
Abstract
Epigenetic treatment has been approved by regulatory agencies for haematological malignancies. The success observed in cutaneous lymphomas represents a proof of principle that similar results may be obtained in solid tumours. Several agents that interfere with DNA methylation-demethylation and histones acetylation/deacetylation have been studied, and some (such as azacytidine, decitabine, valproic acid and vorinostat) are already in clinical use.
The aim of this review is to provide a brief overview of the molecular events underlying the antitumour effects of epigenetic treatments and to summarise data available on clinical trials that tested the use of epigenetic agents against solid tumours. We not only list results but also try to indicate how the proper evaluation of this treatment might result in a better selection of effective agents and in a more rapid development.
We divided compounds in demethylating agents and HDAC inhibitors. For each class, we report the antitumour activity and the toxic side effects. When available, we describe plasma pharmacokinetics and pharmacodynamic evaluation in tumours and in surrogate tissues (generally white blood cells).
Epigenetic treatment is a reality in haematological malignancies and deserves adequate attention in solid tumours. A careful consideration of available clinical data however is required for faster drug development and possibly to re-evaluate some molecules that were perhaps discarded too early.
Similar content being viewed by others


Background
Research on tumour biology has provided conclusive evidence on the primary role of genetic alterations in the initiation and progression of cancer. However, the deregulation of epigenetic processes such as DNA methylation and alterations of “histone code” are equally important oncogenic factors per se [1–4]. Epigenetic processes affect the packaging of chromatin and direct distinct cellular gene expression programmes. They are heritable through cell division and do not involve changes in the DNA sequence [4–6]. Operating at the level of chromatin structure, epigenetic mechanisms play a key role during embryogenesis, X-chromosome silencing, cellular proliferation and differentiation and in disease states [2, 4–6]. They also facilitate a selective readout of the genome, thereby regulating stem cell developmental potential and cell fate. Subtle disturbances of the epigenetic framework in progenitor, differentiating or terminal cells may, besides well-known genetic alterations, promote carcinogenesis [7, 8]. The dynamic and reversible nature of epigenetic mechanisms makes these processes of therapeutic relevance in many diseases including cancer.
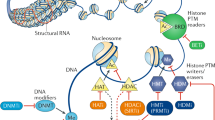
Epigenetic processes involve methylation of DNA and post-translational modification of nucleosomal histones, which contribute to a complex “epigenetic code” that superposes the nucleotide sequence to direct gene expression [4, 9–11] (Fig. 1a).

Schematic representation of gene expression regulation by epigenetic drugs, components of the DNA and chromatin-modifying machinery and ncRNAs. a Epigenetic drugs reported to be effective against cancer cells inhibit the activity of DNA methyltransferases (DNMTi) or histone deacetylases (HDACi). DNMTs add a methyl group (CH3) to the 5′ carbon atom of cytosine in DNA CpG dinucleotides. DNMTs also participate in multiprotein chromatin-modifying complexes containing histone deacetylases (HDACs) and histone methyltransferases (HMTs),which induce post-translational modifications of lysine residues in the amino terminal tails of nucleosomal histones, including deacetylation (HDACs), methylation (HMTs) and acetylation (histone acetyltransferases (HAT). Specific molecular modifications on CpGs and nucleosomal histones affect the higher order of chromatin architecture and function by changing the interaction of histones with DNA or the contact between different histones in adjacent nucleosomes. This allows or denies the accessibility of the transcriptional machinery and DNA-binding proteins to specific sites on genome, resulting in activation or silencing of gene transcription. Ac acetylation, Me methylation. b Short and long ncRNA are emerging as novel regulators of chromatin structure, alternative to DNA-binding proteins. They can act as key specificity determinants for epigenetic regulation of gene expression. In the nucleus, both short and long ncRNAs can bind complementary sequences on DNA or nascent RNA transcripts and guide the Argonaute-containing complexes (Ago) to recruit HDACs, HMTs and DNMTs for gene silencing. Nascent lncRNAs can also be tethered to the locus from which they are transcribed through association with RNA polymerase II (Pol II). In the cytosol, microRNAs and siRNAs act as post-transcriptional regulators of the expression of HDAC and DNMTs through their complementarity with mRNA sequences
DNA methylation results from the transfer of a methyl group from a methyl donor substrate, namely S-adenosyl-L-methionine (AdoMet), to the 5′ position of a cytosine in a CpG context. CpG dinucleotides can be sparse or tend to be gathered in repetitive sequences in or around gene promoters in regions known as CpG islands. The methylation status of CpG moieties within regulatory DNA sequences affects the transcription of the related gene [2, 10]. The creation of DNA methylation patterns during the embryogenesis establishes the compartmentalization of genome into transcriptionally active and inactive domains. DNA methylation is fundamental for a correct expression of imprinted genes, chromosomal dosage compensation (X-chromosome inactivation) and tissue-specific gene expression [2, 3, 12–14].
The oncogenic effect of DNA methylation is mainly related to the formation of a repressive chromatin structure on promoter regions that impairs the constitutive expression of genes involved in cell cycle regulation, DNA repair, apoptosis, differentiation, drug resistance, angiogenesis and metastasis [1–4]. A family of enzymes known as DNA methyltransferases (DNMTs) catalyses the DNA methylation reaction. DNMT1 is a maintenance methylase that recognises and methylates hemi-methylated CpG dinucleotides during DNA replication allowing the propagation and conservation of the DNA methylation patterns through the future generations [14, 15]. DNMT3a and 3b generally act as de novo methylases. They are highly expressed in embryonic stem cells, early embryos and developing germ cells and, at a low rate, in somatic tissues or postnatal animals. DNA methyltransferase-3-like (DNMT3-Like) lacks enzymatic activity but may be essential for the establishment of maternal methylation imprints and the appropriate expression of maternally imprinted genes. The inhibitory effect of CpG island methylation on gene expression is mediated by the involvement of proteins with high affinity for methylated CpGs. These methyl CpG-binding proteins (MeCP1, MeCP2, MBDs and Kaiso) [16–21] exert their function as transcriptional repressors via chromatin modification. Methyl CpG-binding proteins are often part of large repressor complexes as NuRD, NoRC, mSin3A and SWI-SNF. Repressor complexes recruit histone deacetylases (HDACs) and histone methyltransferases (HMTs) on methylated target promoter sequences. These enzymes catalyse covalent post-translational modifications of specific residues on histone 3 (H3) and 4 (H4) N-terminal tails (e.g. deacetylation of lysine (K) 9, demethylation of K4 and methylation of K9 and K27 of H3), inducing a compacted transcriptionally inactive chromatin structure. Histone acetylation status also depends on the contrasting activities of HDACs and histone acetyltransferases (HAT) group of enzymes. The latter are presumed to induce histone tail modifications (e.g. acetylation of K9 and K14 of H3), resulting in a transcriptionally active chromatin state. As for histone acetylation, histone lysine methylation can be dynamically regulated by the recruitment of members of the histone lysine methyltransferases and demethylase class of enzymes, which impose memory on gene transcription [22]. Other histone tail modifications include phosphorylation, sumoylation, ubiquitination and ADP ribosylation. Overall, DNA methylation and histone modifications work together to assemble a chromatin structure, which dynamically shifts from a transcriptionally permissive state to a transcriptionally inactive state and vice versa [2, 14].
Inhibition of HDACs can be achieved in normal conditions by endogenous molecules [23] explaining the plausibility of this process in the normal regulation of gene expression.
Aberrant DNA methylation and chromatin modifications, altering gene transcription states, are common hallmarks of human tumour cells [24]. Studies on leukaemias have provided paradigmatic examples for the functional implications of genetic and epigenetic alterations in cancer development [25, 26]. These studies underline the possibility of reversing disease-associated aberrant epigenetic states by targeting the catalytic activities of chromatin remodelling enzymes. Thus, these enzymes are attractive targets for therapeutic intervention in cancer [27–29]. The possibility of drug development in this field has recently been reviewed [22, 24, 30, 31].
