK2/STAT3 pathway in solid tumors and increase the number of intratumoral CD4+ and CD8+ T cells through a STING-dependent antitumor immunity [236, 237] (Fig. 3). PARPis and the ongoing clinical trials are collected in Table 1.

The antitumor mechanisms of PARP inhibitors. (A) The PARP inhibitor was devised to dampen the repair of DNA damage, which results in “synthetic lethality” in BRCA mutant cancer cells. (B) PARP inhibition can induce PD-L1 expression via JAK2/STAT3 pathway in solid tumors. (C) In BRCA1-deficient ovarian cancer, PARP inhibitor can increase the number of intratumoral CD4+ and CD8.+ T cells through a STING-dependent antitumor immunity (created with BioRender.com)
7.2.1 PARPis in endometrial cancer
Loss function of PTEN, the most frequent mutation in EC, can result in inadequate repair of DSBs and consequent sensitivity to PARPis [238, 239]. Preclinical research has indicated a potential benefit of PARPis in PTEN-deficient cell lines and tumor models [238, 240], and PI3K blockade can sensitize cells to PARPis [239, 241].
The genetic similarities between high-grade serous ovarian cancer and serous endometrial cancer imply that serous endometrial tumors with high copy number, even those lacking BRCA mutation, may also benefit from PARPis. One phase I trial (NCT03586661) is assessing niraparib, a PARPi, with the PI3K inhibitor copanlisib in patients with recurrent endometrial and ovarian malignancies, and other clinical trials are actively evaluating the antitumor effect of a single-agent PARPi.
It is acknowledged that tumors with a lot of mutations also have many neoantigens and PD-1 expression, therefore, immune checkpoint therapy may strengthen the efficacy of PARPis in endometrial malignancies with polymerase-epsilon (POLE) mutation or high microsatellite instability (MSI-H) [231]. Accordingly, several associated clinical trials have been established. A phase II DOMEC trial (NCT03951415) conducted the synergetic efficacy of PARPi olaparib and durvalumab (a selective monoclonal antibody that blocks PD-L1 with high-affinity) in metastatic or recurrent EC patients, and the results showed that it was well tolerated, but did not meet the prespecified 50% 6-month progression-free survival [242].
Similar to OC, EC may benefit from the combination of PARPi and antiangiogenic therapy. In cases of recurrent and/or refractory EC, combinations of rucaparib and bevacizumab (NCT03476798), and olaparib and cediranib (NCT03660826) are being studied.
7.2.2 PARPis in cervical cancer
Preclinical research has demonstrated that PARPis boost apoptotic response and make CC cells more sensitive to cisplatin [243,244,245]. Preclinical research has been done on PARPis in CC, several clinical trials are currently being conducted [246].
The utility of PARPis in advanced-stage CC was examined in two GOG/NRG oncology studies [247, 248]. For the treatment of patients with advanced, persistent, or recurrent CC, PARPi veliparib was coupled with paclitaxel and cisplatin in a phase I trial. The median PFS was 6.2 months (95% CI 2.9–10.1 months), and OS was 14.5 months (95% CI 8.2–19.4 months). The only grade 3 and 4 adverse effects were dyspnea and neutropenia, the maximum tolerable dose was not reached, and the ORR for patients with measurable diseases was 34%. The researchers concluded that the combination therapy was safe and practical in persistent and recurrent CC [247]. In the second clinical research examining the efficacy and tolerance of PARPi veliparib and topotecan (a potent anticancer camptothecin analog), twenty-seven patients with persistent or recurrent CC were examined. Grades 3 and 4 toxicities frequently occurred, including anemia (59%), thrombocytopenia (44%), leukopenia (22%), and neutropenia (19%). Only 2 patients (7%) showed signs of partial response, while 4 experienced disease progression after 6 months of treatment. Overall, this research showed that veliparib with topotecan had serious side effects with little clinical activity. However, a subset of patients with low PARP-1 levels on immunohistochemical staining showed noticeably greater PFS and OS, indicating that PARP-1 may be a viable biomarker for identifying patients who may benefit from this treatment [248].
Additional clinical trials are currently being conducted to further investigate the efficacy of PARPis combination therapies in CC. In patients with metastatic, recurrent, or persistent gynecological cancers, several combination therapies of PARPi are being researched [249]. In phase II research called Clovis-001 (NCT03476798), the combination of rucaparib and bevacizumab is being tested in recurrent cervical or endometrial cancer. PARPis make cancer cells more radiosensitive because radiation causes DNA damage, which may theoretically be increased by preventing DNA repair pathways. Reduced survival was observed in pancreatic cancer cell lines treated with radiation in combination to rucaparib [250]. In the phase I/II research NIVIX (NCT03644342), the combination of niraparib with pelvic radiation is being studied in metastatic stage IV invasive CC.
7.2.3 PARPis in ovarian cancer
Multiple clinical trials have proved that PARPis function as a feasible therapy option in patients with and without HRD, manifested by significantly prolonged PFS [251]. Three PARPis (olaparib, rucaparib, and niraparib) have been approved for OC treatment by the FDA and EMA [252]. Studies on other PARPis, such as veliparib and talazoparib, have exhibited encouraging clinical outcomes and will soon gain licensure (NCT01472783, NCT02470585, NCT01540565, NCT01286987).
In a phase II (NCT00628251) randomized research assessing olaparib plus PLD in patients with gBRCA mutated platinum-resistant or partially platinum-sensitive relapsed OC, researchers compared the efficacy of PARPis as monotherapy and that of chemotherapy, and they found that there was no distinctive difference in ORR or PFS [253]. Later the phase III trial SOLO3 (NCT02282020) was conducted to assess olaparib monotherapy versus nonplatinum chemotherapy (PLD, paclitaxel, gemcitabine, or topotecan) in patients with gBRCA mutated platinum-sensitive relapsed ovarian cancer (PSROC) who had undergone two or more cycles of platinum-based chemotherapy. The ORR was much higher in the olaparib group than that of chemotherapy group in the total 266 patients (72.2% vs. 51.4%; P = 0.002). As for the patients who had received 2 prior lines of platinum treatment, the ORR was 84.6% with olaparib and 61.5% with chemotherapy. Olaparib considerably outperformed chemotherapy in terms of PFS (13.4 v 9.2 months; P = 0.013). Adverse events were consistent with the established safety profiles of olaparib and chemotherapy [254]. It should be highlighted that although olaparib resulted in statistically significant and clinically relevant improvements, the control arm in this trial was a nonplatinum therapy for platinum-sensitive patients; consequently, the practicality for clinical application is doubted.
Based on the condition of gBRCA mutation (gBRCAm) and the level of loss of heterozygosity (LOH), the phase II trial ARIEL2 (NCT01891344) assessed the efficacy of rucaparib monotherapy in patients with three different types of HRD (gBRCAm, non-gBRCAm/LOH high, and non-gBRCAm/LOH low) and the biggest clinical improvements were observed in the gBRCAm cohort with the highest PFS and ORR [255, 256].
Olaparib was tested in a phase II (NCT00679783) multicenter, nonrandomized trial in patients with advanced high-grade serous and/or poorly differentiated OC. Among the 63 patients who had target lesions, the ORR for gBRCAm cohort was 33% and for non-gBRCAm cohort it was 4% [257]. Based on the QUADRA trial (NCT02354586), niraparib was approved in 2020 as the maintenance treatment for recurrent OC patients regardless of genetic profiles [258].
The expressions of PD-1 and neoantigen for possible immune system recognition in gBRCAm or HRD tumors make it possible to improve anticancer efficacy through combination therapy of PARPis and immune checkpoint inhibitors (ICIs). Currently, many ongoing clinical research are being conducted to assess the impact of ICIs with PARPis on OC patients [259]. A Phase I/II Study is actively evaluating the efficacy of MEDI4736, an anti-PD-L1 antibody, in combination with olaparib for advanced OC patients (NCT02734004).
PARPi combined with antiangiogenic treatment is another area with potential value. It has been validated that hypoxia caused the defects in HRR genes, including BRCA [260]. When compared to bevacizumab monotherapy, olaparib combined with bevacizumab maintenance produced a better PFS in newly diagnosed, advanced OC patients, according to a randomized, double-blind PAOLA-1/ENGOT-ov25 trial (NCT02477644) [261]. Niraparib plus bevacizumab significantly improved PFS compared to single-agent niraparib (11.9 vs. 6.4 months; P = 0.0001) (NCT02354131), while olaparib and cediranib together improved PFS by 8.7 months (NCT01116648) in patients with PSROC. These findings suggest that this synergetic strategy is effective in patients with PSROC independent of BRCA status and that the anti-angiogenesis drugs may be especially significant for gBRCA mutated tumors. Accordingly, the phase III trials (NCT03278717, NCT02502266) are now being carried actively to further evaluate the effects of these combination therapies on recurrent OC.
Combination strategies that focus on other genes involved in the HRR pathway are also gaining popularity. It has been validated that PI3K inhibition could decrease the expression of BRCA and improve the efficacy of PARPis [262]. The recommended dosage of the mTOR inhibitor vistusertib plus olaparib showed minimal clinical benefit in OC patients, with 20% RR and 15% RR observed in each group of a phase I trial (NCT02208375). In a phase I research (NCT01623349), olaparib was also examined in combination with the pan-PI3K inhibitor buparlisib in patients with advanced OC. The RR was 29%, and all the patients who responded harbored high-grade serous histology and the majority (8 out of 12 responders) had gBRCAm. In addition, there was no difference in response to the combination regimen between patients who had platinum-sensitive or platinum-resistant disease [263]. Olaparib and alpelisib, a PI3K-alpha inhibitor, were combined in a multicenter, open-label, phase Ib trial in the 28 recurrent EOC patients with high-grade serous histology, and 36% of participants achieved partial response and 50% showed stable disease [264]. Based on the above research, regimen combining olaparib and PI3K inhibitors is feasible and exhibits promising preliminary clinical evidence particularly in patients with advanced EOC, which warrants further investigation.
Furthermore, multiple phase I studies are now being conducted in patients with OC to evaluate the combination therapies of PARPis with TKIs that target ATR, MEK1/2, PI3K, WEE1, and/or PI3K (NCT03162627, NCT04065269, NCT03579316).
8 Immunotherapeutic strategies for gynecological cancers
Infiltrated immune cells serve a crucial role in the progression of tumors and co-constitute heterogeneous microenvironment by combining different cells and extracellular components. It is acknowledged that the numbers of neutrophils, macrophages, and dendritic cells (DCs) are significantly increased in EC, while NK cells are significantly decreased [265]. The fact that tumor-associated macrophages (TAMs) but not regulatory T cells (Tregs) have been proved to be correlated with a poor prognosis and lymph node metastasis of EC [266] together with the preferential enrichment of macrophages and exhausted CD8+ T cells suggests an immunosuppressive microenvironment in EC [267].
Infiltrating macrophages act as a driver of type I EC by sensitizing EC cells to estrogen through upregulating estrogen receptor alpha expression, and it was discovered to be a prognostic biomarker of cancer progression [268]. CCL2-facilitated macrophage infiltration has also been confirmed by in vitro studies [269]. Macrophages also promote metastasis through EMT via CCL18-activated PI3K/AKT/mTOR signaling in EC [270]. In addition, infiltrated macrophages supply interleukin (IL)-8, which might be associated with myometrial invasion and angiogenesis in EC [271].
Cytotoxic T lymphocytes are essential for tumor control, but their functions are compromised by cancer cells and Tregs. The suppression of T cell-mediated antitumor immunity was suggested to be associated with progression and development of EC through B7-H3 [272]. Higher levels of immunosuppressive cytokines, including TGF-β, were involved in EC and CD8+ T cell-mediated cytotoxic capacity was suppressed by downregulating intracellular cytolytic molecules [273]. Tregs were also reported to induce tolerance of immune therapy in EC [274, 275], and the number of FoxP3N Tregs was increased in dMMR EC [276]. The number of CD8+/CD4+ T cells and Tregs, and Treg/CD8+ and Treg/CD4+ ratios were significantly higher in patients with advanced poorly differentiated EC. Accordingly, the disease-free survival of patients with higher Tregs and Treg/CD8+ ratios was significantly worse than that of patients with lower Tregs and Treg/CD8+ ratios [274].
Given the molecular mechanisms and findings from multiple clinical research aiming at evaluating the antitumor efficacy of several promising ICIs (Table 2), the immunotherapeutics exert exciting new frontiers for patients with gynecological cancers.
8.1 Immune checkpoint inhibitors for gynecological cancers
8.1.1 PD-1/PD-L1 blockade
PD-1 is widely expressed in activated T cells, NK cells, B lymphocytes, macrophages, DCs, and monocytes [277, 278]. The activity of PD-1 and its ligands (PD-L1 and PD-L2) is responsible for regulating T cell activity, activating apoptosis of antigen-specific T cells and inhibiting Tregs apoptosis [277]. The PD-1/PD-L1 axis, a target of immunotherapy, plays a pivotal role in promoting immune self-tolerance and cancer escape. It was found that 61.3% of EC were PD‑1 positive, which was almost exclusively in tumor‑infiltrating immune cells [279]. Correspondingly, PD‑L1 was positive in 14.3% of normal endometria and in 17.3% of EC tissues, while PD‑L2 was positive in 20.0% of normal endometria and 37.3% of EC tissues [279].
As a monoclonal IgG4 kappa-isotype antibody against the PD-1 receptor, pembrolizumab was approved for dMMR or MSI-H EC patients and recurrent or metastatic CC patients [280, 281]. In the KEYNOTE-028 study, pembrolizumab monotherapy demonstrated durable antitumor activity in a cohort of patients with PD-L1-positive, advanced EC. The ORR was 13.0% and the median PFS was 1.8 months, with adverse events including fatigue, pruritus, pyrexia, and decreased appetite [282]. In MSI-H or dMMR advanced EC, PD-1 inhibitors avelumab and durvalumab have shown a response rate of 27% and 43%, respectively. While, dostarlimab and pembrolizumab have shown a response rate of 49% and 57%, respectively [283]. Pembrolizumab treatment in patients with recurrent CC also shows considerable clinical benefits with manageable adverse effects [284, 285]. Dostarlimab has recently been approved in the EU and USA for the treatment of patients with dMMR recurrent or advanced EC [286]. The protective effect of dostarlimab is still being evaluated in ongoing clinical trials in advanced CC patients and recurrent OC patients who are not suitable for platinum treatment (NCT03833479 and NCT04679064, respectively) [287, 288]. As for MSI EC, dostarlimab showed a higher affinity against PD-1 than pembrolizumab with encouraging clinical activity, manifested by the preliminary results [289], and it exhibited significant antitumor activity in both dMMR/MSI-H and mismatch repair-proficient (pMMR)/microsatellite stable (MSS) EC with a manageable safety profile, based on the results from a phase I, single-arm study [290].
PD-1/PD-L1 blockade has also been investigated for combination therapies in gynecological cancers (Fig. 4). Preclinical studies demonstrated that lenvatinib, a multiple receptor TKI targeting VEGF and FGF receptors, increased IFN-γ+ and granzyme B+ CD8+ T cells, which improved cancer immunotherapy when combined with PD-1 blockade, and its combination therapy with pembrolizumab has approved by FDA for patients with advanced EC [291]. In the 309/KEYNOTE-775 study, patients treated with pembrolizumab plus lenvatinib showed longer PFS (6.6 months) than those in the chemotherapy arm (3.8 months). However, in addition to adverse events, a higher occurrence rate (88.9%) was found in the combination treatment group, suggesting palliative treatment [292]. The combination of PD-L1 blockers with PARPis has been demonstrated to exert a synergistic effect in patients with recurrent EC [293].

Targeted immunotherapies and the anti-PD-1/PD-L1 combination strategies in gynecological cancers. (A) The promising immunotherapeutic targets for gynecological cancers. (B) Blockade of the PD-1/PD-L1 checkpoint promotes anti-tumor T cell immunity. The synergistic antitumor strategies of anti-PD-1/PD-L1 in combination with PARP inhibitors, anti-CTLA4 antibody, kinase inhibitors, or angiogenesis inhibitors involved in clinical trials (created with BioRender.com)
8.1.2 CTLA-4 blockade
CTLA-4 is expressed exclusively in T cells, where it dampens the activation of T cells by outcompeting CD28 in binding to CD80 and CD86, as well as actively delivering inhibitory signals to the T cells [294]. The expression of CTLA-4 seems to be higher than that of PD-L1 and is associated with a low CD4+/CD8+ ratio and high tumor grade in EC [295].
CTLA-4 blocking antibody (MDX-CTLA4) stimulated extensive tumor necrosis with lymphocyte and granulocyte infiltrates in vaccinated patients with metastatic OC [296]. In a phase II trial (NCT03495882), the CTLA-4 antibody zalifrelimab was combined with a PD-1 antibody (balstilimab) in patients with recurrent and/or metastatic CC, and the ORR was 25.6% [297]. In recurrent BRCA-deficient OC patients, a combination of PARPi and CTLA-4 blockade (tremelimumab) led to decreased tumor size with grade 1 and 2 toxicities [298].
8.1.3 CD47 blockade
CD47 protein, expressed in both healthy and cancer cells, plays a crucial role in blocking the cytotoxic activity of myeloid cells by delivering a “do not eat me signal” upon binding to the signal-regulatory protein alpha (SIRPα) receptor on macrophage cells (Fig. 5) [299].

Mechanisms of CD47 mediated immune tolerance in tumor microenvironment. CD47 expressed in cancer cells activates SIRPα on macrophages, which delivers a “do not eat me signal.” Furthermore, the interactions between CD47 on tumor cells and SIRPα expressed in CD103.+ dendritic cells contribute to restrict NK cell recruitment which elicit anti-tumor effect by orchestrating the expression of granzyme B, IFN-γ, TNF-α, NKG2A, and NKG2D (created with BioRender.com)
CD47 cannot be observed in the normal endometrium, but its expression is increased along with the progression of endometrial hyperplasia to carcinoma, and elevated CD47 expression was also associated with poorer prognosis and higher clinicopathological grade of EC [300]. In EC, CD47 acts as an antiphagocytic signal that promotes tumor resistance against tumor-associated macrophages, and CD47 blockade increased the infiltration of macrophages in vivo and promoted phagocytosis of EC cells by M2 macrophages [301]. Recent studies in endometriosis revealed that targeting CD47 increased macrophage phagocytosis and induced apoptosis of ectopic endometrial stromal cells [302]. These results suggest a dual mechanism of CD47-SIRPα signaling in eradicating targeted cells.
Preclinical research has demonstrated that targeting CD47 is a promising approach for improving OC treatment both as a single-agent therapy or in combination with another agent [303]. CD47/SIRPα inhibitors include magrolimab (Hu5F9-G4), TTI-621, ALX148 and several small-molecule inhibitors, such as RRx-001 [304,305,306]. Although magrolimab has been approved for the treatment of hematopoietic malignancies and several other CD47/SIRPα inhibitors have been assessed in many advanced cancers [307,308,309,310], clinical data about the efficacy of CD47/SIRPα inhibitors in patients with gynecological cancers are rare. Herein, in a first-in-class phase I trial of Hu5F9-G4 in patients with advanced cancers, two patients with ovarian/fallopian tube cancers had partial remissions for 5.2 and 9.2 months and reductions of 50% and 44% in target lesions, respectively [311].
