
Abstract
Purpose
Genetically predisposed breast cancer (BC) patients represent a minor but clinically meaningful subgroup of the disease, with 25% of all cases associated with actionable variants in BRCA1/2. Diagnostic implementation of next-generation sequencing (NGS) resulted in the rare identification of BC patients with double heterozygosity for deleterious variants in genes partaking in homologous recombination repair of DNA. As clinical heterogeneity poses challenges for genetic counseling, this study focused on the occurrence and clinical relevance of double heterozygous BC in South Africa.
Methods
DNA samples were diagnostically screened using the NGS-based Oncomine™ BRCA Expanded Research Assay. Data was generated on the Ion GeneStudio S5 system and analyzed using the Torrent Suite™ and reporter software. The clinical significance of the variants detected was determined using international variant classification guidelines and treatment implications.
Results
Six of 1600 BC patients (0.375%) tested were identified as being bi-allelic for two germline likely pathogenic or pathogenic variants. Most of the variants were present in BRCA1/2, including two founder-related small deletions in three cases, with family-specific variants detected in ATM, BARD1, FANCD2, NBN, and TP53. The scientific interpretation and clinical relevance were based on the clinical and tumor characteristics of each case.
Conclusion
This study increased current knowledge of the risk implications associated with the co-occurrence of more than one pathogenic variant in the BC susceptibility genes, confirmed to be a rare condition in South Africa. Further molecular pathology-based studies are warranted to determine whether clinical decision-making is affected by the detection of a second pathogenic variant in BRCA1/2 and TP53 carriers.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Patients with a genetic predisposition to breast cancer (BC) represent a minor but clinically meaningful subgroup of the disease. This predisposition accounts for 5–15% of the detected malignancies. It is mainly due to germline likely pathogenic (LP) and pathogenic variants (PVs) in high- and moderate-penetrance genes such as BRCA1 and BRCA2, which are involved in homologous recombination repair (HRR) of DNA damage [1]. The frequency of causative variants in these two genes varies and is influenced by specific geographical locations, unique population lineages, and some non-genetic factors. In populations such as these, founder effects may exist, resulting in a higher incidence of causative variants. This could result in more patients being bi-allelic or double heterozygous (DH) for LP/PVs. This is the case for the Ashkenazi Jewish population, where the DH percentage is 1.8% compared to 0.2–0.8% in different non-founder ethnic groups [2]. Based on these low percentages, the probability of carrying more than one PV in a single patient is generally low. Although rare, their presence could result in notable clinical variability in patients depending on the genes involved and their position or role in the HRR pathway.
Le Page et al. [2] reported on various studies that described DHs in the BRCA genes observed in hereditary breast and ovarian cancer syndrome (HBOC) families. With the advent of next-generation sequencing (NGS), more patients are currently being screened for various genes associated with HBOC. More recent reports indicated the involvement of various non-BRCA genes in critical pathways such as DNA repair [3, 4]. Little, however, is known about their phenotype. As clinical heterogeneity among families poses great challenges for genetic counseling when dealing with hereditary cancer syndromes, this study focused on the occurrence and clinical relevance observed of DH BC patients identified in the South African (SA) population using NGS-based panel testing.
Methods
DNA was extracted from peripheral blood, where after the samples were screened using the NGS-based Oncomine™ BRCA Expanded Research Assay (Life Technologies) according to the detailed protocol described by van der Merwe et al. [5]. Variants were filtered and annotated using the Ion Reporter™ Software version 5.18.2 (Thermo Fisher Scientific Inc., USA). The average coverage depth was 936X.
The clinical significance of the variants was determined based on the updated guidelines of the American Society of Medical Genetics and Genomics and the Association for Molecular Pathology [6], including the amended version designed specifically for TP53 [7]. Freely accessible public databases such as ClinVar, ClinGen, LOVC, BRCA Exchange, and the genomic search engine VarSome [8] were used to interrogate and ultimately classify variants.
Patients
Samples of 1600 patients were received at the National Health Laboratory Service (NHLS) Human Genetics laboratory in Bloemfontein, SA. The patients were diagnosed with breast, ovarian or prostate cancer, for whom clinicians requested diagnostic multigene panel screening to confirm/exclude HBOC. All the patients underwent pre-and post-test counseling, during which clinical and family-related cancer information was shared after obtaining informed consent for diagnostic testing, data sharing, and publication of results. Patients' unique identifiers were de-identified for publication purposes.
Results
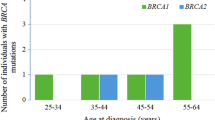
Six female patients (0.375%) were identified as being bi-allelic for two germline LP/PVs. The genetic variants, together with patient and tumor characteristics, are summarized in Table 1. The average age at diagnosis was 37.5 years. Although all the patients reported a family history of breast and/or various cancer types, Cases 5&6 had the highest risk based on the presence of multiple first-degree relatives affected by the disease. A high percentage of BRCA1/2 variants, as expected for HBOC syndrome, were observed (50%, 6/12). Of the non-BRCA genes, TP53 was observed for two cases, with the remaining genes (ATM, BARD1, FANCD2, and NBN) all contributing a single variant (Table 1). The family history of Case 1 exhibited the widest range of cancer types, as expected from a pathogenic TP53 variant, with a family member diagnosed with a cancerous lesion on the chin and deceased at age 29.