Evidence is growing that non-coding RNAs (ncRNAs) are involved in inducing chromatin modifications and act as additional molecular determinants for epigenetic regulation of gene expression also in human cells [32–34] (Fig. 1b). NcRNAs comprise a large and heterogeneous family of RNA molecules differing in length (short, such as microRNAs, and long ncRNAs), which are transcribed from DNA but not translated into proteins. By regulating gene expression at the transcriptional and post-transcriptional level, they affect a broad range of physiologic functions and pathologies such as neoplastic diseases [35, 36]. Both short and long ncRNAs appear to function by guiding the recruitment HDACs, HMTs and DNMTs, and other proteins involved in the epigenetic regulation of transcription, to homology-containing loci on gene promoters and in the genome. Moreover, short ncRNA, as microRNAs and siRNAs, can repress the expression of HDAC, DNMTs and other components of chromatin-modifying complexes at the post-transcriptional level by interacting with their mRNAs [32, 33, 35] (Fig. 1b). Therefore, ncRNAs play direct roles in DNA methylation, heterochromatin formation, histone modification and gene silencing. In turn, they are epigenetically targeted for repression or activation; this can be a valuable way of amplifying changes in the levels of downstream effectors. Knowledge of these emerging regulatory roles of ncRNAs has implication not only in cellular physiology and pathology but also for the development of novel epigenetic drugs that re-establish the correct pattern of gene expression in complex diseases such as cancer.
Currently in the clinical setting, there are two classes of epigenetic drugs, which act through the inhibition of the enzymatic activities responsible for epigenetic transcriptional silencing: DNMTs and HDACs (Fig. 2). DNA methylation inhibitors 5-azacytidine (azacytidine) and 5-aza-2′-deoxycytidine (decitabine) have been approved by the US Food and Drug Administration (FDA) in 2004 and 2006, respectively, for the treatment of patients with myelodysplastic syndromes (MDS). US FDA approved the HDAC inhibitors suberoylanilide hydroxamic acid (SAHA, vorinostat, in 2006) and romidepsin (depsipeptide, in 2009) for the treatment of patients with progressive, persistent or recurrent cutaneous T-cell lymphoma [37]. In 2015, FDA approved panobinostat in combination with bortezomib and dexamethasone for the treatment of patients with multiple myeloma [38] and belinostat for the treatment of patients with peripheral T-cell lymphoma (PTCL) [39].

Chemical structures of different classes of DNMT and HDAC inhibitors. Antimetabolites 5-azacytidine and decitabine (5-aza-2′-deoxycytidine) are cytidine analogues; these nucleoside derivatives are incorporated into DNA leading to covalent adduct formation, thus acting as mechanistic inhibitors. The non-nucleoside DNMT inhibitor hydralazine interacts within the binding pocket of the enzyme interfering with the DNA methylation mechanism
The success of epigenetic therapies in inducing clinical responses in MDS and lymphoma not only gave to this kind of treatment high visibility, but it also suggested that similar results might be obtained in solid tumours and that this line of research deserves proper evaluation. The relevance of epigenetic treatment in haematological malignancies (leukaemias, lymphomas, myelodysplastic syndromes, myeloma) have already been described in detail [40].
In clinical studies, epigenetic treatment was administered alone or in combination with standard anticancer therapies (usually chemotherapy, sometimes radiotherapy) to improve their antitumour activities [41]. In some cases, the aim was more directly related to the control of gene activity and to prevent the development of resistance due to the overexpression of a specific gene, for example thymidylate synthase [42].
In several papers [43, 44], particularly those more recently published [45, 46], a detailed explanation for the combination of epigenetic and standard anticancer treatment is provided together with complete and stimulating results.
The present review will focus on the epigenetic treatment of solid tumours: we collected data from clinical studies available as full papers, data on ongoing studies are available at clinicaltrials.gov site and have been reviewed [47].
In the text, we discuss data on the pharmacology, pharmacokinetics (PK) and pharmacodynamics (PD), on the toxic side effects and on the antitumour activity of epigenetic treatments. In the tables, we give more details for epigenetic treatment alone (Table 1) or in combination with traditional anticancer agents or radiotherapy (Table 2).
Pharmacological aspects
An effective treatment, based on a sound scientific rationale, requires the identification of the relevant target(s) and the demonstration that the target(s) can be inhibited without excessive toxicity, and that the duration of this effect is sufficient to interfere with cell growth.
In the case of epigenetic treatment, it is necessary to prove that one of the different mechanisms that finely regulate gene expression (see above) is altered. Even if this mechanism is well established for many molecules, for some compounds such as CI-994 (N-acetyl-dinaline), an inhibitor of class I HDACs, the actual relevance of an epigenetic mode of action remains uncertain [27, 48, 49].
The list of epigenetic drugs is becoming longer every day and it also includes “old” drugs: Hydralazine, for example, has long been used as an anti-hypertensive agent, but it has recently been recognised as a demethylating agent [50, 51]. This also happens for HDAC inhibitors that include old molecules (such as valproic acid (VPA) [52] and a dozen compounds that have been recently synthesised [53].
Pharmacokinetics
To obtain an authentic effect on epigenetic mechanisms, the choice of the dose and schedule of administration is crucial since, especially for demethylating agents, if the drug concentration is too high this may result in cell toxicity or in a “traditional” antiproliferative effect. Furthermore, when a combination is used, the epigenetic effect must persist during chemotherapy (and possibly also later) in order to obtain an adequate synergism. This concept is guiding recent studies on new schedules of drug administration, as is the case for romidepsin [54].
Detailed data on pharmacokinetics have been published for several drugs (see Tables 1 and 2 for a complete list). The pharmacology of demethylating agents has been originally described in the past, and recent analyses mostly concern their interaction with other agents used in combination, such as temozolomide [55].
The pharmacology of HDAC inhibitors is particularly complex since many of these compounds act as enzyme inducers and may therefore modify their own kinetics when repeated dosing is used, or the pharmacology of associated drugs.
Drug concentrations in plasma are generally low, and a very sensitive assay, such as HPLC coupled to mass spectrometry, is required to obtain reliable data [56].
Individual characteristics may influence the kinetic parameters (pharmacogenetics): in the case of hydralazine, fast or slow metabolism is genetically determined, and in some studies, doses have been escalated or de-escalated according to individual metabolic parameters [57–59].
Pharmacodynamic effect
The most interesting part of the evaluation of anticancer drugs, particularly when dealing with an innovative mechanism of action, is the study of their effect in tumour cells. Several technical problems arise when trying to quantify the effect of epigenetic drugs, especially in solid tumours: it is difficult to decide what to measure, when and where.
Concerning demethylating agents, the effect is generally measured by evaluating global DNA methylation, but some authors determined the methylation status of specific genes that had previously been selected [60–62] and the level of expression of foetal haemoglobin has also been used as a PD marker [63]. The subject of DNA methylation may also be related to drug resistance caused by the activity of O(6)-methylguanine DNA methyltransferase (MGMT) that has been described in cerebral tumours [3, 64]. Decitabine has been used in an attempt to reduce methylation of genes involved in DNA repair in melanoma patients treated with temozolomide [55] but no such effect was evident even if decitabine caused hypomethylation of the HbF gene promoter.
The situation is more complex for HDAC inhibitors since not all drugs inhibit the different enzyme classes that are present in eukaryotic cells to the same extent. Some molecules have a wide inhibiting effect [65]; some are more restricted, and class-specific inhibitors such as CHR-3996, specific for class I HDACs enzymes [45], are being introduced in the clinics. Furthermore, the inhibition of de-acetylating enzymes may result in acetylation not only of histones, but also of other proteins, such as tubulin and Hsp90 that are involved in anticancer drug activity/resistance or in unrelated cellular pathways [66, 67].
An additional difficulty derives from the definition of a cut-off value: some authors, for example, required a doubling of histone acetylation to consider a result as “relevant” [68] but this was not mandatory in many other studies.