8.2 Engineered immune-activating strategies for gynecological cancers
8.2.1 Oncolytic viral immunotherapy
Oncolytic therapy, specifically targeting cancer cells, has provided a revolutionary tool for converting “immune-cold” tumors to “immune-hot” tumors with antitumor responses. The cytotoxicity of oncolytic viruses occurs through a dual mechanism that is dependent on directly triggering tumor cell lysis and the induction of systemic antitumor immunity. For instance, oncolytic virus-induced tumor cell lysis may result in the release of a variety of tumor-associated antigens/neoantigens, danger-associated molecular patterns, and viral pathogen-associated molecular patterns to trigger inflammatory immune responses [312].
Intraperitoneal Olvi-Vec virotherapy, based on a modified vaccinia virus, showed promising safety and clinical activity in platinum-resistant or refractory OC patients through enhancing tumor infiltration of CD8+ T cells and activating tumor-specific T cells in peripheral blood [313]. The safety and efficacy of enadenotucirev, a tumor-selective adenoviral vector, were evaluated in platinum-resistant OC. Enadenotucirev plus paclitaxel demonstrated manageable tolerability and prolonged median PFS by increasing tumor immune-cell infiltration in platinum-resistant OC [314]. Oncolytic reoviral therapy was also evaluated in patients with recurrent ovarian, tubal, or peritoneal cancer. However, the addition of reovirus to paclitaxel did not sufficiently reduce the hazard of progression or death [315]. Oncolytic measles virus has been engineered to express carcinoembryonic antigen and showed dose-dependent biological activity in recurrent OC patients [316]. Adenovirus has also been reported to be feasible and safe for recurrent OC patients, suggesting a potential therapeutic option [317].
8.2.2 Adoptive cell therapies
Adoptive T cell therapy (ACT) has emerged as an effective and potentially curative therapy for malignancies, which utilizes either tumor-infiltrating lymphocyte (TIL)-derived T cells or T cells genetically modified to express tumor-recognizing receptors [318]. Engineered T cells for cancer therapy include antigen-specific transgenic T cell receptor T cells (TCR-T cells) and chimeric antigen receptor T cells (CAR-T cells).
In recent years, numerous clinical trials associated with TCR-T and CAR-T cells have been ongoing. Adoptive transfer of folate receptor-α-redirected autologous T cells showed preliminary activity in patients with recurrent OC [319]. Due to the lack of ideal tumor surface antigens, CAR-T cell therapy showed limited outcomes in treating solid tumors. In contrast, TCR-T cells identify intracellular and cell-surface antigens presented by the major histocompatibility complex (MHC) and thus have the potential to access many more target antigens than CAR-T cells, providing great promise in treating solid tumors [320].
Currently, numerous antigens have been applied for TCR-T-engineered cells in clinical trials, including HPV16-E6/E7, NY-ESO-1, MAGE-A3, MAGE-A4, and mesothelin. Antigens used as CAR-T therapeutic targets include mesothelin, CD70, CD22, CD133, GD2, PSMA, MUC1, MUC16, HER-2, nectin-4, FR-a, ALPP, B7-H3 and TnMUC1 [321]. Although ACT is a promising option for treatment of malignant tumors, several challenges warrant further investigation, such as severe adverse effects, loss of target antigen, and the short persistence of transferred T cells [321]. Engineered modifications and clinical trials need to be further explored to facilitate the use of ACT for gynecologic tumors.
Adoptive NK cell therapy has shown an ORR of 26.7% in gynecological cancers, suggesting a promising treatment modality [322]. The CAR strategy was also applied to NK cells and macrophages, which led to CAR-NK and CAR-M cell therapies. These approaches seem to be alternatives to T cells and safer than CAR-T cells [323, 324], while the promise and challenges of these therapies need to be extensively explored in gynecological cancers.
8.3 Other emerging candidates for immunotherapy: TIM-3, LAG-3, and TIGIT
With the growing knowledge of effective biomarkers, coinhibitory receptors, such as TIM-3, LAG-3, and TIGIT, have been identified for their crucial roles in modulating T cell responses and maintaining immune homeostasis.
TIM-3 is a type I transmembrane protein expressed in T cells, NK cells, monocytes, macrophages, and DCs. Upon interaction with its ligand(s), TIM-3 acts as a negative regulator of antitumor immunity via T cell exhaustion and suppression of innate immune responses [325]. Plasma-soluble TIM-3 has emerged as an inhibitor of sepsis progression, which contrasts with membrane TIM-3 on monocytes [326]. Previous study has demonstrated that TIM-3 is commonly expressed in both MMR-intact and dMMR EC [327]. Therefore, inhibitors targeting TIM-3 may be a potential strategy for EC therapy.
A few clinical trials of TIM-3 inhibitors, such as LY3321367, sabatolimab, and LY3415244, have been performed in patients with advanced solid tumors. The combination therapies of LY3321367 or sabatolimab with anti-PD-L1 antibody both exhibited safety profiles and favorable pharmacokinetics, while sabatolimab, compared to LY3321367, showed a better antitumor activity when administered in combination with spartalizuma [328, 329]. LY3415244, a TIM-3/PD-L1 bispecific antibody, was reported to result in the development of clinically significant anaphylactic infusion-related reactions in 16.7% of patients and treatment-emergent antidrug antibodies in all patients, which suggests unexpected immunogenicity [330]. Thus, the bispecific TIM-3 and PD-L1 format exhibited better promise than monotherapy and combination therapy, while how to lower the immunogenicity risk requires further investigation.
LAG-3, an immunosuppressive checkpoint molecule, expressed in lymphocytes and its ligand GAL-3, has been identified on EC cells, particularly in nonmethylated dMMR cancers, supportting a role for immunotherapies targeting LAG-3 and/or GAL-3 in a subset of EC [331]. In an analysis of 26 cervical tumors, LAG-3 was detected in 65% of cases while PD-L1 in 85% of cases [332]. In OC, LAG-3 was negatively related to the infiltration of a specific CD8+ T cell subset [333].
Ieramilimab (LAG525) is an inhibitor of LAG-3 and according to phase I/II clinical trials, it was tolerated in advanced malignancies with no synergistic antitumor activity in combination with anti-PD-1 spartalizumab (PDR001) [334]. Treatment with the LAG-3 blocking antibody relatlimab plus the PD-1 blocking antibody nivolumab showed better median PFS (10.1 months vs. 4.6 months) and PFS (47.7% vs. 36.0%) than nivolumab monotherapy in patients with previously untreated metastatic or unresectable melanoma [335].
The T cell immunoglobulin and ITIM domain (TIGIT) is a well-known member of the immunoglobulin superfamily that is expressed exclusively on lymphocytes, particularly on CD4+ Tregs, CD8+ T cells, and NKs. Previous studies revealed that TIGIT blockage could lower the immunosuppression induced by CD4+ Tregs in OC mice models, boost functional responsiveness of NKs towards OC, and anti-TIGIT treatment also exerted promising antitumor effect, which suggests that inhibition of TIGIT is a potential therapeutic target in OC patients due to its effect on NK cell suppression and Treg activation [336, 337].
Recently, a phase I clinical trial of the anti-TIGIT antibody vibostolimab alone or in combination with pembrolizumab for patients with advanced solid tumors demonstrated that the combination therapy showed favorable antitumor effect [338].
Accordingly, TIM-3, LAG-3, and TIGIT are the potential targets of emerging immunotherapies especially for patients with advanced solid tumors but have not been well-studied in gynecological cancers, especially in the clinical setting. Therefore, the clinical impact of those targets via monotherapy or combination therapies warrants further investigation.
9 Repurposing of existing drugs in gynecological cancers
Repurposing approved drugs to identify new pharmacological/therapeutic indications is increasingly becoming an attractive proposition because the usage of de-risked compounds requires lower overall costs and shorter development timelines [339, 340]. During the recent decades, several “old” drugs have been found to exert promising antitumor effect in EC.
Aspirin directly inhibits the enzyme cyclooxygenase (COX), and its primary effect is considered to be on the anucleate platelet by inhibiting COX-1 acetylation. While circulating platelets are validated to play a critical role in tumor metastasis via immune evasion. Up to now, a large body of evidence supports the preventive role of aspirin in the development and recurrence of cancers in the observational setting [341]. The STICs and STONEs trial (NCT03480776), a randomized phase II, double-blind, placebo-controlled trial, is currently ongoing to assess the effect of aspirin in the prevention of OC with BRCA1 and BRCA2 mutations.
Metformin is a first-line drug for type 2 diabetes and has beneficial effects on various metabolic syndromes. Most experimental data have revealed that it electively targets CSCs and acts together with chemotherapy to exert antitumor effects in different cancers mainly through AMPK activation and PI3K/AKT/mTOR inhibition. Given the available clinical findings and the molecular mechanisms, it also showed a potential role as an adjuvant therapy for EC [342, 343].
Quinacrine (QC) is an oral, inexpensive drug that was initially used extensively as an antimalarial drug, which exhibited significant antitumor activity in chemo-resistant EC mouse xenografts [344], suggesting its potential role as an important maintenance therapy to standard chemotherapy for patients with chemo-resistant EC.
Pioglitazone, a non-oncological drug, is a peroxisome proliferator-activated receptor gamma (PPAR-γ) agonist. Compared to the standard paclitaxel treatment, it showed significant dose-dependent anticancer activity against EC induced by N-ethyl-N-nitrosourea (ENU) and estradiol hexadrobenzoate (EHB) [345].
Gestrinone, initially designed as a contraceptive, now is being used as a therapy for endometriosis in clinical. According to a recent study, it has the potential to protect against gynecological cancers through regulation of the JNK-P21 axis, repurposing it may be beneficial to patients with gynecological cancers, especially for CC patients [346].
Perphenazine, approved for psychosis therapy, has been identified as a promising drug against both progesterone-sensitive and progesterone-resistant EC [347]. In addition, another antipsychotics drug, chlorpromazine (CPZ), showed a significant antitumor effect via upregulating the expression of progesterone receptor B (PRB) to sensitize progestin-resistant EC cells to MPA, which suggests that CPZ might be a potential candidate drug for conservative treatment for EC, and the combination of CPZ and MPA could act as a therapeutic option for progestin resistant EC patients [348].
These repurposing strategies have provided significant advancements in the treatment for gynecological cancers; however, most data are based on observational level. Therefore, long-term clinical trials are strongly deserved to further investigate the clinical benefits.
10 Future perspectives: challenges and opportunities
Currently, nonsurgical treatments are urgently needed for gynecological malignancies, especially in endometrial cancer, for there are more young patients with a desire to maintain fertility due to the increasing incidence of EC and the younger age of onset [189, 349,350,351]. Therefore, acquired progesterone resistance should be thoroughly studied to provide therapeutic and prognostic targets or combination therapy strategies to overcome hormone therapy resistance in EC. Fortunately, antipsychotic drugs, perphenazine and CPZ, have been identified as promising drug candidates against progesterone-resistant EC [347, 348]. Previous study demonstrated that CPZ could upregulate the expression of PRB, suggesting that it might be a therapeutic option for progestin-resistant EC patients and act as a promising candidate drug for fertility-preserving options when combined with MPA [348]. Therefore, the potential clinical benefits should be further investigated by long-term clinical trials.
During the past decade, based on a further understanding of epigenetics, more epigenetic regulators with promising drug candidates have been validated in gynecological cancers, especially in EC and OC [38, 352]. A phase I study showed a synergetic effect of the combination of entionstat, a HDACi, and MPA in EC patients [93], so a treatment trial should be carried to further investigate the efficacy and toxicity of this combination therapy. Base on the knowledge that DNA methylation inhibitors may exert an effective role for EC treatment, so clinical trials involving these molecules are warranted to wait. Clinical trials of epigenetic monotherapy have proved disappointing in the treatment for OC patients [38, 101], suggesting that preclinical research should be focused on combination strategy with various epigenetic medications.
Although PARPis have made remarkable progress in the treatment of patients with EOC [353, 354], the application is restricted by the primary and acquired resistance. Several mechanisms underlying PARP inhibition resistance in OC have been reported [355,356,357,358], and new combinations of PARPis with antiangiogenic agents or immunotherapies are currently undergoing clinical evaluation [359,360,361], so more emphasis should be shifted to improve clinically applied strategies based on the results of these trials.
Furthermore, immunotherapies, mainly PD-1/PD-L1 inhibitors, have led to significant breakthroughs in personalized treatment for patients with dMMR or MSI EC and recurrent or metastatic CC [362]. However, only 10–30% of OC patients show long-term and durable responses to ICIs targeting PD-1/PD-L1 or CTLA-4, and the remaining majority do not respond [38]. Accordingly, how to tackle acquired resistance and the lack of response to ICIs is critical, and biomarkers, such as other immune checkpoints or coinhibitory receptors are mandatory to be identified. Notably, CD47 has become an immunotherapeutic hotspot and provided insights into new treatment options for patients with OC, based on the promising activities in preclinical models [303], thus related ongoing clinical trial (NCT03558139) are warranted to wait, and additional efforts should be made to facilitate the early application of research results to clinics.
11 Conclusion
Significant breakthroughs have been made in the further understanding and improvement in treatment strategies for gynecological cancers, with a growing number of preclinical research and clinical trials on potential molecular drugs and their corresponding targets are being studied. Hormone receptor-targeted therapies, the signaling pathway molecules (e.g., PI3K/AKT/mTOR), immune-targeted strategies (e.g., anti-PD-1/ PD-L1 agents) might be the promising treatment for EC patients, based on the histological type and molecular subtypes. As for OC, PARPis have revolutionized the management of patients with EOC, with the recommendation of niraparib, rucaparib, and olaparib for maintenance therapy depending on the identification of HRD (e.g., gBRCAm). Immunotherapeutic strategies are expected to be clinically beneficial for CC patients, due to their persistent oncogenic HPV infection. However, challenges still exist for the conservative treatment for EC, improvement of immunotherapy response, and how to overcome acquired resistance of PARPis and ICIs. Therefore, numerous efforts should be made to deepen our understanding of pathogenesis driving gynecological malignancies, so as to revolutionize targeted therapy by exploring new specific biomarkers. Drug repositioning strategy should not be ignored, which has emerged as a promising antitumor treatment. In addition, considerable progresses are being made toward combination therapy in clinical research, so the long-term phase II–III clinical trials involving hotspot drug candidates that we discussed above are strongly warranted to wait.