Most of the tumors were detected using mammography, with their BIRADS classification ranging from suspicious to highly suggestive of malignancy (Table 1). Five of the seven tumors (Case 5 was diagnosed with bilateral synchronous BC) were triple-negative (TNBC), with the remaining two representing a potential luminal molecular subtype identified by immunohistochemistry and FISH for Her2 equivocal cases. Most of the tumors were of grade III, with two of the tumors already progressed to stage IV. All but one patient received neoadjuvant therapy, although Case 3 defaulted after only three of the planned eight cycles due to adverse side effects. Despite the reduced cycles, the tumor responded adequately to perform surgery. All but one of the TNBC cases achieved a pathologic complete response (pCR) post neoadjuvant therapy, thereby improving their outcome and reducing the recurrence risk (Table 1).
For Case 1, patient autonomy was respected, as she opted for breast-conserving therapy due to her young age. A segmentectomy of the right and a reduction of the left breast were performed. Upon disclosure of the genetic test result, the patient was advised to consider a bilateral mastectomy with or without reconstructive surgery. However, the patient defaulted from returning to the clinic for treatment. Of the three BRCA1 mutation carriers, only Case 2 thus far has opted for a risk-reducing bilateral salpingo-oophorectomy (BSO) as part of her treatment plan. Case 3 has recently completed adjuvant treatment and has been referred to discuss and plan her BSO. Segregation and predictive testing of these variants in related family members has only recently been initiated and is underway (Table 1).
Discussion
A mere 0.375% of the patients were bi-allelic for LP/PVs in genes involved in the HRR pathway. Although multiple international reports described DHs for both BRCA1 and BRCA2 before [9,10,11], their combination with non-BRCA genes is limited [3, 4]. The likelihood of being DH for LP/PVs in different dominant cancer-predisposing BC genes is relatively uncommon but is increased in populations where founder effects exist [4], such as SA [12, 13].
Identifying germline LP/PVs in genes associated with hereditary BC has implications for the index patient and related family members, as preventative measures and management strategies can be implemented for mutation carriers to prevent or enhance early cancer detection. The presence of specific variants can impact and change a patient’s treatment plan, for example, the use of poly(ADP-Ribose) polymerase (PARP) inhibitors associated with increased cancer cell sensitivity [14].
Molecular interpretation
BARD1 and TP53 - Case 1
BARD1(NM_000465.4):c.448C > T (p.Arg150Ter) is predicted to cause a loss of normal protein function through either protein truncation or nonsense-mediated mRNA decay. The variant is associated with a personal and/or family history of BC and OVC [15, 16]. Together with BRCA1, the two tumor suppressor proteins form a heterodimeric complex that mediates cell survival and repair of DNA damage by maintaining genomic stability [17]. This occurs through nuclear functions such as DNA damage signaling and repair, transcriptional regulation, and cell cycle control [18]. The ineffective BARD1 protein will lack the critical ankyrin repeat and BRCT domains, thereby abolishing the protein’s interaction with p53 and BARD1-p53-dependent apoptosis [19].
TP53(NM_000546.6):c.695_696insT (p.His233Profs*7) occurred in the most frequently mutated region (exons 5–8), which forms part of the DNA-binding domain [20, 21]. When intact, the p53 protein functions as a cell stress-activated sequence-specific transcription factor that regulates gene expression in essential cellular processes [22]. The highly conserved DNA-binding domain, which includes exon 7, results in the formation of a stable protein with tumor suppressor activities that include the induction of cell cycle arrest, apoptosis, or DNA repair [22, 23]. With premature truncation, the protein will lack canonical p53 tumor suppressor activities, promoting cancer cell proliferation, survival, and potentially metastasis.
BRCA1 and BRCA2 - Cases 2 and 3
Two truncating variants, namely BRCA1(NM_007297.4):c.1374del (p.Asp458Glusfs*17) and BRCA2(NM_000059.4):c.7934del (p.Arg2645Asnfs*3) were observed for Case 2, with BRCA1 (NM_007297.4):c.45dup (p.Asn16Ter) and BRCA2 c.7934del detected for Case 3 (Table 1). Of these, BRCA1 c.1374del and BRCA2 c.7934del represent confirmed SA founder variants [12, 24, 25], with the third representing an SA recurrent variant (BRCA1 c.45dup) [26]. These two cases currently represent the third and fourth occurrence of DH involving founder variants in BRCA1/2 in SA [9].
Although BRCA1/2’s association with breast and OVC was confirmed two centuries ago [27, 28], their involvement in other cancer types, such as prostate and pancreatic cancer [29, 30], has recently come to the forefront. These two proteins play a crucial role in maintaining genomic integrity during DNA double-strand break (DSB) repair by regulating repair [31, 32] through interaction with numerous other proteins involved in the HRR machinery [33].
BRCA1 is involved in the recognition of DNA breaks and is responsible for the recruitment of BRCA2 to the break, where they interact with PALB2 [34]. As exons 11–13 encode the central regions of the protein, truncating variants in the early parts of the gene (Case 3-exon 2; Case 2-exon 10) will result in the loss of multiple binding domains, impacting BRCA1’s function and expression.
To ensure the maintenance of genomic stability upon replication stress, BRCA2 regulates the stalled DNA replication fork by loading and stabilizing polymerized RAD51 onto the DNA. This is facilitated through binding to its BRC repeats, situated toward the C terminal of the protein [35]. In the case of BRCA2 c.7934del, although the RAD51 binding domain constituting the BRC repeats (exon 11) will be intact, the secondary DNA-binding domain (exons 12–26) will be absent, resulting in DNA damage repair defects and tumorigenesis.