The effect of HDAC inhibitors was generally determined in terms of enzymatic activity, but in some cases histone acetylation, particularly H3 and H4, has been used as marker of activity. A consistent increase of H3 acetylation in peripheral blood mononuclear cells (PBMC) at effective doses has been observed with several agents, even if large inter-patient variations were often reported [69], and, more importantly, intratumoral H3 acetylation increase did not always correlate with response [70].
The effect of epigenetic treatment has also been evaluated by looking at specific genes in terms of expression [57, 62] or re-activation [71]. It is becoming clear that many elements are involved, and that it may be difficult to identify a consistent pattern in gene activation-inactivation [72] to be used as a marker of epigenetic activity.
The gold standard of pharmacodynamic studies is the evaluation of the effects in tumours. The study of malignant cells is certainly more complicated in patients with solid tumours than in leukaemia since cancer cells are more difficult to obtain, especially at multiple time points. Some reports on the evaluation of epigenetic therapy in solid tumour tissues have been published and deserve special consideration. The number of evaluable samples was often low, but informative results were obtained and reported [41, 73]. In ovarian cancer, it has been possible to evaluate the activity of demethylating agents on cells obtained from ascitic fluid and a gene-specific reduction of DNA methylation was evident [74]. Serial biopsies were obtained from patients with in head-and-neck carcinomas [70], glioblastoma [75], cervical and breast cancer and other tumours [57, 58, 66].
To overcome the difficulty in obtaining tumour samples, several groups have focused on the identification of surrogate markers. PBMC represent the most commonly used alternative. It is possible to measure HDAC activity and histone acetylation or DNA methylation. PBMC have also been used, in studies of drug combinations, to measure the target of non-epigenetic drug [76]. When PBMC and tumour biopsies were compared, however, results were not always consistent [66]. PBMC represent a “surrogate” tissue, and further improvement is required in order to make results obtained in these cells more representative of what is actually taking place in cancer.
A promising technique is the evaluation of circulating cell-free DNA [77] on which several specific analyses to identify epigenetic modifications can be performed [78]. The activity of demethylating agents has been evaluated by measuring the methylation status of circulating cell-free DNA in plasma in patients with refractory advanced non-small cell lung cancer (NSCLC) [62]. This is an interesting alternative with applications in many aspects of medicine, but further studies are required before it can be considered a reliable marker of epigenetic activity.
To obtain an adequate effect, sufficient drug concentrations must persist in the target cells for an adequate time. If there is a relationship between plasma kinetics and tissue effect (PK/PD relationship), drug dosing may be adapted on the basis of PK parameters that are easier to obtain. The permanence of a sufficient drug concentration or of a measurable effect has been evaluated in different tumour types and provides a rationale for the antitumour activity observed (see Tables 1 and 2 for details). Concerning the PK/PD relationship, data are difficult to interpret: no correspondence was found between plasma levels of valproate and histone acetylation in cervical cancer [79] so that PD assays may be required until we can devise more efficient PK models.
Toxic side effects
One of the most interesting characteristics of many epigenetic drugs is that toxicity, at doses sufficient to achieve effective plasma concentrations, is generally very mild and has been described in detail in many studies of epigenetic agents used alone or in combination with standard anticancer agents (see Tables 1 and 2 for details).
Toxicity of demethylating agents
The best known agents that interfere with DNA methylation are decitabine and 5-azacytidine. More limited data are available for zebularine [46]. Since these agents also have traditional antiproliferative activity, the dose used plays a key role. At high doses, 5-azacytidine can cause neutropenia [68], similar to what is described for traditional anticancer drugs. It is generally assumed that in order to exploit the epigenetic action, it is necessary to use very low doses that are insufficient to cause any antiproliferative effect.
Toxicity of HDAC inhibitors
VPA is the best known molecule in this class since it has been used for many years as an antiepileptic drug: it is extremely well tolerated by patients, and also its long-term effects are well known. The most commonly reported complaints are neurological symptoms (such as dizziness) that are generally transitory and reversible [79, 80]. Neurological symptoms may become excessive when VPA is combined with other agents [63].
When vorinostat (SAHA) was tested in mice, relevant systemic toxicity was observed only at high doses [81]. In patients, anaemia, anorexia, hyperglycaemia, thrombocytopenia, fatigue and nausea have frequently been reported [82]. Similar toxic side effects were described for SB939 [83], for rosminostat [69] and romidepsin [70]. ECG abnormalities of different severity are the most concerning toxic side effects of CHR3996 [45] and of romidepsin [54].
Antitumour activity
In a phase III trial of chemotherapy ± epigenetic treatment in cancer of the uterine cervix [59], there was an increase in progression-free survival, and the analysis of molecular correlates is pending.
Concerning phase II studies, that evaluated the antitumour activity of epigenetic agents alone or in combination with standard anticancer treatment, several studies have been reported. In many cases, results were described in terms of reduction of tumour volume in advanced disease resistant to several lines of anticancer treatments, similarly to what is standard for the evaluation of traditional antiproliferative agents [74, 82, 84, 85]. It is probably not surprising that several trials were reported as negative (see Tables 1 and 2 for details). Responses were evaluated according to Response Evaluation Criteria In Solid Tumors (RECIST): these may be useful for conventional anticancer therapies but do not seem adequate for epigenetic treatments that may result in disease stabilisation rather than in tumour shrinkage.
The evaluation of an epigenetic treatment, which is strictly connected to gene expression, can be performed more accurately in diseases where the genetic influence on the activity of the antitumour treatment is known. For this reason, the choice to evaluate the combination of entinostat and erlotinib in NSCLC was very sound [86]. The addition of the epigenetic agent, however, only improved progression-free survival (PFS) in tumours with high levels of e-cadherin, suggesting that this may represent a selection criterion for further studies.
The neo-adjuvant treatment of breast cancer represents a unique possibility in order to evaluate the activity of new agents. Not only new anticancer agents but also epigenetic therapy (hydralazine and valproate) have been tested in this setting [57], and the analysis of tumour biopsies confirmed that an epigenetic effect in terms of demethylation and of histone acetylation was detectable.
An interesting tactic was to evaluate the addition of epigenetic treatment in order to prevent or to reverse resistance due to the overexpression of a specific gene. This is the case, for example, of the increase in thymidylate synthase induced by fluorouracil treatment [87]. A clinical trial performed to demonstrate that this can be obtained in patients proved that treatment was feasible even if no clinically meaningful effect was obtained and no data were available for PD evaluation [88]. In a different study, patients with tumours progressing during standard chemotherapy were treated with the same regimen with the addition of hydralazine and valproate [58]. Data were analysed in detail, and even if tumour response was limited, there was evidence that adequate plasma concentrations were achieved and that an epigenetic effect was present. A similar approach was used in melanoma patients treated with temozolomide and decitabine: there was no antitumour activity even in the presence of a measurable PD effect [55].
Conclusions
Epigenetic therapy is being more and more recognised as an effective and well-tolerated treatment of cancer. Data in leukaemias and myelodysplastic syndromes are now consistent, and the success obtained in cutaneous lymphomas represents a proof of principle that solid tumours may also respond. This is also supported by preclinical data; still clinical results fall short of expectations: several reasons may explain this discrepancy.
We are convinced that, similarly to what has been observed for tyrosine-kinase inhibitors in cancer [89], we need a better selection of tumours and of patients that may benefit from these treatments. It has already been stated that epigenetic drugs, and HDAC inhibitors in particular, “might be useful only in those tumours in which HDACs are directly involved in the pathogenesis” [27].
It is not surprising that clinical results have generally been disappointing: standard evaluation of anticancer activity, mostly based on tumour volume reduction, may not be an adequate index of activity for epigenetic treatment. Epigenetics is a complex mechanism of gene regulation: it will take time before we can exploit it at its best. We definitely need more appropriate tests to select potentially responding tumours, but we also need agents with demonstrated epigenetic activity and solid data in order to choose the most effective dose and schedule.