References
Crosbie, E. J., Kitson, S. J., McAlpine, J. N., Mukhopadhyay, A., Powell, M. E., & Singh, N. (2022). Endometrial cancer. The Lancet, 399(10333), 1412–1428. https://doi.org/10.1016/s0140-6736(22)00323-3
Makker, V., MacKay, H., Ray-Coquard, I., Levine, D. A., Westin, S. N., Aoki, D., et al. (2021). Endometrial cancer. Nature Reviews Disease Primers, 7(1), 88. https://doi.org/10.1038/s41572-021-00324-8
Cancer Genome Atlas Research, N, Kandoth, C., Schultz, N., Cherniack, A. D., Akbani, R., Liu, Y., et al. (2013). Integrated genomic characterization of endometrial carcinoma. Nature, 497(7447), 67–73. https://doi.org/10.1038/nature12113
Piulats, J. M., Guerra, E., Gil-Martin, M., Roman-Canal, B., Gatius, S., Sanz-Pamplona, R., et al. (2017). Molecular approaches for classifying endometrial carcinoma. Gynecologic Oncology, 145(1), 200–207. https://doi.org/10.1016/j.ygyno.2016.12.015
Gallo, A., Catena, U., Saccone, G., & Di Spiezio Sardo, A. (2021). Conservative surgery in endometrial cancer. Journal of Clinical Medicine, 11(1):183. https://doi.org/10.3390/jcm11010183
Connor, E. V., & Rose, P. G. (2018). Management strategies for recurrent endometrial cancer. Expert Review of Anticancer Therapy, 18(9), 873–885. https://doi.org/10.1080/14737140.2018.1491311
Sung, H., Ferlay, J., Siegel, R. L., Laversanne, M., Soerjomataram, I., Jemal, A., et al. (2021). Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians, 71(3), 209–249. https://doi.org/10.3322/caac.21660
Fabbro, M., Colombo, P. E., Leaha, C. M., Rouanet, P., Carrere, S., Quenet, F., et al. (2020). Conditional probability of survival and prognostic factors in long-term survivors of high-grade serous ovarian cancer. Cancers (Basel), 12(8):2184. https://doi.org/10.3390/cancers12082184
Marchetti, C., Pisano, C., Facchini, G., Bruni, G. S., Magazzino, F. P., Losito, S., et al. (2010). First-line treatment of advanced ovarian cancer: Current research and perspectives. Expert Review of Anticancer Therapy, 10(1), 47–60. https://doi.org/10.1586/era.09.167
Feliu, J., Heredia-Soto, V., Girones, R., Jimenez-Munarriz, B., Saldana, J., Guillen-Ponce, C., et al. (2020). Management of the toxicity of chemotherapy and targeted therapies in elderly cancer patients. Clinical and Translational Oncology, 22(4), 457–467. https://doi.org/10.1007/s12094-019-02167-y
Bast, R. C., Jr., Hennessy, B., & Mills, G. B. (2009). The biology of ovarian cancer: New opportunities for translation. Nature Reviews Cancer, 9(6), 415–428. https://doi.org/10.1038/nrc2644
Hawkins, S. M., & Matzuk, M. M. (2008). The menstrual cycle: Basic biology. Annals of the New York Academy of Sciences, 1135, 10–18. https://doi.org/10.1196/annals.1429.018
Groothuis, P. G., Dassen, H. H., Romano, A., & Punyadeera, C. (2007). Estrogen and the endometrium: Lessons learned from gene expression profiling in rodents and human. Human Reproduction Update, 13(4), 405–417. https://doi.org/10.1093/humupd/dmm009
Young, S. L. (2013). Oestrogen and progesterone action on endometrium: A translational approach to understanding endometrial receptivity. Reproductive Biomedicine Online, 27(5), 497–505. https://doi.org/10.1016/j.rbmo.2013.06.010
Rodriguez, A. C., Blanchard, Z., Maurer, K. A., & Gertz, J. (2019). Estrogen signaling in endometrial cancer: A key oncogenic pathway with several open questions. Hormones and Cancer, 10(2–3), 51–63. https://doi.org/10.1007/s12672-019-0358-9
Nelson, L. R., & Bulun, S. E. (2001). Estrogen production and action. Journal of the American Academy of Dermatology, 45(3 Suppl), S116-124. https://doi.org/10.1067/mjd.2001.117432
Daley-Brown, D., Oprea-Ilies, G. M., Lee, R., Pattillo, R., & Gonzalez-Perez, R. R. (2015). Molecular cues on obesity signals, tumor markers and endometrial cancer. Hormone Molecular Biology and Clinical Investigation, 21(1), 89–106. https://doi.org/10.1515/hmbci-2014-0049
Onstad, M. A., Schmandt, R. E., & Lu, K. H. (2016). Addressing the role of obesity in endometrial cancer risk, prevention, and treatment. Journal of Clinical Oncology, 34(35), 4225–4230. https://doi.org/10.1200/JCO.2016.69.4638
Deligdisch-Schor, L. (2020). Hormone therapy effects on the uterus. Advances in Experimental Medicine and Biology, 1242, 145–177. https://doi.org/10.1007/978-3-030-38474-6_8
Kim, J. J., & Chapman-Davis, E. (2010). Role of progesterone in endometrial cancer. Seminars in Reproductive Medicine, 28(1), 81–90. https://doi.org/10.1055/s-0029-1242998
Westin, S. N., Fellman, B., Sun, C. C., Broaddus, R. R., Woodall, M. L., Pal, N., et al. (2021). Prospective phase II trial of levonorgestrel intrauterine device: Nonsurgical approach for complex atypical hyperplasia and early-stage endometrial cancer. American Journal of Obstetrics and Gynecology, 224(2), 191 e191-191 e115. https://doi.org/10.1016/j.ajog.2020.08.032
Chaudhry, P., & Asselin, E. (2009). Resistance to chemotherapy and hormone therapy in endometrial cancer. Endocrine-Related Cancer, 16(2), 363–380. https://doi.org/10.1677/ERC-08-0266
Mitsushita, J., Toki, T., Kato, K., Fujii, S., & Konishi, I. (2000). Endometrial carcinoma remaining after term pregnancy following conservative treatment with medroxyprogesterone acetate. Gynecologic Oncology, 79(1), 129–132. https://doi.org/10.1006/gyno.2000.5896
Bulun, S. E., & Simpson, E. R. (2008). Aromatase expression in women’s cancers. Advances in Experimental Medicine and Biology, 630, 112–132. https://doi.org/10.1007/978-0-387-78818-0_8
Li, Y. F., Hu, W., Fu, S. Q., Li, J. D., Liu, J. H., & Kavanagh, J. J. (2008). Aromatase inhibitors in ovarian cancer: Is there a role? International Journal of Gynecological Cancer, 18(4), 600–614. https://doi.org/10.1111/j.1525-1438.2007.01075.x
Manna, P. R., Molehin, D., & Ahmed, A. U. (2016). Dysregulation of aromatase in breast, endometrial, and ovarian cancers: An overview of therapeutic strategies. Progress in Molecular Biology and Translational Science, 144, 487–537. https://doi.org/10.1016/bs.pmbts.2016.10.002
Kavanagh, J. J., Hu, W., Fu, S., Deavers, M., Moore, C., Coleman, R. L., et al. (2007). Anti-tumor activity of letrozole in patients with recurrent advanced low malignant potential or low-grade serous ovarian tumors. Journal of Clinical Oncology, 25(18), 5582–5582. https://doi.org/10.1200/jco.2007.25.18_suppl.5582
Colon-Otero, G., Zanfagnin, V., Hou, X., Foster, N. R., Asmus, E. J., Wahner Hendrickson, A., et al. (2020). Phase II trial of ribociclib and letrozole in patients with relapsed oestrogen receptor-positive ovarian or endometrial cancers. ESMO Open, 5(5), e000926. https://doi.org/10.1136/esmoopen-2020-000926
Konstantinopoulos, P. A., Lee, E. K., Xiong, N., Krasner, C., Campos, S., Kolin, D. L., et al. (2023). A phase II, two-stage study of letrozole and abemaciclib in estrogen receptor-positive recurrent endometrial cancer. Journal of Clinical Oncology, 41(3), 599–608. https://doi.org/10.1200/jco.22.00628
Heudel, P., Frenel, J. S., Dalban, C., Bazan, F., Joly, F., Arnaud, A., et al. (2022). Safety and efficacy of the mTOR inhibitor, vistusertib, combined with anastrozole in patients with hormone receptor-positive recurrent or metastatic endometrial cancer: The VICTORIA multicenter, open-label, phase 1/2 randomized clinical trial. JAMA Oncology, 8(7), 1001–1009. https://doi.org/10.1001/jamaoncol.2022.1047
Grundker, C., & Emons, G. (2021). Role of Gonadotropin-releasing hormone (GnRH) in ovarian cancer. Cells, 10(2):437. https://doi.org/10.3390/cells10020437
Emons, G., & Grundker, C. (2021). The Role of Gonadotropin-Releasing Hormone (GnRH) in Endometrial Cancer. Cells, 10(2):292. https://doi.org/10.3390/cells10020292
Xu, D. F., Liu, P. P., Fan, L., Xie, Q., Zhang, Z. Q., Wang, L. Q., et al. (2022). GnRH antagonist weakens endometrial stromal cells growth ability by decreasing c-kit receptor expression. Reproductive Biology and Endocrinology, 20(1), 29. https://doi.org/10.1186/s12958-021-00886-y
Rackow, B. W., Kliman, H. J., & Taylor, H. S. (2008). GnRH antagonists may affect endometrial receptivity. Fertility and Sterility, 89(5), 1234–1239. https://doi.org/10.1016/j.fertnstert.2007.04.060
Matsumoto, T., Suzuki, T., Nakamura, M., Yamamoto, M., Iizuka, T., Ono, M., et al. (2023). Androgen promotes squamous differentiation of atypical cells in cervical intraepithelial neoplasia via an ELF3-dependent pathway. Cancer Medicine. https://doi.org/10.1002/cam4.5824
Li, X., Ma, S., Deng, Y., Yi, P., & Yu, J. (2022). Targeting the RNA m(6)A modification for cancer immunotherapy. Molecular Cancer, 21(1), 76. https://doi.org/10.1186/s12943-022-01558-0
Kumar, A., Emdad, L., Fisher, P. B., & Das, S. K. (2023). Targeting epigenetic regulation for cancer therapy using small molecule inhibitors. Advances in Cancer Research, 158, 73–161. https://doi.org/10.1016/bs.acr.2023.01.001
Chen, S., Xie, P., Cowan, M., Huang, H., Cardenas, H., Keathley, R., et al. (2022). Epigenetic priming enhances antitumor immunity in platinum-resistant ovarian cancer. Journal of Clinical Investigation, 132(14):e158800. https://doi.org/10.1172/jci158800
Robertson, K. D., Uzvolgyi, E., Liang, G., Talmadge, C., Sumegi, J., Gonzales, F. A., et al. (1999). The human DNA methyltransferases (DNMTs) 1, 3a and 3b: Coordinate mRNA expression in normal tissues and overexpression in tumors. Nucleic Acids Research, 27(11), 2291–2298. https://doi.org/10.1093/nar/27.11.2291
Tao, M. H., & Freudenheim, J. L. (2010). DNA methylation in endometrial cancer. Epigenetics, 5(6), 491–498. https://doi.org/10.4161/epi.5.6.12431
Cornel, K. M. C., Wouters, K., Van de Vijver, K. K., van der Wurff, A. A. M., van Engeland, M., Kruitwagen, R., et al. (2019). Gene promoter methylation in endometrial carcinogenesis. Pathology Oncology Research, 25(2), 659–667. https://doi.org/10.1007/s12253-018-0489-2
Multinu, F., Chen, J., Madison, J. D., Torres, M., Casarin, J., Visscher, D., et al. (2020). Analysis of DNA methylation in endometrial biopsies to predict risk of endometrial cancer. Gynecologic Oncology, 156(3), 682–688. https://doi.org/10.1016/j.ygyno.2019.12.023
Yuan, J., Mao, Z., Lu, Q., Xu, P., Wang, C., Xu, X., et al. (2021). Hypermethylated PCDHGB7 as a biomarker for early detection of endometrial cancer in endometrial brush samples and cervical scrapings. Frontiers in Molecular Biosciences, 8, 774215. https://doi.org/10.3389/fmolb.2021.774215
Szalmas, A., & Konya, J. (2009). Epigenetic alterations in cervical carcinogenesis. Seminars in Cancer Biology, 19(3), 144–152. https://doi.org/10.1016/j.semcancer.2009.02.011
Choi, C. H., Lee, K. M., Choi, J. J., Kim, T. J., Kim, W. Y., Lee, J. W., et al. (2007). Hypermethylation and loss of heterozygosity of tumor suppressor genes on chromosome 3p in cervical cancer. Cancer Letters, 255(1), 26–33. https://doi.org/10.1016/j.canlet.2007.03.015
Ki, K. D., Lee, S. K., Tong, S. Y., Lee, J. M., Song, D. H., & Chi, S. G. (2008). Role of 5′-CpG island hypermethylation of the FHIT gene in cervical carcinoma. Journal of Gynecologic Oncology, 19(2), 117–122. https://doi.org/10.3802/jgo.2008.19.2.117
Kim, J. H., Choi, Y. D., Lee, J. S., Lee, J. H., Nam, J. H., & Choi, C. (2010). Assessment of DNA methylation for the detection of cervical neoplasia in liquid-based cytology specimens. Gynecologic Oncology, 116(1), 99–104. https://doi.org/10.1016/j.ygyno.2009.09.032
Yang, N., Nijhuis, E. R., Volders, H. H., Eijsink, J. J., Lendvai, A., Zhang, B., et al. (2010). Gene promoter methylation patterns throughout the process of cervical carcinogenesis. Cellular Oncology, 32(1–2), 131–143. https://doi.org/10.3233/CLO-2009-0510
Yanatatsaneejit, P., Mutirangura, A., & Kitkumthorn, N. (2011). Human papillomavirus’s physical state and cyclin A1 promoter methylation in cervical cancer. International Journal of Gynecological Cancer, 21(5), 902–906. https://doi.org/10.1097/IGC.0b013e3182158683
Kitkumthorn, N., Yanatatsanajit, P., Kiatpongsan, S., Phokaew, C., Triratanachat, S., Trivijitsilp, P., et al. (2006). Cyclin A1 promoter hypermethylation in human papillomavirus-associated cervical cancer. BMC Cancer, 6, 55. https://doi.org/10.1186/1471-2407-6-55
Dong, S., Lu, Q., Xu, P., Chen, L., Duan, X., Mao, Z., et al. (2021). Hypermethylated PCDHGB7 as a universal cancer only marker and its application in early cervical cancer screening. Clinical and Translational Medicine, 11(6), e457. https://doi.org/10.1002/ctm2.457
Aoki, K., & Taketo, M. M. (2007). Adenomatous polyposis coli (APC): A multi-functional tumor suppressor gene. Journal of Cell Science, 120(Pt 19), 3327–3335. https://doi.org/10.1242/jcs.03485
Stafl, A., & Mattingly, R. F. (1975). Angiogenesis of cervical neoplasia. American Journal of Obstetrics and Gynecology, 121(6), 845–852.
Zhang, Z., Huettner, P. C., Nguyen, L., Bidder, M., Funk, M. C., Li, J., et al. (2006). Aberrant promoter methylation and silencing of the POU2F3 gene in cervical cancer. Oncogene, 25(39), 5436–5445. https://doi.org/10.1038/sj.onc.1209530
Overmeer, R. M., Henken, F. E., Snijders, P. J., Claassen-Kramer, D., Berkhof, J., Helmerhorst, T. J., et al. (2008). Association between dense CADM1 promoter methylation and reduced protein expression in high-grade CIN and cervical SCC. The Journal of Pathology, 215(4), 388–397. https://doi.org/10.1002/path.2367
Narayan, G., Arias-Pulido, H., Koul, S., Vargas, H., Zhang, F. F., Villella, J., et al. (2003). Frequent promoter methylation of CDH1, DAPK, RARB, and HIC1 genes in carcinoma of cervix uteri: Its relationship to clinical outcome. Molecular Cancer, 2, 24. https://doi.org/10.1186/1476-4598-2-24
Steenbergen, R. D., Kramer, D., Braakhuis, B. J., Stern, P. L., Verheijen, R. H., Meijer, C. J., et al. (2004). TSLC1 gene silencing in cervical cancer cell lines and cervical neoplasia. Journal of the National Cancer Institute, 96(4), 294–305. https://doi.org/10.1093/jnci/djh031
Sova, P., Feng, Q., Geiss, G., Wood, T., Strauss, R., Rudolf, V., et al. (2006). Discovery of novel methylation biomarkers in cervical carcinoma by global demethylation and microarray analysis. Cancer Epidemiology, Biomarkers & Prevention, 15(1), 114–123. https://doi.org/10.1158/1055-9965.EPI-05-0323
Wisman, G. B., Nijhuis, E. R., Hoque, M. O., Reesink-Peters, N., Koning, A. J., Volders, H. H., et al. (2006). Assessment of gene promoter hypermethylation for detection of cervical neoplasia. International Journal of Cancer, 119(8), 1908–1914. https://doi.org/10.1002/ijc.22060
Cho, S., Cinghu, S., Yu, J. R., & Park, W. Y. (2011). Helicase-like transcription factor confers radiation resistance in cervical cancer through enhancing the DNA damage repair capacity. Journal of Cancer Research and Clinical Oncology, 137(4), 629–637. https://doi.org/10.1007/s00432-010-0925-5
Hesson, L. B., Cooper, W. N., & Latif, F. (2007). The role of RASSF1A methylation in cancer. Disease Markers, 23(1–2), 73–87. https://doi.org/10.1155/2007/291538
Yang, H. J., Liu, V. W., Wang, Y., Chan, K. Y., Tsang, P. C., Khoo, U. S., et al. (2004). Detection of hypermethylated genes in tumor and plasma of cervical cancer patients. Gynecologic Oncology, 93(2), 435–440. https://doi.org/10.1016/j.ygyno.2004.01.039
Kuzmin, I., Liu, L., Dammann, R., Geil, L., Stanbridge, E. J., Wilczynski, S. P., et al. (2003). Inactivation of RAS association domain family 1A gene in cervical carcinomas and the role of human papillomavirus infection. Cancer Research, 63(8), 1888–1893.
Cohen, Y., Singer, G., Lavie, O., Dong, S. M., Beller, U., & Sidransky, D. (2003). The RASSF1A tumor suppressor gene is commonly inactivated in adenocarcinoma of the uterine cervix. Clinical Cancer Research, 9(8), 2981–2984.
Watts, G. S., Futscher, B. W., Holtan, N., Degeest, K., Domann, F. E., & Rose, S. L. (2008). DNA methylation changes in ovarian cancer are cumulative with disease progression and identify tumor stage. BMC Medical Genomics, 1, 47. https://doi.org/10.1186/1755-8794-1-47
Wei, S. H., Balch, C., Paik, H. H., Kim, Y. S., Baldwin, R. L., Liyanarachchi, S., et al. (2006). Prognostic DNA methylation biomarkers in ovarian cancer. Clinical Cancer Research, 12(9), 2788–2794. https://doi.org/10.1158/1078-0432.CCR-05-1551
Li, M., Balch, C., Montgomery, J. S., Jeong, M., Chung, J. H., Yan, P., et al. (2009). Integrated analysis of DNA methylation and gene expression reveals specific signaling pathways associated with platinum resistance in ovarian cancer. BMC Medical Genomics, 2, 34. https://doi.org/10.1186/1755-8794-2-34
Flanagan, J. M., Wilhelm-Benartzi, C. S., Metcalf, M., Kaye, S. B., & Brown, R. (2013). Association of somatic DNA methylation variability with progression-free survival and toxicity in ovarian cancer patients. Annals of Oncology, 24(11), 2813–2818. https://doi.org/10.1093/annonc/mdt370
Flanagan, J. M., Wilson, A., Koo, C., Masrour, N., Gallon, J., Loomis, E., et al. (2017). Platinum-based chemotherapy induces methylation changes in blood dna associated with overall survival in patients with ovarian cancer. Clinical Cancer Research, 23(9), 2213–2222. https://doi.org/10.1158/1078-0432.CCR-16-1754
Parashar, S., Cheishvili, D., Mahmood, N., Arakelian, A., Tanvir, I., Khan, H. A., et al. (2018). DNA methylation signatures of breast cancer in peripheral T-cells. BMC Cancer, 18(1), 574. https://doi.org/10.1186/s12885-018-4482-7
Zhang, Y., Petropoulos, S., Liu, J., Cheishvili, D., Zhou, R., Dymov, S., et al. (2018). The signature of liver cancer in immune cells DNA methylation. Clinical Epigenetics, 10, 8. https://doi.org/10.1186/s13148-017-0436-1
Gui, T., Liu, M., Yao, B., Jiang, H., Yang, D., Li, Q., et al. (2021). TCF3 is epigenetically silenced by EZH2 and DNMT3B and functions as a tumor suppressor in endometrial cancer. Cell Death and Differentiation, 28(12), 3316–3328. https://doi.org/10.1038/s41418-021-00824-w
Verdone, L., Caserta, M., & Di Mauro, E. (2005). Role of histone acetylation in the control of gene expression. Biochemistry and Cell Biology, 83(3), 344–353. https://doi.org/10.1139/o05-041
Gujral, P., Mahajan, V., Lissaman, A. C., & Ponnampalam, A. P. (2020). Histone acetylation and the role of histone deacetylases in normal cyclic endometrium. Reproductive Biology and Endocrinology, 18(1), 84. https://doi.org/10.1186/s12958-020-00637-5
Singh, B. N., Zhang, G., Hwa, Y. L., Li, J., Dowdy, S. C., & Jiang, S. W. (2010). Nonhistone protein acetylation as cancer therapy targets. Expert Review of Anticancer Therapy, 10(6), 935–954. https://doi.org/10.1586/era.10.62
Jenuwein, T., & Allis, C. D. (2001). Translating the histone code. Science, 293(5532), 1074–1080. https://doi.org/10.1126/science.1063127
Ren, J., Zhang, J., Cai, H., Li, Y., Zhang, Y., Zhang, X., et al. (2014). HDAC as a therapeutic target for treatment of endometrial cancers. Current Pharmaceutical Design, 20(11), 1847–1856. https://doi.org/10.2174/13816128113199990528
Singh, B. N., Zhou, H., Li, J., Tipton, T., Wang, B., Shao, G., et al. (2011). Preclinical studies on histone deacetylase inhibitors as therapeutic reagents for endometrial and ovarian cancers. Future Oncology, 7(12), 1415–1428. https://doi.org/10.2217/fon.11.124
Danam, R. P., Howell, S. R., Brent, T. P., & Harris, L. C. (2005). Epigenetic regulation of O6-methylguanine-DNA methyltransferase gene expression by histone acetylation and methyl-CpG binding proteins. Molecular Cancer Therapeutics, 4(1), 61–69.