TP53 and FANCD2 - Case 4
TP(NM_000546.6):c.523C > T (p.Arg175Cys) in exon 5 is also situated in the DNA-binding domain. The Arg-175 residue is a mutational hotspot for hypomorphic variants and is involved in the structural integrity of the p53 DNA-binding domain. The Arg-175 residue forms part of the hydrophobic L3 hairpin that assists in stabilizing the DNA-binding domain, which is affected by the introduction of the new hydrophilic Cys-175 residue [36, 37].
FANCD2 forms part of the Fanconi Anemia (FA) pathway, an uncommon heterogeneous biallelic genetic disorder characterized by progressive aplastic anemia, congenital growth malformations, and tumor susceptibility [38]. This pathway involves more than 22 identified genes and plays a critical role in the repair of genomic instability such as repairing DNA interstrand cross-links needed for homologous DNA repair. Failure to repair these cross-links are closely associated with tumorigenesis [39].
FANCD2, together with BRCA2, is essential for protecting stalled replication forks against nucleolytic degradation through the regulation of responses to replication stress induced by mitomycin and hydroxyurea [40, 41]. Apart from the two DNA-binding domains, the EDGE domain (aa 1427–1430) is in the carboxyl terminus encoded by exon 44. This domain is indispensable for cellular resistance to mitomycin, as it stimulates interstrand crosslinks [42], which prevent DNA unwinding by DNA helicases and stop the progression of the replication and transcription machinery [43, 44].
The truncating FANCD2(NM_033084.6):c.2983G > T (p.Gly995Ter) variant falls within the second DNA-binding domain. The PV variant will not only impact this domain but will also result in the abolition of the EDGE domain. As the interstrand crosslinks are difficult to repair, the absence of a full-length protein, including the indispensable EDGE domain, will increase replication stress and mitomycin’s upregulation. It will ultimately result in more interstrand crosslinks associated with no DNA repair and a predisposition to cancer due to the inactivation of the cell cycle barriers.
As multiple recognized BC genes such as BRCA1 (also known as FANCS), BRCA2 (FANCD1), and PALB2 (FANCN) all form part of both the FA pathway and the HRR machinery needed for double-stranded DNA repair, various studies investigated the role of other FA genes such as FANCD2 with regards to cancer susceptibility, and more specifically BC [45]. Despite numerous studies reporting sensitivity of FA mutation positive tumors being sensitive to DNA damaging agents such as PARPi due to the accumulation of DNA lesions, no consensus on this matter has been reached, mainly due to either small sample sizes or a lack of molecular characterization in previous epidemiological studies [45,46,47].
Recently, Zhu and co-workers [48] screened 1481 Chinese high-risk BC patients for 16 FA genes and reported 38 pathogenic mutation carriers in the 12 unconfirmed BC genes after excluding BRCA1, BRCA2, PALB2, and RAD51C. Of these genes, FANCA was the most commonly mutated (0.54%) followed by FANCD2 (0.41%). Upon investigation, various clinicopathological associations (although not statistically significant, possibly hampered by the small sample size) were reported, pertaining to larger tumor sizes, lower ER and PR positivity rates and lower 3.5 year invasive disease-free survival and distant recurrence-free survival rates in mutation carriers when compared to non-carriers [48].
The risk to mono-allelic FANCD2 carriers to develop cancer were investigated using the relatives of patients with FA enrolled in the National Cancer Institute Bone Marrow Failure Syndrome cohort [49]. Patients with cancer and their unaffected relatives were genotyped where after the rates, types of cancer and their age at diagnosis were evaluated. Although the study indicated that the risk of cancer was not increased among all FA heterozygotes, only two genes were conclusively excluded as potential risk genes, namely FANCA and FANCC (observed versus expected ratios (O/E) of 0.92 and 0.91, respectively). With regards to FANCD2, the O/E for cancer risk and more specifically BC risk, were similar to that of BRCA2 (FANCD1), an established high-risk BC gene. The O/E for all cancer types were 2.8 for BRCA2 and 2.85 for FANCD2, with the risk for BC specifically indicated as 11.0 and 16.3, respectively [49].
Studies evaluating the protein expression and ubiquatination levels of FANCD2 in breast tumors have also identified an association between BRCA1 (another confirmed high-risk BC gene) and FANCD2 protein expression, and in breast tumors an association between FANCD2 protein expression and tumors size, TNM stage, ER status and Ki-67 index and ultimately, disease-free survival rate. Multivariable analysis demonstrated that high levels of FANCD2 expression and low levels of FANCD2 ubiquitination can be considered as independent prognostic factors and of value to BC patients [50,51,52]. These studies suggest that pathogenic variants in FANCD2 would abolish the protective effect of FANCD2 during tumorigenesis when higher expression levels is observed [50,51,52].
Although mono-allelic disruptive variants in FANCD2 is rare, they are now more frequently identified during genetic testing, emphasizing the need for a thorough understanding of the function and epidemiology of this gene with regards to cancer and more specifically BC risk. For these reasons, the case was included in this report despite the fact that there are currently no consensus risks and management options for the disease for mono-allelic mutation carriers.
BRCA1 and NBN - Case 5
Apart from the BRCA1 Afrikaner founder variant, an LP splice site variant was identified in NBN(NM_00248.5):c.896 + 2 T > C (p.?). The change affects a rare donor splice site, which is expected to disrupt RNA splicing and result in protein function loss. The nibrin protein forms part of and regulates the activity of the MRN protein complex involved in the recognition of and coordination of DSB repair through interaction with ATM. Nibrin guides this complex, carrying the two proteins to the DNA-damaged site in the cell’s nucleus [53], where the complex proceeds to mend the damage [54].