Several technical issues remain to be solved and this will keep researchers busy, both in preclinical and clinical settings, for a long time.
Abbreviations
- AdoMet:
-
adenosyl-L-methionine
- DNMT3-Like:
-
DNA methyltransferase-3-like
- DNMTs:
-
DNA methyltransferases
- FDA:
-
Food and Drug Administration
- H3:
-
histone 3
- H4:
-
histone 4
- HATs:
-
histone acetyltransferases
- HDACs:
-
histone deacetylases
- HMTs:
-
histone methyltransferases
- MDS:
-
myelodysplastic syndromes
- MGMT:
-
O(6)-methylguanine DNA methyltransferase
- ncRNAs:
-
non-coding RNAs
- NSCLC:
-
non-small cell lung cancer
- PBMC:
-
peripheral blood mononuclear cells
- PD:
-
pharmacodynamics
- PFS:
-
progression-free survival
- PK:
-
pharmacokinetics
- RECIST:
-
Response Evaluation Criteria In Solid Tumors
- SAHA:
-
suberoylanilide hydroxamic acid (vorinostat)
- VPA:
-
valproic acid
References
Baylin SB, Schuebel KE. Genomic biology: the epigenomic era opens. Nature. 2007;448:548–9.
Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–92.
Rodríguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med. 2011;17:330–9.
Turner BM. Defining an epigenetic code. Nat Cell Biol. 2007;9:2–6.
Baylin SB, Ohm JE. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction? Nat Rev Cancer. 2006;6:107–16.
Clark SJ. Action at a distance: epigenetic silencing of large chromosomal regions in carcinogenesis. Hum Mol Genet. 2007;16(Spec No 1):R88–95.
Meissner A. Epigenetic modifications in pluripotent and differentiated cells. Nat Biotechnol. 2010;28:1079–88.
Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–60.
Bird AP. CpG-rich islands and the function of DNA methylation. Nature. 1986;321:209–13.
Bird AP, Wolffe AP. Methylation-induced repression--belts, braces, and chromatin. Cell. 1999;99:451–4.
Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. 2007;8:286–98.
Li E, Beard C, Jaenisch R. Role for DNA methylation in genomic imprinting. Nature. 1993;366:362–5.
Walsh CP, Bestor TH. Cytosine methylation and mammalian development. Genes Dev. 1999;13:26–34.
Zardo G, Fazi F, Travaglini L, Nervi C. Dynamic and reversibility of heterochromatic gene silencing in human disease. Cell Res. 2005;15:679–90.
Brenner C, Fuks F. DNA methyltransferases: facts, clues, mysteries. Curr Top Microbiol Immunol. 2006;301:45–66.
Prokhortchouk A, Hendrich B, Jørgensen H, Ruzov A, Wilm M, Georgiev G, et al. The p120 catenin partner Kaiso is a DNA methylation-dependent transcriptional repressor. Genes Dev. 2001;15:1613–8.
Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, et al. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19:187–91.
Feng Q, Zhang Y. The MeCP1 complex represses transcription through preferential binding, remodeling, and deacetylating methylated nucleosomes. Genes Dev. 2001;15:827–32.
Hendrich B, Guy J, Ramsahoye B, Wilson VA, Bird A. Closely related proteins MBD2 and MBD3 play distinctive but interacting roles in mouse development. Genes Dev. 2001;15:710–23.
Ng HH, Zhang Y, Hendrich B, Johnson CA, Turner BM, Erdjument-Bromage H, et al. MBD2 is a transcriptional repressor belonging to the MeCP1 histone deacetylase complex. Nat Genet. 1999;23:58–61.
Fuks F, Burgers WA, Brehm A, Hughes-Davies L, Kouzarides T. DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat Genet. 2000;24:88–91.
Højfeldt JW, Agger K, Helin K. Histone lysine demethylases as targets for anticancer therapy. Nat Rev Drug Discov. 2013;12:917–30.
Spiegel S, Milstien S, Grant S. Endogenous modulators and pharmacological inhibitors of histone deacetylases in cancer therapy. Oncogene. 2012;31:537–51.
Easwaran H, Tsai HC, Baylin SB. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell. 2014;54:716–27.
Zardo G, Cimino G, Nervi C. Epigenetic plasticity of chromatin in embryonic and hematopoietic stem/progenitor cells: therapeutic potential of cell reprogramming. Leukemia. 2008;22:1503–18.
Mehdipour P, Santoro F, Minucci S. Epigenetic alterations in acute myeloid leukemias. FEBS J. 2015;282:1786–800.
Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6:38–51.
Leone G, D’Alò F, Zardo G, Voso MT, Nervi C. Epigenetic treatment of myelodysplastic syndromes and acute myeloid leukemias. Curr Med Chem. 2008;15:1274–87.
Rius M, Lyko F. Epigenetic cancer therapy: rationales, targets and drugs. Oncogene. 2012;31:4257–65.
Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov. 2012;11:384–400.
Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 2014;13:673–91.
Holoch D, Moazed D. RNA-mediated epigenetic regulation of gene expression. Nat Rev Genet. 2015;16:71–84.
Roberts TC, Morris KV, Weinberg MS. Perspectives on the mechanism of transcriptional regulation by long non-coding RNAs. Epigenetics. 2014;9:13–20.
Zardo G, Ciolfi A, Vian L, Billi M, Racanicchi S, Grignani F, et al. Transcriptional targeting by microRNA-polycomb complexes: a novel route in cell fate determination. Cell Cycle. 2012;11:3543–9.
Liz J, Esteller M. lncRNAs and microRNAs with a role in cancer development. Biochim Biophys Acta. 2015; doi:10.1016/j.bbagrm.2015.06.015.
Pagano F, De Marinis E, Grignani F, Nervi C. Epigenetic role of miRNAs in normal and leukemic hematopoiesis. Epigenomics. 2013;5:539–52.
Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. 2007;12:1247–52.
San-Miguel JF, Hungria VTM, Yoon SS, Beksac M, Dimopoulos MA, Elghandour A, et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 2014;15:1195–206.
O’Connor OA, Horwitz S, Masszi T, Van Hoof A, Brown P, Doorduijn J, et al. Belinostat in patients with relapsed or refractory peripheral T-cell lymphoma: results of the pivotal phase II BELIEF (CLN-19) study. J Clin Oncol. 2015;33:2492–9.
Santini V, Melnick A, Maciejewski JP, Duprez E, Nervi C, Cocco L, et al. Epigenetics in focus: pathogenesis of myelodysplastic syndromes and the role of hypomethylating agents. Crit Rev Oncol Hematol. 2013;88:231–45.
Ree AH, Dueland S, Folkvord S, Hole KH, Seierstad T, Johansen M, et al. Vorinostat, a histone deacetylase inhibitor, combined with pelvic palliative radiotherapy for gastrointestinal carcinoma: the Pelvic Radiation and Vorinostat (PRAVO) phase 1 study. Lancet Oncol. 2010;11:459–64.
Appleton K, Mackay HJ, Judson I, Plumb JA, McCormick C, Strathdee G, et al. Phase I and pharmacodynamic trial of the DNA methyltransferase inhibitor decitabine and carboplatin in solid tumors. J Clin Oncol. 2007;25:4603–9.
Munster P, Marchion D, Bicaku E, Lacevic M, Kim J, Centeno B, et al. Clinical and biological effects of valproic acid as a histone deacetylase inhibitor on tumor and surrogate tissues: phase I/II trial of valproic acid and epirubicin/FEC. Clin Cancer Res. 2009;15:2488–96.
Munster P, Marchion D, Bicaku E, Schmitt M, Lee JH, DeConti R, et al. Phase I trial of histone deacetylase inhibition by valproic acid followed by the topoisomerase II inhibitor epirubicin in advanced solid tumors: a clinical and translational study. J Clin Oncol. 2007;25:1979–85.