Lee, J., Yoon, Y. S., & Chung, J. H. (2008). Epigenetic silencing of the WNT antagonist DICKKOPF-1 in cervical cancer cell lines. Gynecologic Oncology, 109(2), 270–274. https://doi.org/10.1016/j.ygyno.2008.01.034
Bodily, J. M., Mehta, K. P., & Laimins, L. A. (2011). Human papillomavirus E7 enhances hypoxia-inducible factor 1-mediated transcription by inhibiting binding of histone deacetylases. Cancer Research, 71(3), 1187–1195. https://doi.org/10.1158/0008-5472.CAN-10-2626
Sivakumar, K. K., Stanley, J. A., Behlen, J. C., Wuri, L., Dutta, S., Wu, J., et al. (2022). Inhibition of Sirtuin-1 hyperacetylates p53 and abrogates Sirtuin-1-p53 interaction in Cr(VI)-induced apoptosis in the ovary. Reproductive Toxicology, 109, 121–134. https://doi.org/10.1016/j.reprotox.2022.03.007
Wang, Z., Yan, Y., Lou, Y., Huang, X., Liu, L., Weng, Z., et al. (2023). Diallyl trisulfide alleviates chemotherapy sensitivity of ovarian cancer via the AMPK/SIRT1/PGC1α pathway. Cancer Science, 114(2), 357–369. https://doi.org/10.1111/cas.15627
Chen, F., Kolben, T., Meister, S., Czogalla, B., Kolben, T. M., Hester, A., et al. (2022). The role of resveratrol, sirtuin1 and RXRα as prognostic markers in ovarian cancer. Archives of Gynecology and Obstetrics, 305(6), 1559–1572. https://doi.org/10.1007/s00404-021-06262-w
Lu, T. Y., Kao, C. F., Lin, C. T., Huang, D. Y., Chiu, C. Y., Huang, Y. S., et al. (2009). DNA methylation and histone modification regulate silencing of OPG during tumor progression. Journal of Cellular Biochemistry, 108(1), 315–325. https://doi.org/10.1002/jcb.22256
Zhang, Z., Joh, K., Yatsuki, H., Zhao, W., Soejima, H., Higashimoto, K., et al. (2007). Retinoic acid receptor beta2 is epigenetically silenced either by DNA methylation or repressive histone modifications at the promoter in cervical cancer cells. Cancer Letters, 247(2), 318–327. https://doi.org/10.1016/j.canlet.2006.05.013
Liu, D., Zhou, P., Zhang, L., Gong, W., Huang, G., Zheng, Y., et al. (2012). HDAC1/DNMT3A-containing complex is associated with suppression of Oct4 in cervical cancer cells. Biochemistry (Moscow), 77(8), 934–940. https://doi.org/10.1134/S0006297912080159
Li, B., Lu, W., Qu, J., Zhang, Y., & Wan, X. (2017). DICER1 regulates endometrial carcinoma invasion via histone acetylation and methylation. Journal of Cancer, 8(6), 933–939. https://doi.org/10.7150/jca.17435
Duenas-Gonzalez, A., Medina-Franco, J. L., Chavez-Blanco, A., Dominguez-Gomez, G., & Fernandez-de Gortari, E. (2016). Developmental DNA methyltransferase inhibitors in the treatment of gynecologic cancers. Expert Opinion on Pharmacotherapy, 17(3), 323–338. https://doi.org/10.1517/14656566.2016.1118053
Inoue, F., Sone, K., Toyohara, Y., Takahashi, Y., Kukita, A., Hara, A., et al. (2021). Targeting epigenetic regulators for endometrial cancer therapy: Its molecular biology and potential clinical applications. International Journal of Molecular Sciences, 22(5):2305. https://doi.org/10.3390/ijms22052305
Ihira, K., Dong, P., Xiong, Y., Watari, H., Konno, Y., Hanley, S. J., et al. (2017). EZH2 inhibition suppresses endometrial cancer progression via miR-361/Twist axis. Oncotarget, 8(8), 13509–13520. https://doi.org/10.18632/oncotarget.14586
Oki, S., Sone, K., Oda, K., Hamamoto, R., Ikemura, M., Maeda, D., et al. (2017). Oncogenic histone methyltransferase EZH2: A novel prognostic marker with therapeutic potential in endometrial cancer. Oncotarget, 8(25), 40402–40411. https://doi.org/10.18632/oncotarget.16316
Duska, L. R., Filiaci, V. L., Walker, J. L., Holman, L. L., Hill, E. K., Moore, R. G., et al. (2021). A surgical window trial evaluating medroxyprogesterone acetate with or without entinostat in patients with endometrial cancer and validation of biomarkers of cellular response. Clinical Cancer Research, 27(10), 2734–2741. https://doi.org/10.1158/1078-0432.Ccr-20-4618
Zambrano, P., Segura-Pacheco, B., Perez-Cardenas, E., Cetina, L., Revilla-Vazquez, A., Taja-Chayeb, L., et al. (2005). A phase I study of hydralazine to demethylate and reactivate the expression of tumor suppressor genes. BMC Cancer, 5, 44. https://doi.org/10.1186/1471-2407-5-44
de la Cruz-Hernandez, E., Perez-Cardenas, E., Contreras-Paredes, A., Cantu, D., Mohar, A., Lizano, M., et al. (2007). The effects of DNA methylation and histone deacetylase inhibitors on human papillomavirus early gene expression in cervical cancer, an in vitro and clinical study. Virology Journal, 4, 18. https://doi.org/10.1186/1743-422X-4-18
Chen, J., Ghazawi, F. M., Bakkar, W., & Li, Q. (2006). Valproic acid and butyrate induce apoptosis in human cancer cells through inhibition of gene expression of Akt/protein kinase B. Molecular Cancer, 5, 71. https://doi.org/10.1186/1476-4598-5-71
You, J. S., Kang, J. K., Lee, E. K., Lee, J. C., Lee, S. H., Jeon, Y. J., et al. (2008). Histone deacetylase inhibitor apicidin downregulates DNA methyltransferase 1 expression and induces repressive histone modifications via recruitment of corepressor complex to promoter region in human cervix cancer cells. Oncogene, 27(10), 1376–1386. https://doi.org/10.1038/sj.onc.1210776
Jiang, Y., Wang, Y., Su, Z., Yang, L., Guo, W., Liu, W., et al. (2010). Synergistic induction of apoptosis in HeLa cells by the proteasome inhibitor bortezomib and histone deacetylase inhibitor SAHA. Molecular Medicine Reports, 3(4), 613–619. https://doi.org/10.3892/mmr_00000305
Fu, S., Hu, W., Iyer, R., Kavanagh, J. J., Coleman, R. L., Levenback, C. F., et al. (2011). Phase 1b–2a study to reverse platinum resistance through use of a hypomethylating agent, azacitidine, in patients with platinum-resistant or platinum-refractory epithelial ovarian cancer. Cancer, 117(8), 1661–1669. https://doi.org/10.1002/cncr.25701
Matei, D., Fang, F., Shen, C., Schilder, J., Arnold, A., Zeng, Y., et al. (2012). Epigenetic resensitization to platinum in ovarian cancer. Cancer Research, 72(9), 2197–2205. https://doi.org/10.1158/0008-5472.CAN-11-3909
Nervi, C., De Marinis, E., & Codacci-Pisanelli, G. (2015). Epigenetic treatment of solid tumours: A review of clinical trials. Clinical Epigenetics, 7, 127. https://doi.org/10.1186/s13148-015-0157-2
Matulonis, U. A., Oza, A. M., Ho, T. W., & Ledermann, J. A. (2015). Intermediate clinical endpoints: A bridge between progression-free survival and overall survival in ovarian cancer trials. Cancer, 121(11), 1737–1746. https://doi.org/10.1002/cncr.29082
Jones, P. A., Issa, J. P., & Baylin, S. (2016). Targeting the cancer epigenome for therapy. Nature Reviews Genetics, 17(10), 630–641. https://doi.org/10.1038/nrg.2016.93
Falchook, G. S., Fu, S., Naing, A., Hong, D. S., Hu, W., Moulder, S., et al. (2013). Methylation and histone deacetylase inhibition in combination with platinum treatment in patients with advanced malignancies. Investigational New Drugs, 31(5), 1192–1200. https://doi.org/10.1007/s10637-013-0003-3
Wang, L., Amoozgar, Z., Huang, J., Saleh, M. H., Xing, D., Orsulic, S., et al. (2015). Decitabine enhances lymphocyte migration and function and synergizes with CTLA-4 Blockade in a murine ovarian cancer model. Cancer Immunology Research, 3(9), 1030–1041. https://doi.org/10.1158/2326-6066.CIR-15-0073
Juergens, R. A., Wrangle, J., Vendetti, F. P., Murphy, S. C., Zhao, M., Coleman, B., et al. (2011). Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discovery, 1(7), 598–607. https://doi.org/10.1158/2159-8290.CD-11-0214
Odunsi, K., Matsuzaki, J., James, S. R., Mhawech-Fauceglia, P., Tsuji, T., Miller, A., et al. (2014). Epigenetic potentiation of NY-ESO-1 vaccine therapy in human ovarian cancer. Cancer Immunology Research, 2(1), 37–49. https://doi.org/10.1158/2326-6066.CIR-13-0126
Zhang, Z., Ma, P., Jing, Y., Yan, Y., Cai, M. C., Zhang, M., et al. (2016). BET bromodomain inhibition as a therapeutic strategy in ovarian cancer by downregulating FoxM1. Theranostics, 6(2), 219–230. https://doi.org/10.7150/thno.13178
McCabe, M. T., Graves, A. P., Ganji, G., Diaz, E., Halsey, W. S., Jiang, Y., et al. (2012). Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proceedings of the National Academy of Sciences of the United States of America, 109(8), 2989–2994. https://doi.org/10.1073/pnas.1116418109
Yang, L., Zhang, Y., Shan, W., Hu, Z., Yuan, J., Pi, J., et al. (2017). Repression of BET activity sensitizes homologous recombination-proficient cancers to PARP inhibition. Science Translational Medicine, 9(400):eaal1645. https://doi.org/10.1126/scitranslmed.aal1645
Favier, A., Rocher, G., Larsen, A. K., Delangle, R., Uzan, C., Sabbah, M., et al. (2021). MicroRNA as epigenetic modifiers in endometrial cancer: A systematic review. Cancers (Basel), 13(5):1137. https://doi.org/10.3390/cancers13051137
Yanokura, M., Banno, K., & Aoki, D. (2020). MicroRNA34b expression enhances chemosensitivity of endometrial cancer cells to paclitaxel. International Journal of Oncology, 57(5), 1145–1156. https://doi.org/10.3892/ijo.2020.5127
Ramon, L. A., Braza-Boils, A., Gilabert, J., Chirivella, M., Espana, F., Estelles, A., et al. (2012). microRNAs related to angiogenesis are dysregulated in endometrioid endometrial cancer. Human Reproduction, 27(10), 3036–3045. https://doi.org/10.1093/humrep/des292
Xiao, L., He, Y., Peng, F., Yang, J., & Yuan, C. (2020). Endometrial cancer cells promote M2-like macrophage polarization by delivering exosomal miRNA-21 under hypoxia condition. Journal of Immunology Research, 2020, 9731049. https://doi.org/10.1155/2020/9731049
Fan, J. T., Zhou, Z. Y., Luo, Y. L., Luo, Q., Chen, S. B., Zhao, J. C., et al. (2021). Exosomal lncRNA NEAT1 from cancer-associated fibroblasts facilitates endometrial cancer progression via miR-26a/b-5p-mediated STAT3/YKL-40 signaling pathway. Neoplasia, 23(7), 692–703. https://doi.org/10.1016/j.neo.2021.05.004
Wang, J., Gong, X., Yang, L., Li, L., Gao, X., Ni, T., et al. (2022). Loss of exosomal miR-26a-5p contributes to endometrial cancer lymphangiogenesis and lymphatic metastasis. Clinical and Translational Medicine, 12(5), e846. https://doi.org/10.1002/ctm2.846
Delangle, R., De Foucher, T., Larsen, A. K., Sabbah, M., Azais, H., Bendifallah, S., et al. (2019). The use of microRNAs in the management of endometrial cancer: A meta-analysis. Cancers (Basel), 11(6):832. https://doi.org/10.3390/cancers11060832
Wang, T., Kong, S., Tao, M., & Ju, S. (2020). The potential role of RNA N6-methyladenosine in cancer progression. Molecular Cancer, 19(1), 88. https://doi.org/10.1186/s12943-020-01204-7
Liu, J., Eckert, M. A., Harada, B. T., Liu, S. M., Lu, Z., Yu, K., et al. (2018). m(6)A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nature Cell Biology, 20(9), 1074–1083. https://doi.org/10.1038/s41556-018-0174-4
Zhang, L., Wan, Y., Zhang, Z., Jiang, Y., Lang, J., Cheng, W., et al. (2021). FTO demethylates m6A modifications in HOXB13 mRNA and promotes endometrial cancer metastasis by activating the WNT signalling pathway. RNA Biology, 18(9), 1265–1278. https://doi.org/10.1080/15476286.2020.1841458
Pu, X., Gu, Z., & Gu, Z. (2020). ALKBH5 regulates IGF1R expression to promote the proliferation and tumorigenicity of endometrial cancer. Journal of Cancer, 11(19), 5612–5622. https://doi.org/10.7150/jca.46097
Hong, L., Pu, X., Gan, H., Weng, L., & Zheng, Q. (2021). YTHDF2 inhibit the tumorigenicity of endometrial cancer via downregulating the expression of IRS1 methylated with m(6)A. Journal of Cancer, 12(13), 3809–3818. https://doi.org/10.7150/jca.54527
Zhang, C., Huang, S., Zhuang, H., Ruan, S., Zhou, Z., Huang, K., et al. (2020). YTHDF2 promotes the liver cancer stem cell phenotype and cancer metastasis by regulating OCT4 expression via m6A RNA methylation. Oncogene, 39(23), 4507–4518. https://doi.org/10.1038/s41388-020-1303-7
Zhang, L., Wan, Y., Zhang, Z., Jiang, Y., Gu, Z., Ma, X., et al. (2021). IGF2BP1 overexpression stabilizes PEG10 mRNA in an m6A-dependent manner and promotes endometrial cancer progression. Theranostics, 11(3), 1100–1114. https://doi.org/10.7150/thno.49345
Xue, T., Liu, X., Zhang, M., E, Q., Liu, S., Zou, M., et al. (2021). PADI2-Catalyzed MEK1 Citrullination activates ERK1/2 and promotes IGF2BP1-mediated SOX2 mRNA stability in endometrial cancer. Advanced Science (Weinh), 8(6), 2002831. https://doi.org/10.1002/advs.202002831
Berger, A. A., Dao, F., & Levine, D. A. (2021). Angiogenesis in endometrial carcinoma: Therapies and biomarkers, current options, and future perspectives. Gynecologic Oncology, 160(3), 844–850. https://doi.org/10.1016/j.ygyno.2020.12.016
Ali, S. H., O’Donnell, A. L., Balu, D., Pohl, M. B., Seyler, M. J., Mohamed, S., et al. (2000). Estrogen receptor-alpha in the inhibition of cancer growth and angiogenesis. Cancer Research, 60(24), 7094–7098.
Ali, S. H., O’Donnell, A. L., Mohamed, S., Mousa, S., & Dandona, P. (2004). Overexpression of estrogen receptor-alpha in the endometrial carcinoma cell line Ishikawa: Inhibition of growth and angiogenic factors. Gynecologic Oncology, 95(3), 637–645. https://doi.org/10.1016/j.ygyno.2004.08.034
Tomao, F., Papa, A., Rossi, L., Zaccarelli, E., Caruso, D., Zoratto, F., et al. (2014). Angiogenesis and antiangiogenic agents in cervical cancer. Oncotargets and Therapy, 7, 2237–2248. https://doi.org/10.2147/OTT.S68286
Willmott, L. J., & Monk, B. J. (2009). Cervical cancer therapy: Current, future and anti-angiogensis targeted treatment. Expert Review of Anticancer Therapy, 9(7), 895–903. https://doi.org/10.1586/era.09.58
Nakamura, M., Bodily, J. M., Beglin, M., Kyo, S., Inoue, M., & Laimins, L. A. (2009). Hypoxia-specific stabilization of HIF-1alpha by human papillomaviruses. Virology, 387(2), 442–448. https://doi.org/10.1016/j.virol.2009.02.036
Koukourakis, M. I., Papazoglou, D., Giatromanolaki, A., Bougioukas, G., Maltezos, E., & Sivridis, E. (2004). VEGF gene sequence variation defines VEGF gene expression status and angiogenic activity in non-small cell lung cancer. Lung Cancer, 46(3), 293–298. https://doi.org/10.1016/j.lungcan.2004.04.037
Renner, W., Kotschan, S., Hoffmann, C., Obermayer-Pietsch, B., & Pilger, E. (2000). A common 936 C/T mutation in the gene for vascular endothelial growth factor is associated with vascular endothelial growth factor plasma levels. Journal of Vascular Research, 37(6), 443–448. https://doi.org/10.1159/000054076
Watson, C. J., Webb, N. J., Bottomley, M. J., & Brenchley, P. E. (2000). Identification of polymorphisms within the vascular endothelial growth factor (VEGF) gene: Correlation with variation in VEGF protein production. Cytokine, 12(8), 1232–1235. https://doi.org/10.1006/cyto.2000.0692
Stevens, A., Soden, J., Brenchley, P. E., Ralph, S., & Ray, D. W. (2003). Haplotype analysis of the polymorphic human vascular endothelial growth factor gene promoter. Cancer Research, 63(4), 812–816.
Yetkin-Arik, B., Kastelein, A. W., Klaassen, I., Jansen, C., Latul, Y. P., Vittori, M., et al. (2021). Angiogenesis in gynecological cancers and the options for anti-angiogenesis therapy. Biochimica et Biophysica Acta Reviews on Cancer, 1875(1), 188446. https://doi.org/10.1016/j.bbcan.2020.188446
Granot, D., Addadi, Y., Kalchenko, V., Harmelin, A., Kunz-Schughart, L. A., & Neeman, M. (2007). In vivo imaging of the systemic recruitment of fibroblasts to the angiogenic rim of ovarian carcinoma tumors. Cancer Research, 67(19), 9180–9189. https://doi.org/10.1158/0008-5472.CAN-07-0684
Hira, V. V., Verbovsek, U., Breznik, B., Srdic, M., Novinec, M., Kakar, H., et al. (2017). Cathepsin K cleavage of SDF-1alpha inhibits its chemotactic activity towards glioblastoma stem-like cells. Biochimica et Biophysica Acta Molecular Cell Research, 1864(3), 594–603. https://doi.org/10.1016/j.bbamcr.2016.12.021
Hira, V. V. V., Breznik, B., Vittori, M., Loncq de Jong, A., Mlakar, J., Oostra, R. J., et al. (2020). Similarities between stem cell niches in glioblastoma and bone marrow: Rays of hope for novel treatment strategies. Journal of Histochemistry and Cytochemistry, 68(1), 33–57. https://doi.org/10.1369/0022155419878416
Krishnapriya, S., Sidhanth, C., Manasa, P., Sneha, S., Bindhya, S., Nagare, R. P., et al. (2019). Cancer stem cells contribute to angiogenesis and lymphangiogenesis in serous adenocarcinoma of the ovary. Angiogenesis, 22(3), 441–455. https://doi.org/10.1007/s10456-019-09669-x
Ayala-Dominguez, L., Olmedo-Nieva, L., Munoz-Bello, J. O., Contreras-Paredes, A., Manzo-Merino, J., Martinez-Ramirez, I., et al. (2019). Mechanisms of vasculogenic mimicry in ovarian cancer. Frontiers in Oncology, 9, 998. https://doi.org/10.3389/fonc.2019.00998
Latacz, E., Caspani, E., Barnhill, R., Lugassy, C., Verhoef, C., Grunhagen, D., et al. (2020). Pathological features of vessel co-option versus sprouting angiogenesis. Angiogenesis, 23(1), 43–54. https://doi.org/10.1007/s10456-019-09690-0
Markowska, A., Sajdak, S., Markowska, J., & Huczynski, A. (2017). Angiogenesis and cancer stem cells: New perspectives on therapy of ovarian cancer. European Journal of Medicinal Chemistry, 142, 87–94. https://doi.org/10.1016/j.ejmech.2017.06.030
Burger, R. A., Sill, M. W., Monk, B. J., Greer, B. E., & Sorosky, J. I. (2007). Phase II trial of bevacizumab in persistent or recurrent epithelial ovarian cancer or primary peritoneal cancer: A Gynecologic Oncology Group study. Journal of Clinical Oncology, 25(33), 5165–5171. https://doi.org/10.1200/JCO.2007.11.5345
Tewari, K. S., Sill, M. W., Long, H. J., 3rd., Penson, R. T., Huang, H., Ramondetta, L. M., et al. (2014). Improved survival with bevacizumab in advanced cervical cancer. New England Journal of Medicine, 370(8), 734–743. https://doi.org/10.1056/NEJMoa1309748
Pfaendler, K. S., Liu, M. C., & Tewari, K. S. (2018). Bevacizumab in Cervical Cancer: 5 Years After. Cancer Journal, 24(4), 187–192. https://doi.org/10.1097/PPO.0000000000000324
Burger, R. A., Brady, M. F., Bookman, M. A., Fleming, G. F., Monk, B. J., Huang, H., et al. (2011). Incorporation of bevacizumab in the primary treatment of ovarian cancer. New England Journal of Medicine, 365(26), 2473–2483. https://doi.org/10.1056/NEJMoa1104390
Oza, A. M., Cook, A. D., Pfisterer, J., Embleton, A., Ledermann, J. A., Pujade-Lauraine, E., et al. (2015). Standard chemotherapy with or without bevacizumab for women with newly diagnosed ovarian cancer (ICON7): Overall survival results of a phase 3 randomised trial. The Lancet Oncology, 16(8), 928–936. https://doi.org/10.1016/S1470-2045(15)00086-8
Perren, T. J., Swart, A. M., Pfisterer, J., Ledermann, J. A., Pujade-Lauraine, E., Kristensen, G., et al. (2011). A phase 3 trial of bevacizumab in ovarian cancer. New England Journal of Medicine, 365(26), 2484–2496. https://doi.org/10.1056/NEJMoa1103799
Coleman, R. L., Brady, M. F., Herzog, T. J., Sabbatini, P., Armstrong, D. K., Walker, J. L., et al. (2017). Bevacizumab and paclitaxel-carboplatin chemotherapy and secondary cytoreduction in recurrent, platinum-sensitive ovarian cancer (NRG Oncology/Gynecologic Oncology Group study GOG-0213): A multicentre, open-label, randomised, phase 3 trial. The Lancet Oncology, 18(6), 779–791. https://doi.org/10.1016/S1470-2045(17)30279-6
Aghajanian, C., Blank, S. V., Goff, B. A., Judson, P. L., Teneriello, M. G., Husain, A., et al. (2012). OCEANS: A randomized, double-blind, placebo-controlled phase III trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. Journal of Clinical Oncology, 30(17), 2039–2045. https://doi.org/10.1200/JCO.2012.42.0505
Lorusso, D., Ferrandina, G., Colombo, N., Pignata, S., Pietragalla, A., Sonetto, C., et al. (2019). Carboplatin-paclitaxel compared to carboplatin-paclitaxel-bevacizumab in advanced or recurrent endometrial cancer: MITO END-2 - a randomized phase II trial. Gynecologic Oncology, 155(3), 406–412. https://doi.org/10.1016/j.ygyno.2019.10.013
Aghajanian, C., Filiaci, V., Dizon, D. S., Carlson, J. W., Powell, M. A., Secord, A. A., et al. (2018). A phase II study of frontline paclitaxel/carboplatin/bevacizumab, paclitaxel/carboplatin/temsirolimus, or ixabepilone/carboplatin/bevacizumab in advanced/recurrent endometrial cancer. Gynecologic Oncology, 150(2), 274–281. https://doi.org/10.1016/j.ygyno.2018.05.018
Monk, B. J., & Pandite, L. N. (2011). Survival data from a phase II, open-label study of pazopanib or lapatinib monotherapy in patients with advanced and recurrent cervical cancer. Journal of Clinical Oncology, 29(36), 4845. https://doi.org/10.1200/JCO.2011.38.8777
van der Steen, M. J., de Waal, Y. R., Westermann, A., Tops, B., Leenders, W., & Ottevanger, P. B. (2016). An impressive response to pazopanib in a patient with metastatic endometrial carcinoma. Netherlands Journal of Medicine, 74(9), 410–413.