Disruptive variants in NBN lead to the autosomal recessive Nijemen breakage syndrome associated with several types of disease, particularly cancer [55]. As earlier studies suggested an association with BC (relative risk of 2.7) [56], it resulted in its inclusion in multigene cancer panels aimed at investigating the HRR pathway. Since then, various studies reported PVs in 0.25% of BC patients [57], which even included founder variants [58, 59]. As mutation carriers had variable BC features, Zuntini et al. [57] investigated and, together with the Breast Cancer Association Consortium [60], concluded that most of the PVs in NBN are proven neutral by segregation analyses, case-control studies and/or expression studies. This resulted in the National Comprehensive Cancer Network (NCCN) guidelines not including NBN in their consensus document for BC risk management [61]. The variant is therefore considered LP with regards to Nijemen breakage syndrome, but a variant of unknown clinical significance for BC risk. For this reason, the three offspring of the index patient were only screened for the PV in BRCA1 (Table 1) without disclosing the potential risk for Nijemen breakage syndrome, an incidental finding. Although carrying a single LP/PV NBN variant might not be solely responsible for the disease in Case 5, it may act as a co-contributor in the case of a DH as the absence of both the MRN complex (of which NBN forms a critical part) and the BRCA1 protein will disrupt HRR.
ATM and BRCA2 - C ase 6
The ATM protein is a serine-threonine protein kinase that primarily responds to DNA damage once activated by auto-phosphorylation in the presence of a DSB. It converts the inactive dimers into catalytically active monomers [62], which are recruited to the DNA repair site by the MRN complex [63], a critical step for the activation of ATM [64]. This, in turn, affects the phosphorylation of BRCA1, CHEK2, and p53, all involved in a signaling cascade that responds to DSBs.
The ATM and ATR (ATM- and Rad3-related) kinases are the central regulators of the DNA damage response (DDR) signaling pathway [65]. Although ATM is primarily activated by DSBs, both are activated upon DNA damage and replication stress. These kinases, together with the DNA-PKcs (DNA-dependent protein kinase) kinases, form part of the most upstream DDR kinases, and share a similar domain organization with conserved FAT and FAT carboxy-terminal (FATC) domains flanking their carboxyl termini [66]. These domains are auto-phosphorylated in or near their FAT domains upon damage, which regulates their activation.
ATM(NM_000051.4):c.7978G > T (p.Glu2660Ter) occurred between the conserved FAT domain (spanning aa 1960–2566) and the PI3K/PI4K and FATC domains (aa 2712–2962 and 3024–3056). The premature truncation due to the introduction of a stop codon will deliver a non-functional peptide which will result in protein instability due to markedly reduced kinase activity [67, 68].
The splice-site variant (BRCA2 (NM_000059.4):c.516G > A, p.Lys172 =) will result in the absence of all BRC repeats responsible for the binding of RAD51, together with additional critical domains downstream involved in HRR [69]. In normal circumstances, BRC3/BRC4 binds RAD51 to assist in blocking nucleoprotein filament formation by RAD51. During a DSB, the RAD51-ssDNA nucleoprotein filament engages with duplex DNA to find a sequence homologous to that of the bound ssDNA [70]. Upon location of a homologous segment, the presynaptic filament catalyzes the formation of a DNA joint between the bound DNA molecules, resulting in RAD51-mediated DNA strand invasion and exchange. Their absence is critical as certain tumors with faulty HRR mechanisms may rely on PARP-mediated DNA repair for survival and are sensitive to its inhibition [71].
Clinical relevance
Most of the tumors observed were TNBC, with Case 5 diagnosed with bilateral synchronous disease. This type is aggressive and associated with a high risk of mortality due to high relapse rates and poor overall survival. Treatment is challenging and relies upon tumor markers, with treating physicians often not taking the genetic result into consideration. This scenario is slowly changing as more patients in SA now have access to genetic screening through the NHLS.
Available treatment options include immunotherapy, radiotherapy, surgery, and chemotherapy, with chemotherapy being the most common (Table 1). This high-risk disease is generally treated with systemic chemotherapy using anthracyclines, alkylating agents, and taxanes in neoadjuvant or adjuvant settings [72]. According to Han et al. [73], neoadjuvant chemotherapy is the preferred approach to treat stage II or III TNBC. The group also postulates that the addition of pembrolizumab to taxane-platinum-based chemotherapy followed by anthracycline significantly increased pCR rates and improved survival in early-stage TNBC [73].
There are, however, limitations that revolve around the development of resistance to anticancer drugs and off-target toxicity, as evident from Case 3. Chemo-resistant TNBC disease is genetically very diverse and evolving, which challenges healthcare practitioners to individualize treatment for incomplete responders and patients relapsing [74]. Advances have been made in the development of Food and Drug Administration-approved drugs that either target programmed cell death or involve antibody–drug conjugates [74]. By implementing personalized medicine aimed at key genetic variants involved in specific molecular pathways, these tumors can be targeted using these inhibitors as single agents and/or in combination with standard chemotherapy regimens. Therefore, professionals across healthcare disciplines should work together to develop effective treatment options for TNBCs. Currently, the mainstay of early-stage TNBC includes systemic chemotherapy and surgery, with or without radiation therapy [73].