Banerji U, van Doorn L, Papadatos-Pastos D, Kristeleit R, Debnam P, Tall M, et al. A phase I pharmacokinetic and pharmacodynamic study of CHR-3996, an oral class I selective histone deacetylase inhibitor in refractory solid tumors. Clin Cancer Res. 2012;18:2687–94.
Chen M, Shabashvili D, Nawab A, Yang SX, Dyer LM, Brown KD, et al. DNA methyltransferase inhibitor, zebularine, delays tumor growth and induces apoptosis in a genetically engineered mouse model of breast cancer. Mol Cancer Ther. 2012;11:370–82.
Nebbioso A, Carafa V, Benedetti R, Altucci L. Trials with ‘epigenetic’ drugs: an update. Mol Oncol. 2012;6:657–82.
Prakash S, Foster B, Meyer M, Wozniak A, Heilbrun L, Flaherty L, et al. Chronic oral administration of CI-994: a phase 1 study. Invest New Drugs. 2001;19:1–1.
Undevia SD. A phase I study of the oral combination of CI-994, a putative histone deacetylase inhibitor, and capecitabine. Ann Oncol. 2004;15:1705–11.
Segura-Pacheco B, Trejo-Becerril C, Perez-Cardenas E, Taja-Chayeb L, Mariscal I, Chavez A, et al. Reactivation of tumor suppressor genes by the cardiovascular drugs hydralazine and procainamide and their potential use in cancer therapy. Clin Cancer Res. 2003;9:1596–603.
Singh N, Dueñas-González A, Lyko F, Medina-Franco JL. Molecular modeling and molecular dynamics studies of hydralazine with human DNA methyltransferase 1. ChemMedChem. 2009;4:792–9.
Göttlicher M, Minucci S, Zhu P, Krämer OH, Schimpf A, Giavara S, et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001;20:6969–78.
Lane A, Chabner B. Histone deacetylase inhibitors in cancer therapy. J Clin Oncol. 2009;27:5459–68.
Amiri-Kordestani L, Luchenko V, Peer CJ, Ghafourian K, Reynolds J, Draper D, et al. Phase I trial of a new schedule of romidepsin in patients with advanced cancers. Clin Cancer Res. 2013;19:4499–507.
Tawbi HA, Beumer JH, Tarhini AA, Moschos S, Buch SC, Egorin MJ, et al. Safety and efficacy of decitabine in combination with temozolomide in metastatic melanoma: a phase I/II study and pharmacokinetic analysis. Ann Oncol. 2013;24:1112–9.
Karahoca M, Momparler RL. Pharmacokinetic and pharmacodynamic analysis of 5-aza-2’-deoxycytidine (decitabine) in the design of its dose-schedule for cancer therapy. Clin Epigenetics. 2013;5(1):3. doi:10.1186/1868-7083-5-3.
Arce C, Pérez-Plasencia C, González-Fierro A, de la Cruz-Hernández E, Revilla-Vázquez A, Chávez-Blanco A, et al. A proof-of-principle study of epigenetic therapy added to neoadjuvant doxorubicin cyclophosphamide for locally advanced breast cancer. PLoS One. 2006;1:e98.
Candelaria M, Gallardo-Rincón D, Arce C, Cetina L, Aguilar-Ponce JL, Arrieta O, et al. A phase II study of epigenetic therapy with hydralazine and magnesium valproate to overcome chemotherapy resistance in refractory solid tumors. Ann Oncol. 2007;18:1529–38.
Coronel J, Cetina L, Pacheco I, Trejo-Becerril C, González-Fierro A, de la Cruz-Hernandez E, et al. A double-blind, placebo-controlled, randomized phase III trial of chemotherapy plus epigenetic therapy with hydralazine valproate for advanced cervical cancer. Preliminary results (1). Med Oncol. 2011;28:540–6.
Vansteenkiste J, Van Cutsem E, Dumez H, Chen C, Ricker JL, Randolph SS, et al. Early phase II trial of oral vorinostat in relapsed or refractory breast, colorectal, or non-small cell lung cancer. Invest New Drugs. 2008;26:483–8.
Singal R, Ramachandran K, Gordian E, Quintero C, Zhao W, Reis IM. Phase I/II study of azacitidine, docetaxel, and prednisone in patients with metastatic castration-resistant prostate cancer previously treated with docetaxel-based therapy. Clin Genitourin Cancer. 2015;13:22–31.
Juergens RA, Wrangle J, Vendetti FP, Murphy SC, Zhao M, Coleman B, et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov. 2011;1:598–607.
Chu BF, Karpenko MJ, Liu Z, Aimiuwu J, Villalona-Calero MA, Chan KK, et al. Phase I study of 5-aza-2’-deoxycytidine in combination with valproic acid in non-small-cell lung cancer. Cancer Chemother Pharmacol. 2013;71:115–21.
Gerson SL. MGMT: its role in cancer aetiology and cancer therapeutics. Nat Rev Cancer. 2004;4:296–307.
Marks PA. Discovery and development of SAHA as an anticancer agent. Oncogene. 2007;26:1351–6.
Ramaswamy B, Fiskus W, Cohen B, Pellegrino C, Hershman DL, Chuang E, et al. Phase I-II study of vorinostat plus paclitaxel and bevacizumab in metastatic breast cancer: evidence for vorinostat-induced tubulin acetylation and Hsp90 inhibition in vivo. Breast Cancer Res Treat. 2012;132:1063–72.
Newbold A, Matthews GM, Bots M, Cluse LA, Clarke CJP, Banks KM, et al. Molecular and biologic analysis of histone deacetylase inhibitors with diverse specificities. Mol Cancer Ther. 2013;12:2709–21.
Braiteh F, Soriano AO, Garcia-Manero G, Hong D, Johnson MM, Silva LDP, et al. Phase I study of epigenetic modulation with 5-azacytidine and valproic acid in patients with advanced cancers. Clin Cancer Res. 2008;14:6296–301.
Brunetto AT, Ang JE, Lal R, Olmos D, Molife LR, Kristeleit R, et al. First-in-human, pharmacokinetic and pharmacodynamic phase i study of resminostat, an oral histone deacetylase inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2013;19:5494–504.
Haigentz M, Kim M, Sarta C, Lin J, Keresztes RS, Culliney B, et al. Phase II trial of the histone deacetylase inhibitor romidepsin in patients with recurrent/metastatic head and neck cancer. Oral Oncol. 2012;48:1281–8.
Mohammed TA, Holen KD, Jaskula-Sztul R, Mulkerin D, Lubner SJ, Schelman WR, et al. A pilot phase II study of valproic acid for treatment of low-grade neuroendocrine carcinoma. Oncologist. 2011;16:835–43.
Belinsky SA, Grimes MJ, Picchi MA, Mitchell HD, Stidley CA, Tesfaigzi Y, et al. Combination therapy with vidaza and entinostat suppresses tumor growth and reprograms the epigenome in an orthotopic lung cancer model. Cancer Res. 2011;71:454–62.
Mackay HJ, Hirte H, Colgan T, Covens A, MacAlpine K, Grenci P, et al. Phase II trial of the histone deacetylase inhibitor belinostat in women with platinum resistant epithelial ovarian cancer and micropapillary (LMP) ovarian tumours. Eur J Cancer. 2010;46:1573–9.
Matei D, Fang F, Shen C, Schilder J, Arnold A, Zeng Y, et al. Epigenetic resensitization to platinum in ovarian cancer. Cancer Res. 2012;72:2197–205.
Galanis E, Jaeckle KA, Maurer MJ, Reid JM, Ames MM, Hardwick JS, et al. Phase II trial of vorinostat in recurrent glioblastoma multiforme: a north central cancer treatment group study. J Clin Oncol. 2009;27:2052–8.
Millward M, Price T, Townsend A, Sweeney C, Spencer A, Sukumaran S, et al. Phase 1 clinical trial of the novel proteasome inhibitor marizomib with the histone deacetylase inhibitor vorinostat in patients with melanoma, pancreatic and lung cancer based on in vitro assessments of the combination. Invest New Drugs. 2012;30:2303–17.