du Bois, A., Floquet, A., Kim, J. W., Rau, J., del Campo, J. M., Friedlander, M., et al. (2014). Incorporation of pazopanib in maintenance therapy of ovarian cancer. Journal of Clinical Oncology, 32(30), 3374–3382. https://doi.org/10.1200/JCO.2014.55.7348
Pignata, S., Lorusso, D., Scambia, G., Sambataro, D., Tamberi, S., Cinieri, S., et al. (2015). Pazopanib plus weekly paclitaxel versus weekly paclitaxel alone for platinum-resistant or platinum-refractory advanced ovarian cancer (MITO 11): A randomised, open-label, phase 2 trial. The Lancet Oncology, 16(5), 561–568. https://doi.org/10.1016/S1470-2045(15)70115-4
du Bois, A., Kristensen, G., Ray-Coquard, I., Reuss, A., Pignata, S., Colombo, N., et al. (2016). Standard first-line chemotherapy with or without nintedanib for advanced ovarian cancer (AGO-OVAR 12): A randomised, double-blind, placebo-controlled phase 3 trial. The Lancet Oncology, 17(1), 78–89. https://doi.org/10.1016/S1470-2045(15)00366-6
Ledermann, J. A., Embleton, A. C., Raja, F., Perren, T. J., Jayson, G. C., Rustin, G. J. S., et al. (2016). Cediranib in patients with relapsed platinum-sensitive ovarian cancer (ICON6): A randomised, double-blind, placebo-controlled phase 3 trial. The Lancet, 387(10023), 1066–1074. https://doi.org/10.1016/S0140-6736(15)01167-8
Moore, K. N., Sill, M. W., Tenney, M. E., Darus, C. J., Griffin, D., Werner, T. L., et al. (2015). A phase II trial of trebananib (AMG 386; IND#111071), a selective angiopoietin 1/2 neutralizing peptibody, in patients with persistent/recurrent carcinoma of the endometrium: An NRG/Gynecologic Oncology Group trial. Gynecologic Oncology, 138(3), 513–518. https://doi.org/10.1016/j.ygyno.2015.07.006
Phi, L. T. H., Sari, I. N., Yang, Y. G., Lee, S. H., Jun, N., Kim, K. S., et al. (2018). Cancer stem cells (CSCs) in drug resistance and their therapeutic implications in cancer treatment. Stem Cells International, 2018, 5416923. https://doi.org/10.1155/2018/5416923
Lytle, N. K., Barber, A. G., & Reya, T. (2018). Stem cell fate in cancer growth, progression and therapy resistance. Nature Reviews Cancer, 18(11), 669–680. https://doi.org/10.1038/s41568-018-0056-x
Giannone, G., Attademo, L., Scotto, G., Genta, S., Ghisoni, E., Tuninetti, V., et al. (2019). Endometrial cancer stem cells: Role, characterization and therapeutic implications. Cancers (Basel), 11(11):1820. https://doi.org/10.3390/cancers11111820
Banz-Jansen, C., Helweg, L. P., & Kaltschmidt, B. (2022). Endometrial cancer stem cells: Where do we stand and where should we go? International Journal of Molecular Sciences, 23(6):3412. https://doi.org/10.3390/ijms23063412
Levine, D. A., Bogomolniy, F., Yee, C. J., Lash, A., Barakat, R. R., Borgen, P. I., et al. (2005). Frequent mutation of the PIK3CA gene in ovarian and breast cancers. Clinical Cancer Research, 11(8), 2875–2878. https://doi.org/10.1158/1078-0432.CCR-04-2142
Martorana, F., Motta, G., Pavone, G., Motta, L., Stella, S., Vitale, S. R., et al. (2021). AKT inhibitors: New weapons in the fight against breast cancer? Frontiers in Pharmacology, 12, 662232. https://doi.org/10.3389/fphar.2021.662232
Oh, D. Y., & Bang, Y. J. (2020). HER2-targeted therapies - A role beyond breast cancer. Nature Reviews Clinical Oncology, 17(1), 33–48. https://doi.org/10.1038/s41571-019-0268-3
Wang, C., Tran, D. A., Fu, M. Z., Chen, W., Fu, S. W., & Li, X. (2020). Estrogen receptor, progesterone receptor, and HER2 receptor markers in endometrial cancer. Journal of Cancer, 11(7), 1693–1701. https://doi.org/10.7150/jca.41943
Buza, N. (2021). HER2 testing in endometrial serous carcinoma: Time for standardized pathology practice to meet the clinical demand. Archives of Pathology and Laboratory Medicine, 145(6), 687–691. https://doi.org/10.5858/arpa.2020-0207-RA
Erickson, B. K., Zeybek, B., Santin, A. D., & Fader, A. N. (2020). Targeting human epidermal growth factor receptor 2 (HER2) in gynecologic malignancies. Current Opinion in Obstetrics and Gynecology, 32(1), 57–64. https://doi.org/10.1097/GCO.0000000000000599
Gordon, M. S., Matei, D., Aghajanian, C., Matulonis, U. A., Brewer, M., Fleming, G. F., et al. (2006). Clinical activity of pertuzumab (rhuMAb 2C4), a HER dimerization inhibitor, in advanced ovarian cancer: Potential predictive relationship with tumor HER2 activation status. Journal of Clinical Oncology, 24(26), 4324–4332. https://doi.org/10.1200/JCO.2005.05.4221
Makhija, S., Amler, L. C., Glenn, D., Ueland, F. R., Gold, M. A., Dizon, D. S., et al. (2010). Clinical activity of gemcitabine plus pertuzumab in platinum-resistant ovarian cancer, fallopian tube cancer, or primary peritoneal cancer. Journal of Clinical Oncology, 28(7), 1215–1223. https://doi.org/10.1200/JCO.2009.22.3354
Kaye, S. B., Poole, C. J., Danska-Bidzinska, A., Gianni, L., Del Conte, G., Gorbunova, V., et al. (2013). A randomized phase II study evaluating the combination of carboplatin-based chemotherapy with pertuzumab versus carboplatin-based therapy alone in patients with relapsed, platinum-sensitive ovarian cancer. Annals of Oncology, 24(1), 145–152. https://doi.org/10.1093/annonc/mds282
Kurzeder, C., Bover, I., Marme, F., Rau, J., Pautier, P., Colombo, N., et al. (2016). Double-blind, placebo-controlled, randomized phase III trial evaluating pertuzumab combined with chemotherapy for low tumor human epidermal growth factor receptor 3 mRNA-expressing platinum-resistant ovarian cancer (PENELOPE). Journal of Clinical Oncology, 34(21), 2516–2525. https://doi.org/10.1200/JCO.2015.66.0787
Bookman, M. A., Darcy, K. M., Clarke-Pearson, D., Boothby, R. A., & Horowitz, I. R. (2003). Evaluation of monoclonal humanized anti-HER2 antibody, trastuzumab, in patients with recurrent or refractory ovarian or primary peritoneal carcinoma with overexpression of HER2: A phase II trial of the Gynecologic Oncology Group. Journal of Clinical Oncology, 21(2), 283–290. https://doi.org/10.1200/JCO.2003.10.104
Fleming, G. F., Sill, M. W., Darcy, K. M., McMeekin, D. S., Thigpen, J. T., Adler, L. M., et al. (2010). Phase II trial of trastuzumab in women with advanced or recurrent, HER2-positive endometrial carcinoma: A Gynecologic Oncology Group study. Gynecologic Oncology, 116(1), 15–20. https://doi.org/10.1016/j.ygyno.2009.09.025
Fader, A. N., Roque, D. M., Siegel, E., Buza, N., Hui, P., Abdelghany, O., et al. (2020). Randomized phase II trial of carboplatin-paclitaxel compared with carboplatin-paclitaxel-trastuzumab in advanced (stage III-IV) or recurrent uterine serous carcinomas that overexpress Her2/Neu (NCT01367002): Updated overall survival analysis. Clinical Cancer Research, 26(15), 3928–3935. https://doi.org/10.1158/1078-0432.CCR-20-0953
Tymon-Rosario, J., Siegel, E. R., Bellone, S., Harold, J., Adjei, N., Zeybek, B., et al. (2021). Trastuzumab tolerability in the treatment of advanced (stage III-IV) or recurrent uterine serous carcinomas that overexpress HER2/neu. Gynecologic Oncology, 163(1), 93–99. https://doi.org/10.1016/j.ygyno.2021.07.033
Fader, A. N., Roque, D. M., Siegel, E., Buza, N., Hui, P., Abdelghany, O., et al. (2018). Randomized phase II trial of carboplatin-paclitaxel versus carboplatin-paclitaxel-trastuzumab in uterine serous carcinomas that overexpress human epidermal growth factor receptor 2/neu. Journal of Clinical Oncology, 36(20), 2044–2051. https://doi.org/10.1200/JCO.2017.76.5966
Liu, J. F., Xiong, N., Campos, S. M., Wright, A. A., Krasner, C., Schumer, S., et al. (2021). Phase II study of the WEE1 inhibitor adavosertib in recurrent uterine serous carcinoma. Journal of Clinical Oncology, 39(14), 1531–1539. https://doi.org/10.1200/JCO.20.03167
Jin, M. H., Nam, A. R., Bang, J. H., Oh, K. S., Seo, H. R., Kim, J. M., et al. (2021). WEE1 inhibition reverses trastuzumab resistance in HER2-positive cancers. Gastric Cancer, 24(5), 1003–1020. https://doi.org/10.1007/s10120-021-01176-7
Sand, A., Piacsek, M., Donohoe, D. L., Duffin, A. T., Riddell, G. T., Sun, C., et al. (2020). WEE1 inhibitor, AZD1775, overcomes trastuzumab resistance by targeting cancer stem-like properties in HER2-positive breast cancer. Cancer Letters, 472, 119–131. https://doi.org/10.1016/j.canlet.2019.12.023
Diver, E. J., Foster, R., Rueda, B. R., & Growdon, W. B. (2015). The therapeutic challenge of targeting HER2 in endometrial cancer. The Oncologist, 20(9), 1058–1068. https://doi.org/10.1634/theoncologist.2015-0149
Growdon, W. B., Groeneweg, J., Byron, V., DiGloria, C., Borger, D. R., Tambouret, R., et al. (2015). HER2 over-expressing high grade endometrial cancer expresses high levels of p95HER2 variant. Gynecologic Oncology, 137(1), 160–166. https://doi.org/10.1016/j.ygyno.2015.01.533
Slomovitz, B. M., & Coleman, R. L. (2012). The PI3K/AKT/mTOR pathway as a therapeutic target in endometrial cancer. Clinical Cancer Research, 18(21), 5856–5864. https://doi.org/10.1158/1078-0432.CCR-12-0662
LoPiccolo, J., Blumenthal, G. M., Bernstein, W. B., & Dennis, P. A. (2008). Targeting the PI3K/Akt/mTOR pathway: Effective combinations and clinical considerations. Drug Resistance Updates, 11(1–2), 32–50. https://doi.org/10.1016/j.drup.2007.11.003
Liao, J., Chen, H., Qi, M., Wang, J., & Wang, M. (2022). MLLT11-TRIL complex promotes the progression of endometrial cancer through PI3K/AKT/mTOR signaling pathway. Cancer Biology & Therapy, 23(1), 211–224. https://doi.org/10.1080/15384047.2022.2046450
Zhao, Y., Sun, H., Feng, M., Zhao, J., Zhao, X., Wan, Q., et al. (2018). Metformin is associated with reduced cell proliferation in human endometrial cancer by inbibiting PI3K/AKT/mTOR signaling. Gynecological Endocrinology, 34(5), 428–432. https://doi.org/10.1080/09513590.2017.1409714
Liu, H., Zhang, L., Zhang, X., & Cui, Z. (2017). PI3K/AKT/mTOR pathway promotes progestin resistance in endometrial cancer cells by inhibition of autophagy. Oncotargets and Therapy, 10, 2865–2871. https://doi.org/10.2147/OTT.S95267
Cai, J., Xu, L., Tang, H., Yang, Q., Yi, X., Fang, Y., et al. (2014). The role of the PTEN/PI3K/Akt pathway on prognosis in epithelial ovarian cancer: A meta-analysis. The Oncologist, 19(5), 528–535. https://doi.org/10.1634/theoncologist.2013-0333
Deng, J., Bai, X., Feng, X., Ni, J., Beretov, J., Graham, P., et al. (2019). Inhibition of PI3K/Akt/mTOR signaling pathway alleviates ovarian cancer chemoresistance through reversing epithelial-mesenchymal transition and decreasing cancer stem cell marker expression. BMC Cancer, 19(1), 618. https://doi.org/10.1186/s12885-019-5824-9
Slomovitz, B. M., Lu, K. H., Johnston, T., Coleman, R. L., Munsell, M., Broaddus, R. R., et al. (2010). A phase 2 study of the oral mammalian target of rapamycin inhibitor, everolimus, in patients with recurrent endometrial carcinoma. Cancer, 116(23), 5415–5419. https://doi.org/10.1002/cncr.25515
Taylor, S. E., Chu, T., Elvin, J. A., Edwards, R. P., & Zorn, K. K. (2020). Phase II study of everolimus and bevacizumab in recurrent ovarian, peritoneal, and fallopian tube cancer. Gynecologic Oncology, 156(1), 32–37. https://doi.org/10.1016/j.ygyno.2019.10.029
Wheler, J. J., Moulder, S. L., Naing, A., Janku, F., Piha-Paul, S. A., Falchook, G. S., et al. (2014). Anastrozole and everolimus in advanced gynecologic and breast malignancies: Activity and molecular alterations in the PI3K/AKT/mTOR pathway. Oncotarget, 5(10), 3029–3038. https://doi.org/10.18632/oncotarget.1799
Rubinstein, M. M., Hyman, D. M., Caird, I., Won, H., Soldan, K., Seier, K., et al. (2020). Phase 2 study of LY3023414 in patients with advanced endometrial cancer harboring activating mutations in the PI3K pathway. Cancer, 126(6), 1274–1282. https://doi.org/10.1002/cncr.32677
Del Campo, J. M., Birrer, M., Davis, C., Fujiwara, K., Gollerkeri, A., Gore, M., et al. (2016). A randomized phase II non-comparative study of PF-04691502 and gedatolisib (PF-05212384) in patients with recurrent endometrial cancer. Gynecologic Oncology, 142(1), 62–69. https://doi.org/10.1016/j.ygyno.2016.04.019
Oza, A. M., Pignata, S., Poveda, A., McCormack, M., Clamp, A., Schwartz, B., et al. (2015). Randomized phase II Trial of ridaforolimus in advanced endometrial carcinoma. Journal of Clinical Oncology, 33(31), 3576–3582. https://doi.org/10.1200/JCO.2014.58.8871
Tsoref, D., Welch, S., Lau, S., Biagi, J., Tonkin, K., Martin, L. A., et al. (2014). Phase II study of oral ridaforolimus in women with recurrent or metastatic endometrial cancer. Gynecologic Oncology, 135(2), 184–189. https://doi.org/10.1016/j.ygyno.2014.06.033
Oza, A. M., Elit, L., Tsao, M. S., Kamel-Reid, S., Biagi, J., Provencher, D. M., et al. (2011). Phase II study of temsirolimus in women with recurrent or metastatic endometrial cancer: A trial of the NCIC Clinical Trials Group. Journal of Clinical Oncology, 29(24), 3278–3285. https://doi.org/10.1200/JCO.2010.34.1578
Tinker, A. V., Ellard, S., Welch, S., Moens, F., Allo, G., Tsao, M. S., et al. (2013). Phase II study of temsirolimus (CCI-779) in women with recurrent, unresectable, locally advanced or metastatic carcinoma of the cervix. A trial of the NCIC Clinical Trials Group (NCIC CTG IND 199). Gynecologic Oncology, 130(2), 269–274. https://doi.org/10.1016/j.ygyno.2013.05.008
Behbakht, K., Sill, M. W., Darcy, K. M., Rubin, S. C., Mannel, R. S., Waggoner, S., et al. (2011). Phase II trial of the mTOR inhibitor, temsirolimus and evaluation of circulating tumor cells and tumor biomarkers in persistent and recurrent epithelial ovarian and primary peritoneal malignancies: A Gynecologic Oncology Group study. Gynecologic Oncology, 123(1), 19–26. https://doi.org/10.1016/j.ygyno.2011.06.022
Bedard, P. L., Tabernero, J., Janku, F., Wainberg, Z. A., Paz-Ares, L., Vansteenkiste, J., et al. (2015). A phase Ib dose-escalation study of the oral pan-PI3K inhibitor buparlisib (BKM120) in combination with the oral MEK1/2 inhibitor trametinib (GSK1120212) in patients with selected advanced solid tumors. Clinical Cancer Research, 21(4), 730–738. https://doi.org/10.1158/1078-0432.CCR-14-1814
Wang, Z., Li, Y., Banerjee, S., & Sarkar, F. H. (2009). Emerging role of Notch in stem cells and cancer. Cancer Letters, 279(1), 8–12. https://doi.org/10.1016/j.canlet.2008.09.030
Kopan, R. (2012). Notch signaling. Cold Spring Harbor Perspectives in Biology, 4(10):a011213. https://doi.org/10.1101/cshperspect.a011213
Venkatesh, V., Nataraj, R., Thangaraj, G. S., Karthikeyan, M., Gnanasekaran, A., Kaginelli, S. B., et al. (2018). Targeting Notch signalling pathway of cancer stem cells. Stem Cell Investigation, 5, 5. https://doi.org/10.21037/sci.2018.02.02
Shang, C., Lang, B., & Meng, L. R. (2018). Blocking NOTCH pathway can enhance the effect of EGFR inhibitor through targeting CD133+ endometrial cancer cells. Cancer Biology & Therapy, 19(2), 113–119. https://doi.org/10.1080/15384047.2016.1250985
Diaz-Padilla, I., Wilson, M. K., Clarke, B. A., Hirte, H. W., Welch, S. A., Mackay, H. J., et al. (2015). A phase II study of single-agent RO4929097, a gamma-secretase inhibitor of Notch signaling, in patients with recurrent platinum-resistant epithelial ovarian cancer: A study of the Princess Margaret, Chicago and California phase II consortia. Gynecologic Oncology, 137(2), 216–222. https://doi.org/10.1016/j.ygyno.2015.03.005
Wen, W., Liang, W., Wu, J., Kowolik, C. M., Buettner, R., Scuto, A., et al. (2014). Targeting JAK1/STAT3 signaling suppresses tumor progression and metastasis in a peritoneal model of human ovarian cancer. Molecular Cancer Therapeutics, 13(12), 3037–3048. https://doi.org/10.1158/1535-7163.MCT-14-0077
Sabaawy, H. E., Ryan, B. M., Khiabanian, H., & Pine, S. R. (2021). JAK/STAT of all trades: Linking inflammation with cancer development, tumor progression and therapy resistance. Carcinogenesis, 42(12), 1411–1419. https://doi.org/10.1093/carcin/bgab075
Zou, S., Tong, Q., Liu, B., Huang, W., Tian, Y., & Fu, X. (2020). Targeting STAT3 in cancer immunotherapy. Molecular Cancer, 19(1), 145. https://doi.org/10.1186/s12943-020-01258-7
Moskowitz, A. J., Ghione, P., Jacobsen, E., Ruan, J., Schatz, J. H., Noor, S., et al. (2021). A phase 2 biomarker-driven study of ruxolitinib demonstrates effectiveness of JAK/STAT targeting in T-cell lymphomas. Blood, 138(26), 2828–2837. https://doi.org/10.1182/blood.2021013379
Beatty, G. L., Shahda, S., Beck, T., Uppal, N., Cohen, S. J., Donehower, R., et al. (2019). A phase Ib/II study of the JAK1 inhibitor, itacitinib, plus nab-paclitaxel and gemcitabine in advanced solid tumors. The Oncologist, 24(1), 14-e10. https://doi.org/10.1634/theoncologist.2017-0665
Pardanani, A., Gotlib, J. R., Jamieson, C., Cortes, J. E., Talpaz, M., Stone, R. M., et al. (2011). Safety and efficacy of TG101348, a selective JAK2 inhibitor, in myelofibrosis. Journal of Clinical Oncology, 29(7), 789–796. https://doi.org/10.1200/JCO.2010.32.8021
Dellinger, T. H., Planutis, K., Tewari, K. S., & Holcombe, R. F. (2012). Role of canonical Wnt signaling in endometrial carcinogenesis. Expert Review of Anticancer Therapy, 12(1), 51–62. https://doi.org/10.1586/era.11.194