Confirming a diagnosis of HBOC is critical to both the patient and related at-risk family members where clinical management is concerned. According to the NCCN guidelines, screening options for BRCA1/2 mutation carriers should include bi-annual clinical breast examinations with magnetic resonance imaging (MRI) or mammogram annually from age 25, as well as prophylactic risk-reducing bilateral mastectomy with or without reconstruction. For carriers with LP/PVs in the moderate penetrance genes such as ATM, annual screening with mammography or MRI is recommended from age 40 with a prophylactic mastectomy only if the family history warrants it. In cases where there is also a strong OVC association, the guidelines recommend a prophylactic risk-reducing BSO from the age of 40 years (Table 2). There is scarce literature documenting the clinical management of DHs, as these patients are managed for the combined cancer burden across both genes. When both genes confer a risk for the same cancer type, the patient is managed according to the guidelines associated with the gene that conveys the highest absolute risk. At-risk related family members should be offered predictive genetic testing for both LP/PVs involved if considered clinically actionable. In the instance of Cases 4&5, predictive genetic testing for NBN and FANCD2 is currently not clinically indicated according to the guidelines due to their unknown associated risk and a lack of evidence regarding clinical management for BC and OVC (Table 2).
Genetic counseling is an important resource for affected individuals and their families, as counselors specialize in helping individuals understand their hereditary risk and facilitate informed, often emotional, decision-making during testing. Post-test counseling of these women and their related family members is crucial to ensure that patients and healthcare providers understand the risks and management associated with genetic conditions. For DHs, counseling on recurrence risks will be more complex as more than one gene might be involved, with some individuals inheriting one or both or neither of these variants.
The mutational status of the DHs carrying LP/PVs in genes involved in the HRR machinery (such as BRCA1/2, TP53, and ATM) has implications for the use of adjuvant radiotherapy (ART). As DNA damage will occur due to ART, it will severely impact cell cycle recruitment, proliferation, and apoptosis, resulting in impaired DSBs, consequently increasing the risk of second malignant neoplasms occurring [75,76,77]. Unfortunately, the genetic results of Cases 1&4 were not yet available at the time of treatment planning. Therefore, knowledge of their germline TP53 variants could not be considered during treatment planning.
The management of patients with especially germline TP53 (Cases 1&4) variants is complex as clinical decision-making needs to weigh the risks of normal tissue toxicity and radiosensitivity against the tumor’s response (radioresistance and radio curability) [78]. Although ART should be avoided as far as possible in TP53-associated disease, according to the guidelines of the American Society for Radiation Oncology (ASTRO) and the American Society of Clinical Oncology (ASCO) [79], the overall prognosis might be poor. Since Case 4 presented with locally advanced BC (stage IIIb), a strong indication for post-mastectomy ATR [79], knowledge about the TP53 variant would not have affected the treatment decision of ATR for this patient at risk of locoregional disease relapse. Despite the contraindication for ART based on the detection of a pathogenic germline TP53 variant [76,77,78], the tumor stage and histopathology results can override the risk associated with a germline TP53 variant, as evident from achieving pCR post-therapy (Table 1).
Conclusion
This study detected a low frequency (0.375%) of bi-allelic BC patients in the SA population, driven by BRCA1/2 founder variants. Additionally, family-specific variants were detected in ATM, BARD1, FANCD2, NBN, and TP53. Although the scientific interpretation and clinical relevance were based on the clinical and tumor characteristics of the DHs, the detection of an incidental NBN variant caused a dilemma for report writing and genetic counseling regarding family follow-ups. The study highlighted the potential consequences of treatment initiation that preceded genetic test results due to sample batching to reduce laboratory costs. This resulted in patients potentially receiving risk-enhancing treatments such as radiotherapy associated with induced risk in TP53 carriers. Despite the small sample size, valuable insights were gained, which highlighted the importance of tumor pathology in bringing genomics into the clinical domain [80]. There is a need for harmonious cooperation between geneticists and clinicians to address multiple challenges associated with the merging of traditional boundaries between diagnosis and treatment in cancer management.
Data availability
The genetic data referred to in this publication is available on ClinVar (submission number SUB11378633 (SCV002504693–SCV002505346)).
References