Diaz LA, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol. 2014;32:579–86.
Wittenberger T, Sleigh S, Reisel D, Zikan M, Wahl B, Alunni-Fabbroni M, et al. DNA methylation markers for early detection of women’s cancer: promise and challenges. Epigenomics. 2014;6:311–27.
Chavez-Blanco A, Segura-Pacheco B, Perez-Cardenas E, Taja-Chayeb L, Cetina L, Candelaria M, et al. Histone acetylation and histone deacetylase activity of magnesium valproate in tumor and peripheral blood of patients with cervical cancer. A phase I study. Mol Cancer. 2005;4:22.
Atmaca A, Al-Batran SE, Maurer A, Neumann A, Heinzel T, Hentsch B, et al. Valproic acid (VPA) in patients with refractory advanced cancer: a dose escalating phase I clinical trial. Br J Cancer. 2007;97:177–82.
Butler L, Agus D, Scher H, Higgins B, Rose A, Cordon-Cardo C, et al. Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, suppresses the growth of prostate cancer cells in vitro and in vivo. Cancer Res. 2000;60:5165–70.
Blumenschein GR, Kies MS, Papadimitrakopoulou VA, Lu C, Kumar AJ, Ricker JL, et al. Phase II trial of the histone deacetylase inhibitor vorinostat (Zolinza, suberoylanilide hydroxamic acid, SAHA) in patients with recurrent and/or metastatic head and neck cancer. Invest New Drugs. 2008;26:81–7.
Razak ARA, Hotte SJ, Siu LL, Chen EX, Hirte HW, Powers J, et al. Phase I clinical, pharmacokinetic and pharmacodynamic study of SB939, an oral histone deacetylase (HDAC) inhibitor, in patients with advanced solid tumours. Br J Cancer. 2011;104:756–62.
Munster PN, Thurn KT, Thomas S, Raha P, Lacevic M, Miller A, et al. A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Br J Cancer. 2011;104:1828–35.
Ramalingam SS, Maitland ML, Frankel P, Argiris AE, Koczywas M, Gitlitz B, et al. Carboplatin and Paclitaxel in combination with either vorinostat or placebo for first-line therapy of advanced non-small-cell lung cancer. J Clin Oncol. 2010;28:56–62.
Witta SE, Jotte RM, Konduri K, Neubauer MA, Spira AI, Ruxer RL, et al. Randomized phase II trial of Erlotinib with and without entinostat in patients with advanced non-small-cell lung cancer who progressed on prior chemotherapy. J Clin Oncol. 2012;30:2248–55.
Peters GJ, van der Wilt CL, van Triest B, Codacci-Pisanelli G, Johnston PG, van Groeningen CJ, et al. Thymidylate synthase and drug resistance. Eur J Cancer. 1995;31A:1299–305.
Fakih MG, Pendyala L, Fetterly G, Toth K, Zwiebel JA, Espinoza-Delgado I, et al. A phase I, pharmacokinetic and pharmacodynamic study on vorinostat in combination with 5-fluorouracil, leucovorin, and oxaliplatin in patients with refractory colorectal cancer. Clin Cancer Res. 2009;15:3189–95.
Codacci-Pisanelli G, Frati L, Mini E. Three cheers for targeted therapy in non-small cell lung cancer… When we hit the target! J Chemother. 2011;23:245–6.
Fouliard S, Robert R, Jacquet-Bescond A, du Rieu QC, Balasubramanian S, Loury D, et al. Pharmacokinetic/pharmacodynamic modelling-based optimisation of administration schedule for the histone deacetylase inhibitor abexinostat (S78454/PCI-24781) in phase I. Eur J Cancer. 2013;49:2791–7.
Lin J, Gilbert J, Rudek MA, Zwiebel JA, Gore S, Jiemjit A, et al. A phase I dose-finding study of 5-azacytidine in combination with sodium phenylbutyrate in patients with refractory solid tumors. Clin Cancer Res. 2009;15:6241–9.
Giaccone G, Rajan A, Berman A, Kelly RJ, Szabo E, Lopez-Chavez A, et al. Phase II study of belinostat in patients with recurrent or refractory advanced thymic epithelial tumors. J Clin Oncol. 2011;29:2052–9.
Yeo W, Chung HC, Chan SL, Wang LZ, Lim R, Picus J, et al. Epigenetic therapy using belinostat for patients with unresectable hepatocellular carcinoma: a multicenter phase I/II study with biomarker and pharmacokinetic analysis of tumors from patients in the Mayo Phase II Consortium and the Cancer Therapeutics Research Group. J Clin Oncol. 2012;30:3361–7.
Ramalingam SS, Belani CP, Ruel C, Frankel P, Gitlitz B, Koczywas M, et al. Phase II study of belinostat (PXD101), a histone deacetylase inhibitor, for second line therapy of advanced malignant pleural mesothelioma. J Thorac Oncol. 2009;4:97–101.
Steele NL, Plumb JA, Vidal L, Tjørnelund J, Knoblauch P, Buhl-Jensen P, et al. Pharmacokinetic and pharmacodynamic properties of an oral formulation of the histone deacetylase inhibitor Belinostat (PXD101). Cancer Chemother Pharmacol. 2011;67:1273–9.
Siu LL, Pili R, Duran I, Messersmith WA, Chen EX, Sullivan R, et al. Phase I study of MGCD0103 given as a three-times-per-week oral dose in patients with advanced solid tumors. J Clin Oncol. 2008;26:1940–7.
Ryan QC, Headlee D, Acharya M, Sparreboom A, Trepel JB, Ye J, et al. Phase I and pharmacokinetic study of MS-275, a histone deacetylase inhibitor, in patients with advanced and refractory solid tumors or lymphoma. J Clin Oncol. 2005;23:3912–22.
Shapiro GI, Frank R, Dandamudi UB, Hengelage T, Zhao L, Gazi L, et al. The effect of food on the bioavailability of panobinostat, an orally active pan-histone deacetylase inhibitor, in patients with advanced cancer. Cancer Chemother Pharmacol. 2012;69:555–62.
Cassier PA, Lefranc AY, Amela E, Chevreau C, Bui BN, Lecesne A, et al. A phase II trial of panobinostat in patients with advanced pretreated soft tissue sarcoma. A study from the French Sarcoma Group. Br J Cancer. 2013;109:909–14.
Rathkopf DE, Picus J, Hussain A, Ellard S, Chi KN, Nydam T, et al. A phase 2 study of intravenous panobinostat in patients with castration-resistant prostate cancer. Cancer Chemother Pharmacol. 2013;72:537–44.
Clive S, Woo MM, Nydam T, Kelly L, Squier M, Kagan M. Characterizing the disposition, metabolism, and excretion of an orally active pan-deacetylase inhibitor, panobinostat, via trace radiolabeled 14C material in advanced cancer patients. Cancer Chemother Pharmacol. 2012;70:513–22.
Reid T, Valone F, Lipera W, Irwin D, Paroly W, Natale R, et al. Phase II trial of the histone deacetylase inhibitor pivaloyloxymethyl butyrate (Pivanex, AN-9) in advanced non-small cell lung cancer. Lung Cancer. 2004;45:381–6.
Venugopal B, Baird R, Kristeleit RS, Plummer R, Cowan R, Stewart A, et al. A phase I study of quisinostat (JNJ-26481585), an oral hydroxamate histone deacetylase inhibitor with evidence of target modulation and antitumor activity, in patients with advanced solid tumors. Clin Cancer Res. 2013;19:4262–72.
Molife LR, Attard G, Fong PC, Karavasilis V, Reid AHM, Patterson S, et al. Phase II, two-stage, single-arm trial of the histone deacetylase inhibitor (HDACi) romidepsin in metastatic castration-resistant prostate cancer (CRPC). Ann Oncol. 2010;21:109–13.