Lu, H., Ju, D. D., Yang, G. D., Zhu, L. Y., Yang, X. M., Li, J., et al. (2019). Targeting cancer stem cell signature gene SMOC-2 overcomes chemoresistance and inhibits cell proliferation of endometrial carcinoma. EBioMedicine, 40, 276–289. https://doi.org/10.1016/j.ebiom.2018.12.044
Nguyen, V. H. L., Hough, R., Bernaudo, S., & Peng, C. (2019). Wnt/beta-catenin signalling in ovarian cancer: Insights into its hyperactivation and function in tumorigenesis. Journal of Ovarian Research, 12(1), 122. https://doi.org/10.1186/s13048-019-0596-z
Zannoni, G. F., Angelico, G., & Santoro, A. (2020). Aberrant non-canonical WNT pathway as key-driver of high-grade serous ovarian cancer development. Virchows Archiv, 477(2), 321–322. https://doi.org/10.1007/s00428-020-02760-5
Matt, S., & Hofmann, T. G. (2016). The DNA damage-induced cell death response: A roadmap to kill cancer cells. Cellular and Molecular Life Sciences, 73(15), 2829–2850. https://doi.org/10.1007/s00018-016-2130-4
Wallace, N. A., & Galloway, D. A. (2014). Manipulation of cellular DNA damage repair machinery facilitates propagation of human papillomaviruses. Seminars in Cancer Biology, 26, 30–42. https://doi.org/10.1016/j.semcancer.2013.12.003
Xue, Y., Toh, S. Y., He, P., Lim, T., Lim, D., Pang, C. L., et al. (2015). HPV16-E2 induces prophase arrest and activates the cellular DNA damage response in vitro and in precursor lesions of cervical carcinoma. Oncotarget, 6(33), 34979–34991. https://doi.org/10.18632/oncotarget.5512
McKinney, C. C., Hussmann, K. L., & McBride, A. A. (2015). The role of the DNA damage response throughout the papillomavirus life cycle. Viruses, 7(5), 2450–2469. https://doi.org/10.3390/v7052450
Ojesina, A. I., Lichtenstein, L., Freeman, S. S., Pedamallu, C. S., Imaz-Rosshandler, I., Pugh, T. J., et al. (2014). Landscape of genomic alterations in cervical carcinomas. Nature, 506(7488), 371–375. https://doi.org/10.1038/nature12881
Rieckmann, T., Tribius, S., Grob, T. J., Meyer, F., Busch, C. J., Petersen, C., et al. (2013). HNSCC cell lines positive for HPV and p16 possess higher cellular radiosensitivity due to an impaired DSB repair capacity. Radiotherapy and Oncology, 107(2), 242–246. https://doi.org/10.1016/j.radonc.2013.03.013
Lindel, K., Burri, P., Studer, H. U., Altermatt, H. J., Greiner, R. H., & Gruber, G. (2005). Human papillomavirus status in advanced cervical cancer: Predictive and prognostic significance for curative radiation treatment. International Journal of Gynecological Cancer, 15(2), 278–284. https://doi.org/10.1111/j.1525-1438.2005.15216.x
Schildkraut, J. M., Iversen, E. S., Wilson, M. A., Clyde, M. A., Moorman, P. G., Palmieri, R. T., et al. (2010). Association between DNA damage response and repair genes and risk of invasive serous ovarian cancer. PLoS One, 5(4), e10061. https://doi.org/10.1371/journal.pone.0010061
Yuan, Z., Cao, K., Lin, C., Li, L., Liu, H. Y., Zhao, X. Y., et al. (2011). The p53 upregulated modulator of apoptosis (PUMA) chemosensitizes intrinsically resistant ovarian cancer cells to cisplatin by lowering the threshold set by Bcl-x(L) and Mcl-1. Molecular Medicine, 17(11–12), 1262–1274. https://doi.org/10.2119/molmed.2011.00176
Yang, J., Zhao, X., Tang, M., Li, L., Lei, Y., Cheng, P., et al. (2017). The role of ROS and subsequent DNA-damage response in PUMA-induced apoptosis of ovarian cancer cells. Oncotarget, 8(14), 23492–23506. https://doi.org/10.18632/oncotarget.15626
You, J., Liu, J., Bao, Y., Wang, L., Yu, Y., Wang, L., et al. (2017). SEI1 induces genomic instability by inhibiting DNA damage response in ovarian cancer. Cancer Letters, 385, 271–279. https://doi.org/10.1016/j.canlet.2016.09.032
Johnson, S. W., Laub, P. B., Beesley, J. S., Ozols, R. F., & Hamilton, T. C. (1997). Increased platinum-DNA damage tolerance is associated with cisplatin resistance and cross-resistance to various chemotherapeutic agents in unrelated human ovarian cancer cell lines. Cancer Research, 57(5), 850–856.
Stefanou, D. T., Bamias, A., Episkopou, H., Kyrtopoulos, S. A., Likka, M., Kalampokas, T., et al. (2021). Correction: Aberrant DNA damage response pathways may predict the outcome of platinum chemotherapy in ovarian cancer. PLoS One, 16(8), e0256051. https://doi.org/10.1371/journal.pone.0256051
Morice, P., Leary, A., Creutzberg, C., Abu-Rustum, N., & Darai, E. (2016). Endometrial cancer. The Lancet, 387(10023), 1094–1108. https://doi.org/10.1016/S0140-6736(15)00130-0
Lord, C. J., & Ashworth, A. (2012). The DNA damage response and cancer therapy. Nature, 481(7381), 287–294. https://doi.org/10.1038/nature10760
Farmer, H., McCabe, N., Lord, C. J., Tutt, A. N., Johnson, D. A., Richardson, T. B., et al. (2005). Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature, 434(7035), 917–921. https://doi.org/10.1038/nature03445
Bryant, H. E., Schultz, N., Thomas, H. D., Parker, K. M., Flower, D., Lopez, E., et al. (2005). Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature, 434(7035), 913–917. https://doi.org/10.1038/nature03443
Gadducci, A., Guarneri, V., Peccatori, F. A., Ronzino, G., Scandurra, G., Zamagni, C., et al. (2019). Current strategies for the targeted treatment of high-grade serous epithelial ovarian cancer and relevance of BRCA mutational status. Journal of Ovarian Research, 12(1), 9. https://doi.org/10.1186/s13048-019-0484-6
Wang, Y., Zheng, K., Xiong, H., Huang, Y., Chen, X., Zhou, Y., et al. (2021). PARP inhibitor upregulates PD-L1 expression and provides a new combination therapy in pancreatic cancer. Frontiers in Immunology, 12, 762989. https://doi.org/10.3389/fimmu.2021.762989
Ding, L., Kim, H. J., Wang, Q., Kearns, M., Jiang, T., Ohlson, C. E., et al. (2018). PARP inhibition elicits STING-dependent antitumor immunity in Brca1-deficient ovarian cancer. Cell Reports, 25(11), 2972-2980.e2975. https://doi.org/10.1016/j.celrep.2018.11.054
Dedes, K. J., Wetterskog, D., Mendes-Pereira, A. M., Natrajan, R., Lambros, M. B., Geyer, F. C., et al. (2010). PTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitors. Science Translational Medicine, 2(53), 53ra75. https://doi.org/10.1126/scitranslmed.3001538
Bian, X., Gao, J., Luo, F., Rui, C., Zheng, T., Wang, D., et al. (2018). PTEN deficiency sensitizes endometrioid endometrial cancer to compound PARP-PI3K inhibition but not PARP inhibition as monotherapy. Oncogene, 37(3), 341–351. https://doi.org/10.1038/onc.2017.326
Forster, M. D., Dedes, K. J., Sandhu, S., Frentzas, S., Kristeleit, R., Ashworth, A., et al. (2011). Treatment with olaparib in a patient with PTEN-deficient endometrioid endometrial cancer. Nature Reviews Clinical Oncology, 8(5), 302–306. https://doi.org/10.1038/nrclinonc.2011.42
Philip, C. A., Laskov, I., Beauchamp, M. C., Marques, M., Amin, O., Bitharas, J., et al. (2017). Inhibition of PI3K-AKT-mTOR pathway sensitizes endometrial cancer cell lines to PARP inhibitors. BMC Cancer, 17(1), 638. https://doi.org/10.1186/s12885-017-3639-0
Post, C. C. B., Westermann, A. M., Boere, I. A., Witteveen, P. O., Ottevanger, P. B., Sonke, G. S., et al. (2022). Efficacy and safety of durvalumab with olaparib in metastatic or recurrent endometrial cancer (phase II DOMEC trial). Gynecologic Oncology, 165(2), 223–229. https://doi.org/10.1016/j.ygyno.2022.02.025
Prasad, C. B., Prasad, S. B., Yadav, S. S., Pandey, L. K., Singh, S., Pradhan, S., et al. (2017). Olaparib modulates DNA repair efficiency, sensitizes cervical cancer cells to cisplatin and exhibits anti-metastatic property. Science and Reports, 7(1), 12876. https://doi.org/10.1038/s41598-017-13232-3
Bianchi, A., Lopez, S., Altwerger, G., Bellone, S., Bonazzoli, E., Zammataro, L., et al. (2019). PARP-1 activity (PAR) determines the sensitivity of cervical cancer to olaparib. Gynecologic Oncology, 155(1), 144–150. https://doi.org/10.1016/j.ygyno.2019.08.010
Mann, M., Kumar, S., Sharma, A., Chauhan, S. S., Bhatla, N., Kumar, S., et al. (2019). PARP-1 inhibitor modulate beta-catenin signaling to enhance cisplatin sensitivity in cancer cervix. Oncotarget, 10(42), 4262–4275. https://doi.org/10.18632/oncotarget.27008
Cohen, A. C., Roane, B. M., & Leath, C. A., 3rd. (2020). Novel therapeutics for recurrent cervical cancer: Moving towards personalized therapy. Drugs, 80(3), 217–227. https://doi.org/10.1007/s40265-019-01249-z
Thaker, P. H., Salani, R., Brady, W. E., Lankes, H. A., Cohn, D. E., Mutch, D. G., et al. (2017). A phase I trial of paclitaxel, cisplatin, and veliparib in the treatment of persistent or recurrent carcinoma of the cervix: An NRG Oncology Study (NCT#01281852). Annals of Oncology, 28(3), 505–511. https://doi.org/10.1093/annonc/mdw635
Kunos, C., Deng, W., Dawson, D., Lea, J. S., Zanotti, K. M., Gray, H. J., et al. (2015). A phase I-II evaluation of veliparib (NSC #737664), topotecan, and filgrastim or pegfilgrastim in the treatment of persistent or recurrent carcinoma of the uterine cervix: An NRG Oncology/Gynecologic Oncology Group study. International Journal of Gynecological Cancer, 25(3), 484–492. https://doi.org/10.1097/IGC.0000000000000380
Jackson, C. G., Moore, K. N., Cantrell, L., Erickson, B. K., Duska, L. R., Richardson, D. L., et al. (2022). A phase II trial of bevacizumab and rucaparib in recurrent carcinoma of the cervix or endometrium. Gynecologic Oncology. https://doi.org/10.1016/j.ygyno.2022.04.016
Chatterjee, P., Choudhary, G. S., Sharma, A., Singh, K., Heston, W. D., Ciezki, J., et al. (2013). PARP inhibition sensitizes to low dose-rate radiation TMPRSS2-ERG fusion gene-expressing and PTEN-deficient prostate cancer cells. PLoS One, 8(4), e60408. https://doi.org/10.1371/journal.pone.0060408
Lightfoot, M., Montemorano, L., & Bixel, K. (2020). PARP inhibitors in gynecologic cancers: What is the next big development? Current Oncology Reports, 22(3), 29. https://doi.org/10.1007/s11912-020-0873-4
Kaufman, B., Shapira-Frommer, R., Schmutzler, R. K., Audeh, M. W., Friedlander, M., Balmana, J., et al. (2015). Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. Journal of Clinical Oncology, 33(3), 244–250. https://doi.org/10.1200/JCO.2014.56.2728
Kaye, S. B., Lubinski, J., Matulonis, U., Ang, J. E., Gourley, C., Karlan, B. Y., et al. (2012). Phase II, open-label, randomized, multicenter study comparing the efficacy and safety of olaparib, a poly (ADP-ribose) polymerase inhibitor, and pegylated liposomal doxorubicin in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer. Journal of Clinical Oncology, 30(4), 372–379. https://doi.org/10.1200/JCO.2011.36.9215
Penson, R. T., Valencia, R. V., Cibula, D., Colombo, N., Leath, C. A., 3rd., Bidziński, M., et al. (2020). Olaparib versus nonplatinum chemotherapy in patients with platinum-sensitive relapsed ovarian cancer and a germline BRCA1/2 mutation (SOLO3): A randomized phase III trial. Journal of Clinical Oncology, 38(11), 1164–1174. https://doi.org/10.1200/jco.19.02745
Swisher, E. M., Lin, K. K., Oza, A. M., Scott, C. L., Giordano, H., Sun, J., et al. (2017). Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. The Lancet Oncology, 18(1), 75–87. https://doi.org/10.1016/S1470-2045(16)30559-9
Dal Molin, G. Z., Omatsu, K., Sood, A. K., & Coleman, R. L. (2018). Rucaparib in ovarian cancer: An update on safety, efficacy and place in therapy. Therapeutic Advances in Medical Oncology, 10, 1758835918778483. https://doi.org/10.1177/1758835918778483
Gelmon, K. A., Tischkowitz, M., Mackay, H., Swenerton, K., Robidoux, A., Tonkin, K., et al. (2011). Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: A phase 2, multicentre, open-label, non-randomised study. The Lancet Oncology, 12(9), 852–861. https://doi.org/10.1016/S1470-2045(11)70214-5
Liu, M. C., Sutedja, J., & Tewari, K. S. (2021). Niraparib in the maintenance treatment of advanced epithelial ovarian, fallopian tube, or primary peritoneal cancer: Safety and efficacy. Expert Review of Anticancer Therapy, 21(5), 475–480. https://doi.org/10.1080/14737140.2021.1880326
Konstantinopoulos, P. A., Waggoner, S., Vidal, G. A., Mita, M., Moroney, J. W., Holloway, R., et al. (2019). Single-arm phases 1 and 2 trial of niraparib in combination with pembrolizumab in patients with recurrent platinum-resistant ovarian carcinoma. JAMA Oncology, 5(8), 1141–1149. https://doi.org/10.1001/jamaoncol.2019.1048
Liu, J. F., Barry, W. T., Birrer, M., Lee, J. M., Buckanovich, R. J., Fleming, G. F., et al. (2014). Combination cediranib and olaparib versus olaparib alone for women with recurrent platinum-sensitive ovarian cancer: A randomised phase 2 study. The Lancet Oncology, 15(11), 1207–1214. https://doi.org/10.1016/S1470-2045(14)70391-2
Harter, P., Mouret-Reynier, M. A., Pignata, S., Cropet, C., González-Martín, A., Bogner, G., et al. (2022). Efficacy of maintenance olaparib plus bevacizumab according to clinical risk in patients with newly diagnosed, advanced ovarian cancer in the phase III PAOLA-1/ENGOT-ov25 trial. Gynecologic Oncology, 164(2), 254–264. https://doi.org/10.1016/j.ygyno.2021.12.016
Rehman, F. L., Lord, C. J., & Ashworth, A. (2012). The promise of combining inhibition of PI3K and PARP as cancer therapy. Cancer Discovery, 2(11), 982–984. https://doi.org/10.1158/2159-8290.CD-12-0433
Matulonis, U. A., Wulf, G. M., Barry, W. T., Birrer, M., Westin, S. N., Farooq, S., et al. (2017). Phase I dose escalation study of the PI3kinase pathway inhibitor BKM120 and the oral poly (ADP ribose) polymerase (PARP) inhibitor olaparib for the treatment of high-grade serous ovarian and breast cancer. Annals of Oncology, 28(3), 512–518. https://doi.org/10.1093/annonc/mdw672
Konstantinopoulos, P. A., Barry, W. T., Birrer, M., Westin, S. N., Cadoo, K. A., Shapiro, G. I., et al. (2019). Olaparib and alpha-specific PI3K inhibitor alpelisib for patients with epithelial ovarian cancer: A dose-escalation and dose-expansion phase 1b trial. The Lancet Oncology, 20(4), 570–580. https://doi.org/10.1016/S1470-2045(18)30905-7
Wallace, A. E., Gibson, D. A., Saunders, P. T., & Jabbour, H. N. (2010). Inflammatory events in endometrial adenocarcinoma. Journal of Endocrinology, 206(2), 141–157. https://doi.org/10.1677/JOE-10-0072
Kubler, K., Ayub, T. H., Weber, S. K., Zivanovic, O., Abramian, A., Keyver-Paik, M. D., et al. (2014). Prognostic significance of tumor-associated macrophages in endometrial adenocarcinoma. Gynecologic Oncology, 135(2), 176–183. https://doi.org/10.1016/j.ygyno.2014.08.028
Guo, Y. E., Li, Y., Cai, B., He, Q., Chen, G., Wang, M., et al. (2021). Phenotyping of immune and endometrial epithelial cells in endometrial carcinomas revealed by single-cell RNA sequencing. Aging (Albany NY), 13(5), 6565–6591. https://doi.org/10.18632/aging.202288
Ning, C., Xie, B., Zhang, L., Li, C., Shan, W., Yang, B., et al. (2016). Infiltrating macrophages induce ERalpha expression through an IL17A-mediated epigenetic mechanism to sensitize endometrial cancer cells to estrogen. Cancer Research, 76(6), 1354–1366. https://doi.org/10.1158/0008-5472.CAN-15-1260
Liu, B., Chen, P., Xi, D., Zhu, H., & Gao, Y. (2017). ATF4 regulates CCL2 expression to promote endometrial cancer growth by controlling macrophage infiltration. Experimental Cell Research, 360(2), 105–112. https://doi.org/10.1016/j.yexcr.2017.08.031
Jing, X., Peng, J., Dou, Y., Sun, J., Ma, C., Wang, Q., et al. (2019). Macrophage ERalpha promoted invasion of endometrial cancer cell by mTOR/KIF5B-mediated epithelial to mesenchymal transition. Immunology and Cell Biology, 97(6), 563–576. https://doi.org/10.1111/imcb.12245
Fujimoto, J., Aoki, I., Khatun, S., Toyoki, H., & Tamaya, T. (2002). Clinical implications of expression of interleukin-8 related to myometrial invasion with angiogenesis in uterine endometrial cancers. Annals of Oncology, 13(3), 430–434. https://doi.org/10.1093/annonc/mdf078
Brunner, A., Hinterholzer, S., Riss, P., Heinze, G., & Brustmann, H. (2012). Immunoexpression of B7–H3 in endometrial cancer: Relation to tumor T-cell infiltration and prognosis. Gynecologic Oncology, 124(1), 105–111. https://doi.org/10.1016/j.ygyno.2011.09.012
Patel, M. V., Shen, Z., Rodriguez-Garcia, M., Usherwood, E. J., Tafe, L. J., & Wira, C. R. (2021). Endometrial cancer suppresses CD8+ T cell-mediated cytotoxicity in postmenopausal women. Frontiers in Immunology, 12, 657326. https://doi.org/10.3389/fimmu.2021.657326
Yamagami, W., Susumu, N., Tanaka, H., Hirasawa, A., Banno, K., Suzuki, N., et al. (2011). Immunofluorescence-detected infiltration of CD4+FOXP3+ regulatory T cells is relevant to the prognosis of patients with endometrial cancer. International Journal of Gynecological Cancer, 21(9), 1628–1634. https://doi.org/10.1097/IGC.0b013e31822c271f
Li, L., Li, Y., Yin, Z., Zhu, J., Yan, D., & Lou, H. (2019). Increased frequency of regulatory T cells in the peripheral blood of patients with endometrioid adenocarcinoma. Oncology Letters, 18(2), 1424–1430. https://doi.org/10.3892/ol.2019.10452
Asaka, S., Yen, T. T., Wang, T. L., Shih, I. M., & Gaillard, S. (2019). T cell-inflamed phenotype and increased Foxp3 expression in infiltrating T-cells of mismatch-repair deficient endometrial cancers. Modern Pathology, 32(4), 576–584. https://doi.org/10.1038/s41379-018-0172-x
Han, Y., Liu, D., & Li, L. (2020). PD-1/PD-L1 pathway: Current researches in cancer. American Journal of Cancer Research, 10(3), 727–742.