Yoshida K, Miki Y (2004) Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Sci 95(11):866–871. https://doi.org/10.1111/j.1349-7006.2004.tb02195.x
Le Page C, Rahimi K, Rodrigues M et al (2020) Clinicopathological features of women with epithelial ovarian cancer and double heterozygosity for BRCA1 and BRCA2: a systematic review and case report analysis. Gynecol Oncol 156:377–386. https://doi.org/10.1016/j.ygyno.2019.11.019
Laish I, Friedman E, Levi-Reznick G, Kedar I, Katz L, Levi Z, Halpern N, Parnasa S, Abu-Shatya A, Half E, Goldberg Y (2021) Double heterozygotes of BRCA1/BRCA2 and mismatch repair gene pathogenic variants: case series and clinical implications. Breast Cancer Res Treat 188(3):685–694. https://doi.org/10.1007/s10549-021-06258-9
Infante M, Arranz-Ledo M, Lastra E, Abella LE, Ferreira R, Orozco M, Hernández L, Martínez N, Durán M (2022) Increased co-occurrence of pathogenic variants in hereditary breast and ovarian cancer and lynch syndromes: a consequence of multigene panel genetic testing? Int J Mol Sci 23(19):11499. https://doi.org/10.3390/ijms231911499
Van der Merwe NC, Combrink HMVE, Ntaita KS, Oosthuizen J (2022) Prevalence of clinically relevant germline BRCA variants in a large unselected South African breast and ovarian cancer cohort: the public sector experience. Front Genet 13:834265. https://doi.org/10.3389/fonc.2022.938561
Richards S, Aziz N, Bale S et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–424. https://doi.org/10.1038/gim.2015.30
Fortuno C, Lee K, Olivier M et al (2021) Specifications of the ACMG/AMP variant interpretation guidelines for germline TP53 variants. Hum Mutat 42:223–236. https://doi.org/10.1002/humu.24150
Kopanos C, Tsiolkas V, Kouris A et al (2019) VarSome: the human genomic variant search engine. Bioinformatics 35:1978–1980. https://doi.org/10.1093/bioinformatics/bty892
Loubser F, de Villiers JNP, van der Merwe NC (2012) Two double heterozygotes in a South-African Afrikaner family: implications for BRCA1 and BRCA2 predictive testing. Clin Genet 82:599–600. https://doi.org/10.1111/j.1399-0004.2012.01878.x
Nomizu T, Matsuzaki M, Katagata N et al (2015) A case of familial breast cancer with double heterozygosity for BRCA1 and BRCA2 genes. Breast Cancer 22:557–561. https://doi.org/10.1007/s12282-012-0432-4
Vietri MT, Caliendo G, D’Elia G, Resse M, Casamassimi A, Minucci PB, Dello Ioio C, Cioffi M, Molinari AM (2020) Five Italian families with two mutations in BRCA genes. Genes (Basel) 11(12):1451. https://doi.org/10.3390/genes11121451
Reeves MD, Yawitch TM, van der Merwe NC, van den Berg HJ, Dreyer G, van Rensburg EJ (2004) BRCA1 mutations in South African breast and/or ovarian cancer families: evidence of a novel founder mutation in Afrikaner families. Int J Cancer 110(5):677–682. https://doi.org/10.1002/ijc.20186
Van der Merwe NC, Hamel N, Schneider SR, Apffeltaedt JP, Wijnen JT, Foulkes WD (2012) A founder BRCA2 mutation in non-Afrikaner breast cancer patients of the Western Cape of South Africa. Clin Genet 81:179–184. https://doi.org/10.1111/j.1399-0004.2010.01617.x
Cerrato A, Morra F, Celetti A (2016) Use of poly ADP-ribose polymerase [PARP] inhibitors in cancer cells bearing DDR defects: the rationale for their inclusion in the clinic. J Exp Clin Cancer Res 35(179):1–13. https://doi.org/10.1186/s13046-016-0456-2
Lilyquist J, LaDuca H, Polley E, Davis BT, Shimelis H, Hu C, Hart SN, Dolinsky JS, Couch FJ, Goldgar DE (2017) Frequency of mutations in a large series of clinically ascertained ovarian cancer cases tested on multi-gene panels compared to reference controls. Gynecol Oncol 147(2):375–380. https://doi.org/10.1016/j.ygyno.2017.08.030
Kaneyasu T, Mori S, Yamauchi H et al (2020) Prevalence of disease-causing genes in Japanese patients with BRCA1/2-wildtype hereditary breast and ovarian cancer syndrome. npj Breast Cancer 6(25):1–13. https://doi.org/10.1038/s41523-020-0163-1
Fox D 3rd, Le Trong I, Rajagopal P, Brzovic PS, Stenkamp RE, Klevit RE (2008) Crystal structure of the BARD1 ankyrin repeat domain and its functional consequences. J Biol Chem 283(30):21179–21186. https://doi.org/10.1074/jbc.M802333200
Irminger-Finger I, Jefford CE (2006) Is there more to BARD1 than BRCA1? Nat Rev Cancer 6(5):382–391. https://doi.org/10.1038/nrc1878
Jefford CE, Feki A, Harb J, Krause KH, Irminger-Finger I (2004) Nuclear-cytoplasmic translocation of BARD1 is linked to its apoptotic activity. Oncogene 23(20):3509–3520. https://doi.org/10.1038/sj.onc.1207427
Zerbino DR, Achuthan P, Akanni W et al (2018) Ensembl 2018. Nucleic Acids Res 46(D1):D754–D761. https://doi.org/10.1093/nar/gkx1098
Lasham A, Knowlton N, Mehta SY, Braithwaite AW, Print CG (2021) Breast cancer patient prognosis is determined by the interplay between TP53 mutation and alternative transcript expression: insights from TP53 long amplicon digital PCR assays. Cancers (Basel) 13(7):1531. https://doi.org/10.3390/cancers13071531
Robles AI, Jen J, Harris CC (2016) Clinical outcomes of TP53 mutations in cancers. Cold Spring Harb Perspect Med 6:a026294. https://doi.org/10.1101/cshperspect.a026294