Bradley D, Rathkopf D, Dunn R, Stadler WM, Liu G, Smith DC, et al. Vorinostat in advanced prostate cancer patients progressing on prior chemotherapy (National Cancer Institute Trial 6862): trial results and interleukin-6 analysis: a study by the Department of Defense Prostate Cancer Clinical Trial Consortium and University of Chicago Phase 2 Consortium. Cancer. 2009;115:5541–9.
Luu TH, Morgan RJ, Leong L, Lim D, McNamara M, Portnow J, et al. A phase II trial of vorinostat (suberoylanilide hydroxamic acid) in metastatic breast cancer: a California Cancer Consortium study. Clin Cancer Res. 2008;14:7138–42.
Modesitt SC, Sill M, Hoffman JS, Bender DP, Gynecologic Oncology Group. A phase II study of vorinostat in the treatment of persistent or recurrent epithelial ovarian or primary peritoneal carcinoma: a Gynecologic Oncology Group study. Gynecol Oncol. 2008;109:182–6.
Traynor AM, Dubey S, Eickhoff JC, Kolesar JM, Schell K, Huie MS, et al. Vorinostat (NSC# 701852) in patients with relapsed non-small cell lung cancer: a Wisconsin Oncology Network phase II study. J Thorac Oncol. 2009;4:522–6.
Woyach JA, Kloos RT, Ringel MD, Arbogast D, Collamore M, Zwiebel JA, et al. Lack of therapeutic effect of the histone deacetylase inhibitor vorinostat in patients with metastatic radioiodine-refractory thyroid carcinoma. J Clin Endocrinol Metab. 2009;94:164–70.
Doi T, Hamaguchi T, Shirao K, Chin K, Hatake K, Noguchi K, et al. Evaluation of safety, pharmacokinetics, and efficacy of vorinostat, a histone deacetylase inhibitor, in the treatment of gastrointestinal (GI) cancer in a phase I clinical trial. Int J Clin Oncol. 2013;18:87–95.
Fujiwara Y, Yamamoto N, Yamada Y, Yamada K, Otsuki T, Kanazu S, et al. Phase I and pharmacokinetic study of vorinostat (suberoylanilide hydroxamic acid) in Japanese patients with solid tumors. Cancer Sci. 2009;100:1728–34.
Krug LM, Curley T, Schwartz L, Richardson S, Marks P, Chiao J, et al. Potential role of histone deacetylase inhibitors in mesothelioma: clinical experience with suberoylanilide hydroxamic acid. Clin Lung Cancer. 2006;7:257–61.
Kelly WK, Richon VM, O’Connor O, Curley T, MacGregor-Curtelli B, Tong W, et al. Phase I clinical trial of histone deacetylase inhibitor: suberoylanilide hydroxamic acid administered intravenously. Clin Cancer Res. 2003;9:3578–88.
Kelly WK, O’Connor OA, Krug LM, Chiao JH, Heaney M, Curley T, et al. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J Clin Oncol. 2005;23:3923–31.
Stearns V, Jacobs LK, Fackler M, Tsangaris TN, Rudek MA, Higgins M, et al. Biomarker modulation following short-term vorinostat in women with newly diagnosed primary breast cancer. Clin Cancer Res. 2013;19:4008–16.
Haas NB, Quirt I, Hotte S, McWhirter E, Polintan R, Litwin S, et al. Phase II trial of vorinostat in advanced melanoma. Invest New Drugs. 2014;32:526–34.
Krug LM, Kindler HL, Calvert H, Manegold C, Tsao AS, Fennell D, et al. Vorinostat in patients with advanced malignant pleural mesothelioma who have progressed on previous chemotherapy (VANTAGE-014): a phase 3, double-blind, randomised, placebo-controlled trial. Lancet Oncol. 2015;16:447–56.
Yong W, Goh B, Soo R, Toh H, Ethirajulu K, Wood J, et al. Phase I and pharmacodynamic study of an orally administered novel inhibitor of histone deacetylases, SB939, in patients with refractory solid malignancies. Ann Oncol. 2011;22:2516–22.
Bauman J, Verschraegen C, Belinsky S, Muller C, Rutledge T, Fekrazad M, et al. A phase I study of 5-azacytidine and erlotinib in advanced solid tumor malignancies. Cancer Chemother Pharmacol. 2012;69:547–54.
Fu S, Hu W, Iyer R, Kavanagh JJ, Coleman RL, Levenback CF, et al. Phase 1b-2a study to reverse platinum resistance through use of a hypomethylating agent, azacitidine, in patients with platinum-resistant or platinum-refractory epithelial ovarian cancer. Cancer. 2011;117:1661–9.
Choy E, Flamand Y, Balasubramanian S, Butrynski JE, Harmon DC, George S, et al. Phase 1 study of oral abexinostat, a histone deacetylase inhibitor, in combination with doxorubicin in patients with metastatic sarcoma. Cancer. 2015;121:1223–30.
Dizon DS, Blessing JA, Penson RT, Drake RD, Walker JL, Johnston CM, et al. A phase II evaluation of belinostat and carboplatin in the treatment of recurrent or persistent platinum-resistant ovarian, fallopian tube, or primary peritoneal carcinoma: a Gynecologic Oncology Group study. Gynecol Oncol. 2012;125:367–71.
Thomas A, Rajan A, Szabo E, Tomita Y, Carter CA, Scepura B, et al. A phase I/II trial of belinostat in combination with cisplatin, doxorubicin, and cyclophosphamide in thymic epithelial tumors: a clinical and translational study. Clin Cancer Res. 2014;20:5392–402.
Hainsworth JD, Daugaard G, Lesimple T, Hübner G, Greco FA, Stahl MJ, et al. Paclitaxel/carboplatin with or without belinostat as empiric first-line treatment for patients with carcinoma of unknown primary site: a randomized, phase 2 trial. Cancer. 2015;121:1654–61.
Richards DA. Gemcitabine plus CI-994 offers no advantage over gemcitabine alone in the treatment of patients with advanced pancreatic cancer: results of a phase II randomized, double-blind, placebo-controlled, multicenter study. Ann Oncol. 2006;17:1096–102.
Fang F, Balch C, Schilder J, Breen T, Zhang S, Shen C, et al. A phase 1 and pharmacodynamic study of decitabine in combination with carboplatin in patients with recurrent, platinum-resistant, epithelial ovarian cancer. Cancer. 2010;116:4043–53.
Glasspool RM, Brown R, Gore ME, Rustin GJS, McNeish IA, Wilson RH, et al. A randomised, phase II trial of the DNA-hypomethylating agent 5-aza-2’-deoxycytidine (decitabine) in combination with carboplatin vs carboplatin alone in patients with recurrent, partially platinum-sensitive ovarian cancer. Br J Cancer. 2014;110:1923–9.
Xia C, Leon-Ferre R, Laux D, Deutsch J, Smith BJ, Frees M, et al. Treatment of resistant metastatic melanoma using sequential epigenetic therapy (decitabine and panobinostat) combined with chemotherapy (temozolomide). Cancer Chemother Pharmacol. 2014;74(4):691–7.
Pili R, Salumbides B, Zhao M, Altiok S, Qian D, Zwiebel J, et al. Phase I study of the histone deacetylase inhibitor entinostat in combination with 13-cis retinoic acid in patients with solid tumours. Br J Cancer. 2012;106:77–84.
Yardley DA, Ismail-Khan RR, Melichar B, Lichinitser M, Munster PN, Klein PM, et al. Randomized phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor. J Clin Oncol. 2013;31:2128–35.
Ngamphaiboon N, Dy GK, Ma WW, Zhao Y, Reungwetwattana T, DePaolo D, et al. A phase I study of the histone deacetylase (HDAC) inhibitor entinostat, in combination with sorafenib in patients with advanced solid tumors. Invest New Drugs. 2015;33:225–32.
Rathkopf D, Wong BY, Ross RW, Anand A, Tanaka E, Woo MM, et al. A phase I study of oral panobinostat alone and in combination with docetaxel in patients with castration-resistant prostate cancer. Cancer Chemother Pharmacol. 2010;66:181–9.