Yi, M., Zheng, X., Niu, M., Zhu, S., Ge, H., & Wu, K. (2022). Combination strategies with PD-1/PD-L1 blockade: Current advances and future directions. Molecular Cancer, 21(1), 28. https://doi.org/10.1186/s12943-021-01489-2
Mo, Z., Liu, J., Zhang, Q., Chen, Z., Mei, J., Liu, L., et al. (2016). Expression of PD-1, PD-L1 and PD-L2 is associated with differentiation status and histological type of endometrial cancer. Oncology Letters, 12(2), 944–950. https://doi.org/10.3892/ol.2016.4744
Gomez-Raposo, C., Merino Salvador, M., Aguayo Zamora, C., Garcia de Santiago, B., & Casado Saenz, E. (2021). Immune checkpoint inhibitors in endometrial cancer. Critical Reviews in Oncology Hematology, 161, 103306. https://doi.org/10.1016/j.critrevonc.2021.103306
De Felice, F., Giudice, E., Bolomini, G., Distefano, M. G., Scambia, G., Fagotti, A., et al. (2021). Pembrolizumab for advanced cervical cancer: Safety and efficacy. Expert Review of Anticancer Therapy, 21(2), 221–228. https://doi.org/10.1080/14737140.2021.1850279
Ott, P. A., Bang, Y. J., Berton-Rigaud, D., Elez, E., Pishvaian, M. J., Rugo, H. S., et al. (2017). Safety and antitumor activity of pembrolizumab in advanced programmed death ligand 1-positive endometrial cancer: Results from the KEYNOTE-028 study. Journal of Clinical Oncology, 35(22), 2535–2541. https://doi.org/10.1200/JCO.2017.72.5952
Green, A. K., Feinberg, J., & Makker, V. (2020). A review of immune checkpoint blockade therapy in endometrial cancer. American Society of Clinical Oncology Educational Book, 40, 1–7. https://doi.org/10.1200/EDBK_280503
Kranawetter, M., Rohrich, S., Mullauer, L., Obermair, H., Reinthaller, A., Grimm, C., et al. (2018). Activity of pembrolizumab in recurrent cervical cancer: Case series and review of published data. International Journal of Gynecological Cancer, 28(6), 1196–1202. https://doi.org/10.1097/IGC.0000000000001291
Borcoman, E., & Le Tourneau, C. (2020). Keynote-158 study, FDA granted accelerated approval of pembrolizumab for the treatment of patients with advanced PD-L1-positive cervical cancer. Annals of Translational Medicine, 8(23), 1611. https://doi.org/10.21037/atm-20-2656
Markham, A. (2021). Dostarlimab: First Approval. Drugs, 81(10), 1213–1219. https://doi.org/10.1007/s40265-021-01539-5
Garcia-Duran, C., Grau, F., Villacampa, G., & Oaknin, A. (2022). ATOMICC trial: A randomized, open-label, phase II trial of anti-PD1, dostarlimab, as maintenance therapy for patients with high-risk locally advanced cervical cancer after chemoradiation. International Journal of Gynecological Cancer. https://doi.org/10.1136/ijgc-2022-003370
Musacchio, L., Salutari, V., Pignata, S., Braicu, E., Cibula, D., Colombo, N., et al. (2021). Randomized phase III trial on niraparib-TSR-042 (dostarlimab) versus physician’s choice chemotherapy in recurrent ovarian, fallopian tube, or primary peritoneal cancer patients not candidate for platinum retreatment: NItCHE trial (MITO 33). International Journal of Gynecological Cancer, 31(10), 1369–1373. https://doi.org/10.1136/ijgc-2021-002593
Rubio-Perez, J., Hernandez, R., Hernandez, T., Doger, B., Casado, V., & Moreno, V. (2021). Dostarlimab for the treatment of endometrium cancer and other solid tumors. Drugs of Today (Barcelona, Spain), 57(3), 187–197. https://doi.org/10.1358/dot.2021.57.3.3233363
Oaknin, A., Gilbert, L., Tinker, A. V., Brown, J., Mathews, C., Press, J., et al. (2022). Safety and antitumor activity of dostarlimab in patients with advanced or recurrent DNA mismatch repair deficient/microsatellite instability-high (dMMR/MSI-H) or proficient/stable (MMRp/MSS) endometrial cancer: interim results from GARNET-a phase I, single-arm study. Journal for Immunotherapy of Cancer, 10(1):e003777. https://doi.org/10.1136/jitc-2021-003777
Kato, Y., Tabata, K., Kimura, T., Yachie-Kinoshita, A., Ozawa, Y., Yamada, K., et al. (2019). Lenvatinib plus anti-PD-1 antibody combination treatment activates CD8+ T cells through reduction of tumor-associated macrophage and activation of the interferon pathway. PLoS One, 14(2), e0212513. https://doi.org/10.1371/journal.pone.0212513
Makker, V., Colombo, N., Casado Herraez, A., Santin, A. D., Colomba, E., Miller, D. S., et al. (2022). Lenvatinib plus pembrolizumab for advanced endometrial cancer. New England Journal of Medicine, 386(5), 437–448. https://doi.org/10.1056/NEJMoa2108330
Post, C. C. B., Westermann, A. M., Bosse, T., Creutzberg, C. L., & Kroep, J. R. (2020). PARP and PD-1/PD-L1 checkpoint inhibition in recurrent or metastatic endometrial cancer. Critical Reviews in Oncology Hematology, 152, 102973. https://doi.org/10.1016/j.critrevonc.2020.102973
Pardoll, D. M. (2012). The blockade of immune checkpoints in cancer immunotherapy. Nature Reviews Cancer, 12(4), 252–264. https://doi.org/10.1038/nrc3239
Khalifa, R., Elsese, N., El-Desouky, K., Shaair, H., & Helal, D. (2022). Immune checkpoint proteins (PD-L1 and CTLA-4) in endometrial carcinoma: Prognostic role and correlation with CD4(+)/CD8(+) tumor infiltrating lymphocytes (TILs) ratio. Journal of Immunoassay & Immunochemistry, 43(2), 192–212. https://doi.org/10.1080/15321819.2021.1981377
Hodi, F. S., Mihm, M. C., Soiffer, R. J., Haluska, F. G., Butler, M., Seiden, M. V., et al. (2003). Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proceedings of the National Academy of Sciences of the United States of America, 100(8), 4712–4717. https://doi.org/10.1073/pnas.0830997100
O’Malley, D. M., Neffa, M., Monk, B. J., Melkadze, T., Huang, M., Kryzhanivska, A., et al. (2022). Dual PD-1 and CTLA-4 checkpoint blockade using balstilimab and zalifrelimab combination as second-line treatment for advanced cervical cancer: An open-label phase II study. Journal of Clinical Oncology, 40(7), 762–771. https://doi.org/10.1200/JCO.21.02067
Adams, S. F., Rixe, O., Lee, J.-H., McCance, D. J., Westgate, S., Eberhardt, S. C., et al. (2017). Phase I study combining olaparib and tremelimumab for the treatment of women with BRCA-deficient recurrent ovarian cancer. Journal of Clinical Oncology, 35(15), e17052-e17052. https://doi.org/10.1200/JCO.2017.35.15_suppl.e17052
Logtenberg, M. E. W., Scheeren, F. A., & Schumacher, T. N. (2020). The CD47-SIRPalpha immune checkpoint. Immunity, 52(5), 742–752. https://doi.org/10.1016/j.immuni.2020.04.011
Yang, M., Jiang, C., Li, L., Xing, H., & Hong, L. (2022). Expression of CD47 in endometrial cancer and its clinicopathological significance. Journal of Oncology, 2022, 7188972. https://doi.org/10.1155/2022/7188972
Gu, S., Ni, T., Wang, J., Liu, Y., Fan, Q., Wang, Y., et al. (2018). CD47 Blockade inhibits tumor progression through promoting phagocytosis of tumor cells by M2 polarized macrophages in endometrial cancer. Journal of Immunology Research, 2018, 6156757. https://doi.org/10.1155/2018/6156757
Li, J., Yan, S., Li, Q., Huang, Y., Ji, M., Jiao, X., et al. (2022). Macrophage-associated immune checkpoint CD47 blocking ameliorates endometriosis. Molecular Human Reproduction, 28(5):gaac010. https://doi.org/10.1093/molehr/gaac010
Tian, L., Xu, B., Teng, K. Y., Song, M., Zhu, Z., Chen, Y., et al. (2022). Targeting Fc receptor-mediated effects and the “don’t eat me” signal with an oncolytic virus expressing an anti-CD47 antibody to treat metastatic ovarian cancer. Clinical Cancer Research, 28(1), 201–214. https://doi.org/10.1158/1078-0432.Ccr-21-1248
Yu, W. B., Ye, Z. H., Chen, X., Shi, J. J., & Lu, J. J. (2021). The development of small-molecule inhibitors targeting CD47. Drug Discovery Today, 26(2), 561–568. https://doi.org/10.1016/j.drudis.2020.11.003
Huang, B., Bai, Z., Ye, X., Zhou, C., Xie, X., Zhong, Y., et al. (2021). Structural analysis and binding sites of inhibitors targeting the CD47/SIRPalpha interaction in anticancer therapy. Computational and Structural Biotechnology Journal, 19, 5494–5503. https://doi.org/10.1016/j.csbj.2021.09.036
Oronsky, B., Carter, C., Reid, T., Brinkhaus, F., & Knox, S. J. (2020). Just eat it: A review of CD47 and SIRP-alpha antagonism. Seminars in Oncology, 47(2–3), 117–124. https://doi.org/10.1053/j.seminoncol.2020.05.009
Advani, R., Flinn, I., Popplewell, L., Forero, A., Bartlett, N. L., Ghosh, N., et al. (2018). CD47 blockade by Hu5F9-G4 and rituximab in non-Hodgkin’s lymphoma. New England Journal of Medicine, 379(18), 1711–1721. https://doi.org/10.1056/NEJMoa1807315
Johnson, L. D. S., Banerjee, S., Kruglov, O., Viller, N. N., Horwitz, S. M., Lesokhin, A., et al. (2019). Targeting CD47 in Sezary syndrome with SIRPalphaFc. Blood Advances, 3(7), 1145–1153. https://doi.org/10.1182/bloodadvances.2018030577
Ansell, S. M., Maris, M. B., Lesokhin, A. M., Chen, R. W., Flinn, I. W., Sawas, A., et al. (2021). Phase I study of the CD47 blocker TTI-621 in patients with relapsed or refractory hematologic malignancies. Clinical Cancer Research, 27(8), 2190–2199. https://doi.org/10.1158/1078-0432.CCR-20-3706
Lakhani, N. J., Chow, L. Q. M., Gainor, J. F., LoRusso, P., Lee, K. W., Chung, H. C., et al. (2021). Evorpacept alone and in combination with pembrolizumab or trastuzumab in patients with advanced solid tumours (ASPEN-01): A first-in-human, open-label, multicentre, phase 1 dose-escalation and dose-expansion study. The Lancet Oncology, 22(12), 1740–1751. https://doi.org/10.1016/S1470-2045(21)00584-2
Sikic, B. I., Lakhani, N., Patnaik, A., Shah, S. A., Chandana, S. R., Rasco, D., et al. (2019). First-in-human, first-in-class phase I trial of the anti-CD47 antibody Hu5F9-G4 in patients with advanced cancers. Journal of Clinical Oncology, 37(12), 946–953. https://doi.org/10.1200/JCO.18.02018
Duan, Q., Zhang, H., Zheng, J., & Zhang, L. (2020). Turning cold into hot: Firing up the tumor microenvironment. Trends Cancer, 6(7), 605–618. https://doi.org/10.1016/j.trecan.2020.02.022
Manyam, M., Stephens, A. J., Kennard, J. A., LeBlanc, J., Ahmad, S., Kendrick, J. E., et al. (2021). A phase 1b study of intraperitoneal oncolytic viral immunotherapy in platinum-resistant or refractory ovarian cancer. Gynecologic Oncology, 163(3), 481–489. https://doi.org/10.1016/j.ygyno.2021.10.069
Moreno, V., Barretina-Ginesta, M. P., Garcia-Donas, J., Jayson, G. C., Roxburgh, P., Vazquez, R. M., et al. (2021). Safety and efficacy of the tumor-selective adenovirus enadenotucirev with or without paclitaxel in platinum-resistant ovarian cancer: A phase 1 clinical trial. Journal for Immunotherapy of Cancer, 9(12):e003645. https://doi.org/10.1136/jitc-2021-003645.