Rivlin N, Brosh R, Oren M, Rotter V (2011) Mutations in the p53 tumor suppressor gene: important milestones at the various steps of tumorigenesis. Genes Cancer 2:466–474. https://doi.org/10.1177/1947601911408889
Van der Merwe NC, van Rensburg EJ (2009) Hereditary breast/ovarian cancer and BRCA mutations: a South African perspective. Curr Oncol 16:91–110. https://doi.org/10.3747/co.vi6i5.529
Oosthuizen J, Kotze MJ, van der Merwe N, Myburgh EJ, Bester P, van der Merwe NC (2021) Globally rare BRCA2 variants with founder haplotypes in the South African population: implications for point-of-care testing based on a single-institution BRCA1/2 next-generation sequencing study. Front Oncol 10:619469. https://doi.org/10.3389/fonc.2020.619469
Smith D, Gerber J, Loubser F, Gardiner SA, Conradie M, Raimond P (2019) New recurring BRCA1 variant: an additional South African founder mutation? SAMJ 109(8):1–4
Miki Y, Swensen J, Shattuck-Eidenset D et al (1994) A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 266(5182):66–71. https://doi.org/10.1126/science.7545954
Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, Collins N, Gregory S, Gumbs C, Micklem G (1995) Identification of the breast cancer susceptibility gene BRCA2. Nature 378(6559):789–792. https://doi.org/10.1038/378789a0.Erratum.In:Nature1996379(6567):749
Cavanagh H, Rogers KMA (2015) The role of BRCA1 and BRCA2 mutations in prostate, pancreatic and stomach cancers. Hered Cancer Clin Pract 13(1):16. https://doi.org/10.1186/s13053-015-0038-x
Mersch J, Jackson MA, Park M, Nebgen D, Peterson SK, Singletary C, Arun BK, Litton JK (2015) Cancers associated with BRCA1 and BRCA2 mutations other than breast and ovarian. Cancer 121(2):269–275. https://doi.org/10.1002/cncr.29041
Lord CJ, Ashworth A (2012) The DNA damage response and cancer therapy. Nature 481(7381):287–294. https://doi.org/10.1038/nature10760
Venkitaraman AR (2014) Cancer suppression by the chromosome custodians, BRCA1 and BRCA2. Science 343(6178):1470–1475. https://doi.org/10.1126/science.1252230
Mao Z, Bozzella M, Seluanov A, Gorbunova V (2008) Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair 7(10):1765–1771. https://doi.org/10.1016/j.dnarep.2008.06.018
Zhang F, Ma J, Wu J, Ye L, Cai H, Xia B, Yu X (2009) PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr Biol 19(6):524–529. https://doi.org/10.1016/j.cub.2009.02.018
Lomonosov M, Anand S, Sangrithi M, Davies R, Venkitaraman AR (2003) Stabilization of stalled DNA replication forks by the BRCA2 breast cancer susceptibility protein. Genes Dev 17(24):3017–3022. https://doi.org/10.1101/gad.279003
Joerger AC, Ang HC, Fersht AR (2006) Structural basis for understanding oncogenic p53 mutations and designing rescue drugs. Proc Natl Acad Sci USA 103(41):15056–15061. https://doi.org/10.1073/pnas.0607286103
Kitayner M, Rozenberg H, Kessler N, Rabinovich D, Shaulov L, Haran TE, Shakked Z (2006) Structural basis of DNA recognition by p53 tetramers. Mol Cell 22(6):741–753. https://doi.org/10.1016/j.molcel.2006.05.015
Mamrak NE, Shimamura A, Howlett NG (2017) Recent discoveries in the molecular pathogenesis of the inherited bone marrow failure syndrome Fanconi anemia. Blood Rev 31(3):93–99. https://doi.org/10.1016/j.blre.2016.10.002
Fang CB, Wu HT, Zhang ML, Liu J, Zhang GJ (2020) Fanconi anemia pathway: mechanisms of breast cancer predisposition development and potential therapeutic targets. Front Cell Dev Biol 8:160. https://doi.org/10.3389/fcell.2020.00160
Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M (2011) Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145(4):529–542. https://doi.org/10.1016/j.cell.2011.03.041
Schlacher K, Wu H, Jasin M (2012) A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 22(1):106–116. https://doi.org/10.1016/j.ccr.2012.05.015
Shakeel S, Rajendra E, Alcón P et al (2019) Structure of the Fanconi anaemia monoubiquitin ligase complex. Nature 575(7781):234–237. https://doi.org/10.1038/s41586-019-1703-4
Schärer OD (2005) DNA interstrand crosslinks: natural and drug-induced DNA adducts that induce unique cellular responses. ChemBioChem 6:27–32. https://doi.org/10.1002/cbic.200400287
Niraj J, Caron MC, Drapeau K, Bérubé S, Guitton-Sert L, Coulombe Y, Couturier AM, Masson JY (2017) The identification of FANCD2 DNA binding domains reveals nuclear localization sequences. Nucleic Acids Res 45(14):8341–8357. https://doi.org/10.1093/nar/gkx543
Gianni P, Matenoglou E, Geropoulos G et al (2022) The Fanconi anemia pathway and breast cancer: a comprehensive review of clinical data. Clin Breast Cancer 22(1):10–25. https://doi.org/10.1016/j.clbc.2021.08.001
Liu W, Polaczek P, Roubal I et al (2021) FANCD2 and RAD51 recombinase directly inhibit DNA2 nuclease at stalled replication forks and FANCD2 acts as a novel RAD51 mediator in strand exchange to promote genome stability. bioRxiv. https://doi.org/10.1101/2021.07.08.450798
Simoneau A, Xiong R, Zou L (2021) The trans cell cycle effects of PARP inhibitors underlie their selectivity toward BRCA1/2-deficient cells. Genes Dev 35(17–18):1271–1289. https://doi.org/10.1101/gad.348479.121
Zhu QY, Li PC, Zhu YF et al (2023) A comprehensive analysis of Fanconi anemia genes in Chinese patients with high-risk hereditary breast cancer. J Cancer Res Clin Oncol 149(15):14303–14313. https://doi.org/10.1007/s00432-023-05236-6