Drappatz J, Lee EQ, Hammond S, Grimm SA, Norden AD, Beroukhim R, et al. Phase I study of panobinostat in combination with bevacizumab for recurrent high-grade glioma. J Neurooncol. 2012;107:133–8.
Strickler JH, Starodub AN, Jia J, Meadows KL, Nixon AB, Dellinger A, et al. Phase I study of bevacizumab, everolimus, and panobinostat (LBH-589) in advanced solid tumors. Cancer Chemother Pharmacol. 2012;70:251–8.
Gray JE, Haura E, Chiappori A, Tanvetyanon T, Williams CC, Pinder-Schenck M, et al. A phase I, pharmacokinetic, and pharmacodynamic study of panobinostat, an HDAC inhibitor, combined with erlotinib in patients with advanced aerodigestive tract tumors. Clin Cancer Res. 2014;20:1644–55.
Bauer S, Hilger RA, Mühlenberg T, Grabellus F, Nagarajah J, Hoiczyk M, et al. Phase I study of panobinostat and imatinib in patients with treatment-refractory metastatic gastrointestinal stromal tumors. Br J Cancer. 2014;110:1155–62.
Jones SF, Infante JR, Thompson DS, Mohyuddin A, Bendell JC, Yardley DA, et al. A phase I trial of oral administration of panobinostat in combination with paclitaxel and carboplatin in patients with solid tumors. Cancer Chemother Pharmacol. 2012;70:471–5.
Bratland A, Dueland S, Hollywood D, Flatmark K, Ree AH. Gastrointestinal toxicity of vorinostat: reanalysis of phase 1 study results with emphasis on dose-volume effects of pelvic radiotherapy. Radiat Oncol. 2011;6:33.
Fakih MG, Fetterly G, Egorin MJ, Muindi JR, Espinoza-Delgado I, Zwiebel JA, et al. A phase I, pharmacokinetic, and pharmacodynamic study of two schedules of vorinostat in combination with 5-fluorouracil and leucovorin in patients with refractory solid tumors. Clin Cancer Res. 2010;16:3786–94.
Ramalingam SS, Parise RA, Ramanathan RK, Ramananthan RK, Lagattuta TF, Musguire LA, et al. Phase I and pharmacokinetic study of vorinostat, a histone deacetylase inhibitor, in combination with carboplatin and paclitaxel for advanced solid malignancies. Clin Cancer Res. 2007;13:3605–10.
Wilson PM, El-Khoueiry A, Iqbal S, Fazzone W, LaBonte MJ, Groshen S, et al. A phase I/II trial of vorinostat in combination with 5-fluorouracil in patients with metastatic colorectal cancer who previously failed 5-FU-based chemotherapy. Cancer Chemother Pharmacol. 2010;65:979–88.
Friday BB, Anderson SK, Buckner J, Yu C, Giannini C, Geoffroy F, et al. Phase II trial of vorinostat in combination with bortezomib in recurrent glioblastoma: a north central cancer treatment group study. Neuro Oncol. 2012;14:215–21.
Schneider BJ, Kalemkerian GP, Bradley D, Smith DC, Egorin MJ, Daignault S, et al. Phase I study of vorinostat (suberoylanilide hydroxamic acid, NSC 701852) in combination with docetaxel in patients with advanced and relapsed solid malignancies. Invest New Drugs. 2012;30:249–57.
Munster PN, Marchion D, Thomas S, Egorin M, Minton S, Springett G, et al. Phase I trial of vorinostat and doxorubicin in solid tumours: histone deacetylase 2 expression as a predictive marker. Br J Cancer. 2009;101:1044–50.
Jones DR, Moskaluk CA, Gillenwater HH, Petroni GR, Burks SG, Philips J, et al. Phase I trial of induction histone deacetylase and proteasome inhibition followed by surgery in non-small-cell lung cancer. J Thorac Oncol. 2012;7:1683–90.
Kolesar JM, Traynor AM, Holen KD, Hoang T, Seo S, Kim K, et al. Vorinostat in combination with bortezomib in patients with advanced malignancies directly alters transcription of target genes. Cancer Chemother Pharmacol. 2013;72:661–7.
Reguart N, Rosell R, Cardenal F, Cardona AF, Isla D, Palmero R, et al. Phase I/II trial of vorinostat (SAHA) and erlotinib for non-small cell lung cancer (NSCLC) patients with epidermal growth factor receptor (EGFR) mutations after erlotinib progression. Lung Cancer. 2014;84:161–7.
Yoo C, Ryu MH, Na YS, Ryoo BY, Lee CW, Maeng J, et al. Phase I and pharmacodynamic study of vorinostat combined with capecitabine and cisplatin as first-line chemotherapy in advanced gastric cancer. Invest New Drugs. 2014;32:271–8.
Dasari A, Gore L, Messersmith WA, Diab S, Jimeno A, Weekes CD, et al. A phase I study of sorafenib and vorinostat in patients with advanced solid tumors with expanded cohorts in renal cell carcinoma and non-small cell lung cancer. Invest New Drugs. 2013;31:115–25.
Deming DA, Ninan J, Bailey HH, Kolesar JM, Eickhoff J, Reid JM, et al. A phase I study of intermittently dosed vorinostat in combination with bortezomib in patients with advanced solid tumors. Invest New Drugs. 2014;32:323–9.
Schelman WR, Traynor AM, Holen KD, Kolesar JM, Attia S, Hoang T, et al. A phase I study of vorinostat in combination with bortezomib in patients with advanced malignancies. Invest New Drugs. 2013;31:1539–46.
Fu S, Hou MM, Naing A, Janku F, Hess K, Zinner R, et al. Phase I study of pazopanib and vorinostat: a therapeutic approach for inhibiting mutant p53-mediated angiogenesis and facilitating mutant p53 degradation. Ann Oncol. 2015;26:1012–8.
Han JY, Lee SH, Lee GK, Yun T, Lee YJ, Hwang KH, et al. Phase I/II study of gefitinib (Iressa(®)) and vorinostat (IVORI) in previously treated patients with advanced non-small cell lung cancer. Cancer Chemother Pharmacol. 2015;75:475–83.
Daud AI, Dawson J, DeConti RC, Bicaku E, Marchion D, Bastien S, et al. Potentiation of a topoisomerase I inhibitor, karenitecin, by the histone deacetylase inhibitor valproic acid in melanoma: translational and phase I/II clinical trial. Clin Cancer Res. 2009;15:2479–87.
Rocca A, Minucci S, Tosti G, Croci D, Contegno F, Ballarini M, et al. A phase I-II study of the histone deacetylase inhibitor valproic acid plus chemoimmunotherapy in patients with advanced melanoma. Br J Cancer. 2009;100:28–36.
Scherpereel A, Berghmans T, Lafitte JJ, Colinet B, Richez M, Bonduelle Y, et al. Valproate-doxorubicin: promising therapy for progressing mesothelioma. A phase II study. Eur Respir J. 2011;37:129–35.
Acknowledgements
We apologise to the researchers whose works are not cited here due to space limitations. The work was supported in part by the Italian Association for Cancer Research (AIRC IG11949 to C.N.), Italian Foundation for Cancer Research (Fellowship for Italy 15142 to E.D.M) and University of Rome “La Sapienza”.
This work is dedicated to the late Professor Carlo Nervi, for us an example of how, in Oncology, clinical practice should be combined with scientific curiosity.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors have no relevant affiliations or financial involvement with any organisation or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript.
Authors’ contributions
CN and EDM wrote the part on the molecular mechanisms of epigenetic regulation, and GC-P collected the published papers and analysed them. All authors share full responsibility for writing the paper. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Nervi, C., De Marinis, E. & Codacci-Pisanelli, G. Epigenetic treatment of solid tumours: a review of clinical trials. Clin Epigenet 7, 127 (2015). https://doi.org/10.1186/s13148-015-0157-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-015-0157-2