Cohn, D. E., Sill, M. W., Walker, J. L., O’Malley, D., Nagel, C. I., Rutledge, T. L., et al. (2017). Randomized phase IIB evaluation of weekly paclitaxel versus weekly paclitaxel with oncolytic reovirus (Reolysin(R)) in recurrent ovarian, tubal, or peritoneal cancer: An NRG Oncology/Gynecologic Oncology Group study. Gynecologic Oncology, 146(3), 477–483. https://doi.org/10.1016/j.ygyno.2017.07.135
Galanis, E., Hartmann, L. C., Cliby, W. A., Long, H. J., Peethambaram, P. P., Barrette, B. A., et al. (2010). Phase I trial of intraperitoneal administration of an oncolytic measles virus strain engineered to express carcinoembryonic antigen for recurrent ovarian cancer. Cancer Research, 70(3), 875–882. https://doi.org/10.1158/0008-5472.CAN-09-2762
Kim, K. H., Dmitriev, I. P., Saddekni, S., Kashentseva, E. A., Harris, R. D., Aurigemma, R., et al. (2013). A phase I clinical trial of Ad5/3-Delta24, a novel serotype-chimeric, infectivity-enhanced, conditionally-replicative adenovirus (CRAd), in patients with recurrent ovarian cancer. Gynecologic Oncology, 130(3), 518–524. https://doi.org/10.1016/j.ygyno.2013.06.003
Met, O., Jensen, K. M., Chamberlain, C. A., Donia, M., & Svane, I. M. (2019). Principles of adoptive T cell therapy in cancer. Seminars in Immunopathology, 41(1), 49–58. https://doi.org/10.1007/s00281-018-0703-z
Kandalaft, L. E., Powell, D. J., Jr., & Coukos, G. (2012). A phase I clinical trial of adoptive transfer of folate receptor-alpha redirected autologous T cells for recurrent ovarian cancer. Journal of Translational Medicine, 10, 157. https://doi.org/10.1186/1479-5876-10-157
Chen, L., Qiao, D., Wang, J., Tian, G., & Wang, M. (2019). Cancer immunotherapy with lymphocytes genetically engineered with T cell receptors for solid cancers. Immunology Letters, 216, 51–62. https://doi.org/10.1016/j.imlet.2019.10.002
Xu, Y., Jiang, J., Wang, Y., Wang, W., Li, H., Lai, W., et al. (2021). Engineered T cell therapy for gynecologic malignancies: Challenges and opportunities. Frontiers in Immunology, 12, 725330. https://doi.org/10.3389/fimmu.2021.725330
Son, J., George, G. C., Nardo, M., Krause, K. J., Jazaeri, A. A., Biter, A. B., et al. (2022). Adoptive cell therapy in gynecologic cancers: A systematic review and meta-analysis. Gynecologic Oncology. https://doi.org/10.1016/j.ygyno.2022.03.013
Xie, G., Dong, H., Liang, Y., Ham, J. D., Rizwan, R., & Chen, J. (2020). CAR-NK cells: A promising cellular immunotherapy for cancer. EBioMedicine, 59, 102975. https://doi.org/10.1016/j.ebiom.2020.102975
Chen, Y., Yu, Z., Tan, X., Jiang, H., Xu, Z., Fang, Y., et al. (2021). CAR-macrophage: A new immunotherapy candidate against solid tumors. Biomedicine & Pharmacotherapy, 139, 111605. https://doi.org/10.1016/j.biopha.2021.111605
Das, M., Zhu, C., & Kuchroo, V. K. (2017). Tim-3 and its role in regulating anti-tumor immunity. Immunological Reviews, 276(1), 97–111. https://doi.org/10.1111/imr.12520
Ren, F., Li, J., Jiang, X., Xiao, K., Zhang, D., Zhao, Z., et al. (2015). Plasma soluble Tim-3 emerges as an inhibitor in sepsis: Sepsis contrary to membrane Tim-3 on monocytes. Tissue Antigens, 86(5), 325–332. https://doi.org/10.1111/tan.12653
Moore, M., Ring, K. L., & Mills, A. M. (2019). TIM-3 in endometrial carcinomas: An immunotherapeutic target expressed by mismatch repair-deficient and intact cancers. Modern Pathology, 32(8), 1168–1179. https://doi.org/10.1038/s41379-019-0251-7
Harding, J. J., Moreno, V., Bang, Y. J., Hong, M. H., Patnaik, A., Trigo, J., et al. (2021). Blocking TIM-3 in treatment-refractory advanced solid tumors: A Phase Ia/b study of LY3321367 with or without an Anti-PD-L1 antibody. Clinical Cancer Research, 27(8), 2168–2178. https://doi.org/10.1158/1078-0432.CCR-20-4405
Curigliano, G., Gelderblom, H., Mach, N., Doi, T., Tai, D., Forde, P. M., et al. (2021). Phase I/Ib clinical trial of sabatolimab, an Anti-TIM-3 antibody, alone and in combination with spartalizumab, an anti-PD-1 antibody, in advanced solid tumors. Clinical Cancer Research, 27(13), 3620–3629. https://doi.org/10.1158/1078-0432.CCR-20-4746
Hellmann, M. D., Bivi, N., Calderon, B., Shimizu, T., Delafontaine, B., Liu, Z. T., et al. (2021). Safety and immunogenicity of LY3415244, a bispecific antibody against TIM-3 and PD-L1, in patients with advanced solid tumors. Clinical Cancer Research, 27(10), 2773–2781. https://doi.org/10.1158/1078-0432.CCR-20-3716
Friedman, L. A., Ring, K. L., & Mills, A. M. (2020). LAG-3 and GAL-3 in endometrial carcinoma: Emerging candidates for immunotherapy. International Journal of Gynecological Pathology, 39(3), 203–212. https://doi.org/10.1097/PGP.0000000000000608
Zhang, Q., Salzler, R., Dore, A., Yang, J., Ma, D., Olson, W. C., et al. (2018). Multiplex immuno-liquid chromatography-mass spectrometry-parallel reaction monitoring (LC-MS-PRM) quantitation of CD8A, CD4, LAG3, PD1, PD-L1, and PD-L2 in frozen human tissues. Journal of Proteome Research, 17(11), 3932–3940. https://doi.org/10.1021/acs.jproteome.8b00605
Matsuzaki, J., Gnjatic, S., Mhawech-Fauceglia, P., Beck, A., Miller, A., Tsuji, T., et al. (2010). Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proceedings of the National Academy of Sciences of the United States of America, 107(17), 7875–7880. https://doi.org/10.1073/pnas.1003345107
Schoffski, P., Tan, D. S. W., Martin, M., Ochoa-de-Olza, M., Sarantopoulos, J., Carvajal, R. D., et al. (2022). Phase I/II study of the LAG-3 inhibitor ieramilimab (LAG525) +/- anti-PD-1 spartalizumab (PDR001) in patients with advanced malignancies. Journal for Immunotherapy of Cancer, 10(2):e003776. https://doi.org/10.1136/jitc-2021-003776
Tawbi, H. A., Schadendorf, D., Lipson, E. J., Ascierto, P. A., Matamala, L., Castillo Gutierrez, E., et al. (2022). Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. New England Journal of Medicine, 386(1), 24–34. https://doi.org/10.1056/NEJMoa2109970
Maas, R. J., Hoogstad-van Evert, J. S., Van der Meer, J. M., Mekers, V., Rezaeifard, S., Korman, A. J., et al. (2020). TIGIT blockade enhances functionality of peritoneal NK cells with altered expression of DNAM-1/TIGIT/CD96 checkpoint molecules in ovarian cancer. Oncoimmunology, 9(1), 1843247. https://doi.org/10.1080/2162402X.2020.1843247
Chen, F., Xu, Y., Chen, Y., & Shan, S. (2020). TIGIT enhances CD4(+) regulatory T-cell response and mediates immune suppression in a murine ovarian cancer model. Cancer Medicine, 9(10), 3584–3591. https://doi.org/10.1002/cam4.2976
Niu, J., Maurice-Dror, C., Lee, D. H., Kim, D. W., Nagrial, A., Voskoboynik, M., et al. (2022). First-in-human phase 1 study of the anti-TIGIT antibody vibostolimab as monotherapy or with pembrolizumab for advanced solid tumors, including non-small-cell lung cancer. Annals of Oncology, 33(2), 169–180. https://doi.org/10.1016/j.annonc.2021.11.002
Pushpakom, S., Iorio, F., Eyers, P. A., Escott, K. J., Hopper, S., Wells, A., et al. (2019). Drug repurposing: Progress, challenges and recommendations. Nature Reviews Drug Discovery, 18(1), 41–58. https://doi.org/10.1038/nrd.2018.168
Tan, G. S. Q., Sloan, E. K., Lambert, P., Kirkpatrick, C. M. J., & Ilomäki, J. (2023). Drug repurposing using real-world data. Drug Discovery Today, 28(1), 103422. https://doi.org/10.1016/j.drudis.2022.103422
Joharatnam-Hogan, N., Cafferty, F. H., Macnair, A., Ring, A., & Langley, R. E. (2020). The role of aspirin in the prevention of ovarian, endometrial and cervical cancers. Womens Health (Lond), 16, 1745506520961710. https://doi.org/10.1177/1745506520961710
Gadducci, A., Biglia, N., Tana, R., Cosio, S., & Gallo, M. (2016). Metformin use and gynecological cancers: A novel treatment option emerging from drug repositioning. Critical Reviews in Oncology Hematology, 105, 73–83. https://doi.org/10.1016/j.critrevonc.2016.06.006
Lee, T. Y., Martinez-Outschoorn, U. E., Schilder, R. J., Kim, C. H., Richard, S. D., Rosenblum, N. G., et al. (2018). Metformin as a therapeutic target in endometrial cancers. Frontiers in Oncology, 8, 341. https://doi.org/10.3389/fonc.2018.00341
Kalogera, E., Roy, D., Khurana, A., Mondal, S., Weaver, A. L., He, X., et al. (2017). Quinacrine in endometrial cancer: Repurposing an old antimalarial drug. Gynecologic Oncology, 146(1), 187–195. https://doi.org/10.1016/j.ygyno.2017.04.022
Kumari, G. K., Kiran, A., & Krishnamurthy, P. T. (2021). Preliminary evaluation on the beneficial effects of pioglitazone in the treatment of endometrial cancer. Medical Oncology, 38(6), 71. https://doi.org/10.1007/s12032-021-01521-x
Ciou, H. H., Lee, T. H., Wang, H. C., Ding, Y. R., Tseng, C. J., Wang, P. H., et al. (2022). Repurposing gestrinone for tumor suppressor through P21 reduction regulated by JNK in gynecological cancer. Translational Research, 243, 21–32. https://doi.org/10.1016/j.trsl.2021.12.002
Chen, Y., Cui, Y., Sun, X., Wu, H., Ou, M., Tang, Y., et al. (2020). Repurposing of antipsychotics perphenazine for the treatment of endometrial cancer. Bioorganic & Medicinal Chemistry Letters, 30(14), 127239. https://doi.org/10.1016/j.bmcl.2020.127239
Cui, Y., Wu, H., Yang, L., Huang, T., Li, J., Gong, X., et al. (2021). Chlorpromazine sensitizes progestin-resistant endometrial cancer cells to MPA by upregulating PRB. Frontiers in Oncology, 11, 665832. https://doi.org/10.3389/fonc.2021.665832
Bejar, F. G., Oaknin, A., Williamson, C., Mayadev, J., Peters, P. N., Secord, A. A., et al. (2022). Novel therapies in gynecologic cancer. American Society of Clinical Oncology Educational Book, 42, 1–17. https://doi.org/10.1200/edbk_351294
Wang, Q., Peng, H., Qi, X., Wu, M., & Zhao, X. (2020). Targeted therapies in gynecological cancers: A comprehensive review of clinical evidence. Signal Transduction and Targeted Therapy, 5(1), 137. https://doi.org/10.1038/s41392-020-0199-6
Fan, R., Wang, Y., Wang, Y., Wei, L., & Zheng, W. (2017). Mechanism of progestin resistance in endometrial precancer/cancer through Nrf2-survivin pathway. American Journal of Translational Research, 9(3), 1483–1491.
Herzog, C., Marín, F., Jones, A., Evans, I., Reisel, D., Redl, E., et al. (2022). A simple cervicovaginal epigenetic test for screening and rapid triage of women with suspected endometrial cancer: Validation in several cohort and case/control sets. Journal of Clinical Oncology, 40(33), 3828–3838. https://doi.org/10.1200/jco.22.00266
Tew, W. P., Lacchetti, C., Ellis, A., Maxian, K., Banerjee, S., Bookman, M., et al. (2020). PARP inhibitors in the management of ovarian cancer: ASCO guideline. Journal of Clinical Oncology, 38(30), 3468–3493. https://doi.org/10.1200/JCO.20.01924
Mirza, M. R., Coleman, R. L., Gonzalez-Martin, A., Moore, K. N., Colombo, N., Ray-Coquard, I., et al. (2020). The forefront of ovarian cancer therapy: Update on PARP inhibitors. Annals of Oncology, 31(9), 1148–1159. https://doi.org/10.1016/j.annonc.2020.06.004
Jiang, X., Li, X., Li, W., Bai, H., & Zhang, Z. (2019). PARP inhibitors in ovarian cancer: Sensitivity prediction and resistance mechanisms. Journal of Cellular and Molecular Medicine, 23(4), 2303–2313. https://doi.org/10.1111/jcmm.14133
Li, H., Liu, Z. Y., Wu, N., Chen, Y. C., Cheng, Q., & Wang, J. (2020). PARP inhibitor resistance: The underlying mechanisms and clinical implications. Molecular Cancer, 19(1), 107. https://doi.org/10.1186/s12943-020-01227-0
Nesic, K., Kondrashova, O., Hurley, R. M., McGehee, C. D., Vandenberg, C. J., Ho, G. Y., et al. (2021). Acquired RAD51C promoter methylation loss causes PARP inhibitor resistance in high-grade serous ovarian carcinoma. Cancer Research, 81(18), 4709–4722. https://doi.org/10.1158/0008-5472.CAN-21-0774
Kondrashova, O., Nguyen, M., Shield-Artin, K., Tinker, A. V., Teng, N. N. H., Harrell, M. I., et al. (2017). Secondary somatic mutations restoring RAD51C and RAD51D associated with acquired resistance to the PARP inhibitor rucaparib in high-grade ovarian carcinoma. Cancer Discovery, 7(9), 984–998. https://doi.org/10.1158/2159-8290.CD-17-0419
Lampert, E. J., Zimmer, A., Padget, M., Cimino-Mathews, A., Nair, J. R., Liu, Y., et al. (2020). Combination of PARP inhibitor olaparib, and PD-L1 inhibitor durvalumab, in recurrent ovarian cancer: A proof-of-concept phase II study. Clinical Cancer Research, 26(16), 4268–4279. https://doi.org/10.1158/1078-0432.CCR-20-0056
Gonzalez-Martin, A., Desauw, C., Heitz, F., Cropet, C., Gargiulo, P., Berger, R., et al. (2022). Maintenance olaparib plus bevacizumab in patients with newly diagnosed advanced high-grade ovarian cancer: Main analysis of second progression-free survival in the phase III PAOLA-1/ENGOT-ov25 trial. European Journal of Cancer, 174, 221–231. https://doi.org/10.1016/j.ejca.2022.07.022
Hardesty, M. M., Krivak, T. C., Wright, G. S., Hamilton, E., Fleming, E. L., Belotte, J., et al. (2022). OVARIO phase II trial of combination niraparib plus bevacizumab maintenance therapy in advanced ovarian cancer following first-line platinum-based chemotherapy with bevacizumab. Gynecologic Oncology, 166(2), 219–229. https://doi.org/10.1016/j.ygyno.2022.05.020
Kim, T. K., Vandsemb, E. N., Herbst, R. S., & Chen, L. (2022). Adaptive immune resistance at the tumour site: Mechanisms and therapeutic opportunities. Nature Reviews Drug Discovery, 21(7), 529–540. https://doi.org/10.1038/s41573-022-00493-5
Konstantinopoulos, P. A., Luo, W., Liu, J. F., Gulhan, D. C., Krasner, C., Ishizuka, J. J., et al. (2019). Phase II study of avelumab in patients with mismatch repair deficient and mismatch repair proficient recurrent/persistent endometrial cancer. Journal of Clinical Oncology, 37(30), 2786–2794. https://doi.org/10.1200/JCO.19.01021
Antill, Y., Kok, P. S., Robledo, K., Yip, S., Cummins, M., Smith, D., et al. (2021). Clinical activity of durvalumab for patients with advanced mismatch repair-deficient and repair-proficient endometrial cancer. A nonrandomized phase 2 clinical trial. Journal for Immunotherapy of Cancer, 9(6):e002255. https://doi.org/10.1136/jitc-2020-002255
Oaknin, A., Tinker, A. V., Gilbert, L., Samouelian, V., Mathews, C., Brown, J., et al. (2020). Clinical activity and safety of the anti-programmed death 1 monoclonal antibody dostarlimab for patients with recurrent or advanced mismatch repair-deficient endometrial cancer: A nonrandomized phase 1 clinical trial. JAMA Oncology, 6(11), 1766–1772. https://doi.org/10.1001/jamaoncol.2020.4515
Makker, V., Taylor, M. H., Aghajanian, C., Oaknin, A., Mier, J., Cohn, A. L., et al. (2020). Lenvatinib plus pembrolizumab in patients with advanced endometrial cancer. Journal of Clinical Oncology, 38(26), 2981–2992. https://doi.org/10.1200/JCO.19.02627
O’Malley, D. M., Bariani, G. M., Cassier, P. A., Marabelle, A., Hansen, A. R., De Jesus Acosta, A., et al. (2022). Pembrolizumab in patients with microsatellite instability-high advanced endometrial cancer: Results from the KEYNOTE-158 study. Journal of Clinical Oncology, 40(7), 752–761. https://doi.org/10.1200/JCO.21.01874
Varga, A., Piha-Paul, S., Ott, P. A., Mehnert, J. M., Berton-Rigaud, D., Morosky, A., et al. (2019). Pembrolizumab in patients with programmed death ligand 1-positive advanced ovarian cancer: Analysis of KEYNOTE-028. Gynecologic Oncology, 152(2), 243–250. https://doi.org/10.1016/j.ygyno.2018.11.017
Nishio, S., Matsumoto, K., Takehara, K., Kawamura, N., Hasegawa, K., Takeshima, N., et al. (2020). Pembrolizumab monotherapy in Japanese patients with advanced ovarian cancer: Subgroup analysis from the KEYNOTE-100. Cancer Science, 111(4), 1324–1332. https://doi.org/10.1111/cas.14340
Zamarin, D., Burger, R. A., Sill, M. W., Powell, D. J., Jr., Lankes, H. A., Feldman, M. D., et al. (2020). Randomized phase II trial of nivolumab versus nivolumab and ipilimumab for recurrent or persistent ovarian cancer: An NRG oncology study. Journal of Clinical Oncology, 38(16), 1814–1823. https://doi.org/10.1200/JCO.19.02059
Liu, J. F., Herold, C., Gray, K. P., Penson, R. T., Horowitz, N., Konstantinopoulos, P. A., et al. (2019). Assessment of combined nivolumab and bevacizumab in relapsed ovarian cancer: A phase 2 clinical trial. JAMA Oncology, 5(12), 1731–1738. https://doi.org/10.1001/jamaoncol.2019.3343
Moroney, J. W., Powderly, J., Lieu, C. H., Bendell, J. C., Eckhardt, S. G., Chang, C. W., et al. (2020). Safety and clinical activity of atezolizumab plus bevacizumab in patients with ovarian cancer: A phase Ib study. Clinical Cancer Research, 26(21), 5631–5637. https://doi.org/10.1158/1078-0432.CCR-20-0477
Tewari, K. S., Monk, B. J., Vergote, I., Miller, A., de Melo, A. C., Kim, H. S., et al. (2022). Survival with cemiplimab in recurrent cervical cancer. New England Journal of Medicine, 386(6), 544–555. https://doi.org/10.1056/NEJMoa2112187
O’Malley, D. M., Oaknin, A., Monk, B. J., Selle, F., Rojas, C., Gladieff, L., et al. (2021). Phase II study of the safety and efficacy of the anti-PD-1 antibody balstilimab in patients with recurrent and/or metastatic cervical cancer. Gynecologic Oncology, 163(2), 274–280. https://doi.org/10.1016/j.ygyno.2021.08.018
Santin, A. D., Deng, W., Frumovitz, M., Buza, N., Bellone, S., Huh, W., et al. (2020). Phase II evaluation of nivolumab in the treatment of persistent or recurrent cervical cancer (NCT02257528/NRG-GY002). Gynecologic Oncology, 157(1), 161–166. https://doi.org/10.1016/j.ygyno.2019.12.034
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, C., Sheng, Y., Sun, X. et al. New insights for gynecological cancer therapies: from molecular mechanisms and clinical evidence to future directions. Cancer Metastasis Rev 42, 891–925 (2023). https://doi.org/10.1007/s10555-023-10113-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-023-10113-2