McReynolds LJ, Giri N, Leathwood L, Risch MO, Carr AG, Alter BP (2022) Risk of cancer in heterozygous relatives of patients with Fanconi anemia. Genet Med 24(1):245–250. https://doi.org/10.1016/j.gim.2021.08.013
van der Groep P, Hoelzel M, Buerger H, Joenje H, de Winter JP, van Diest PJ (2008) Loss of expression of FANCD2 protein in sporadic and hereditary breast cancer. Breast Cancer Res Treat 107(1):41–47. https://doi.org/10.1007/s10549-007-9534-7
Fagerholm R, Sprott K, Heikkinen T et al (2013) Overabundant FANCD2, alone and combined with NQO1, is a sensitive marker of adverse prognosis in breast cancer. Ann Oncol 24(11):2780–2785. https://doi.org/10.1093/annonc/mdt290
Feng L, Jin F (2019) Expression and prognostic significance of Fanconi anemia group D2 protein and breast cancer type 1 susceptibility protein in familial and sporadic breast cancer. Oncol Lett 17(4):3687–3700. https://doi.org/10.3892/ol.2019.10046
Hsu HMWH, Wang HC, Chen ST, Hsu GC, Shen CY, Yu JC (2007) Breast cancer risk is associated with the genes encoding the DNA double-strand break repair Mre11/Rad50/Nbs1 complex. Cancer Epidemiol Biomarkers Prev 16:2024–2032. https://doi.org/10.1158/1055-9965
Uzunoglu H, Korak T, Ergul E, Uren N, Sazci A, Utkan NZ, Kargi E, Triyaki Ç, Yirmibesoglu O (2016) Association of the nibrin gene (NBN) variants with breast cancer. Biomed Rep 4(3):369–373. https://doi.org/10.3892/br.2016.579
Varon R, Vissinga C, Platzer M et al (1998) Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell 93(3):467–476. https://doi.org/10.1016/s0092-8674(00)81174-5
Zhang G, Zeng Y, Liu Z, Wei W (2013) Significant association between Nijmegen breakage syndrome 1 657del5 polymorphism and breast cancer risk. Tumour Biol 34(5):2753–2757. https://doi.org/10.1007/s13277-013-0830-z
Zuntini R, Bonora E, Pradella LM (2021) Detecting variants in the NBN gene while testing for hereditary breast cancer: what to do next? Int J Mol Sci 22(11):5832. https://doi.org/10.3390/ijms22115832
Górski B, Debniak T, Masojć B, Mierzejewski M, Medrek K, Cybulski C, Jakubowska A, Kurzawski G, Chosia M, Scott R, Lubiński J (2003) Germline 657del5 mutation in the NBS1 gene in breast cancer patients. Int J Cancer 106(3):379–381. https://doi.org/10.1002/ijc.11231.Erratum.In:IntJCancer.2003Oct10;106(6):984
Roznowski K, Januszkiewicz-Lewandowska D, Mosor M, Pernak M, Litwiniuk M, Nowak J (2008) I171V germline mutation in the NBS1 gene significantly increases risk of breast cancer. Breast Cancer Res Treat 10(2):343–348. https://doi.org/10.1007/s10549-007-9734-1
Breast Cancer Association Consortium, Dorling L, Carvalho S et al (2021) Breast cancer risk genes—association analysis in more than 113,000 women. N Engl J Med 384(5):428–439. https://doi.org/10.1056/NEJMoa1913948
National Comprehensive Cancer Network (NCCN) (2024) NCCN guidelines version 3.2024. https://www.nccn.org/guidelines/guidelines-detail?category=2&id=1503. Accessed 14 Jan 2023
Bakkenist CJ, Kastan MB (2003) DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421(6922):499–506. https://doi.org/10.1038/nature01368
Stracker TH, Petrini JH (2011) The MRE11 complex: starting from the ends. Nat Rev Mol Cell Biol 12(2):90–103. https://doi.org/10.1038/nrm3047
Lee JH, Paull TT (2005) ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 308(5721):551–554. https://doi.org/10.1126/science.1108297.Erratum.In:Science.2005Jun24;308(5730):1870
Maréchal A, Zou L (2013) DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol 5(9):a012716. https://doi.org/10.1101/cshperspect.a012716
Lempiäinen H, Halazonetis TD (2009) Emerging common themes in regulation of PIKKs and PI3Ks. EMBO J 28(20):3067–3073. https://doi.org/10.1038/emboj.2009.281
Lakin ND, Weber P, Stankovic T, Rottinghaus ST, Taylor AM, Jackson SP (1996) Analysis of the ATM protein in wild-type and ataxia telangiectasia cells. Oncogene 13:2707–2716
Sandoval N, Platzer M, Rosenthal A et al (1999) Characterization of ATM gene mutations in 66 ataxia telangiectasia families. Hum Mol Genet 8(1):69–79. https://doi.org/10.1093/hmg/8.1.69
Andreassen PR, Seo J, Wiek C, Hanenberg H (2021) Understanding BRCA2 function as a tumor suppressor based on domain-specific activities in DNA damage responses. Genes (Basel) 12(7):1034. https://doi.org/10.3390/genes12071034
Zhao L, Xu J, Zhao W, Sung P, Wang HW (2018) Determining the RAD51-DNA nucleoprotein filament structure and function by cryo-electron microscopy. Methods Enzymol 600:179–199. https://doi.org/10.1016/bs.mie.2017.12.002
Morales J, Li L, Fattah FJ, Dong Y, Bey EA, Patel M, Gao J, Boothman DA (2014) Review of poly (ADP-ribose) polymerase (PARP) mechanisms of action and rationale for targeting in cancer and other diseases. Crit Rev Eukaryot Gene Expr 24(1):15–28. https://doi.org/10.1615/critreveukaryotgeneexpr.2013006875
Obidiro O, Battogtokh G, Akala EO (2023) Triple negative breast cancer treatment options and limitations: future outlook. Pharmaceutics 15(7):1796. https://doi.org/10.3390/pharmaceutics15071796
Han T, Shi M, Chen Q, Chen D, Hao J (2023) Effect of adjuvant radiotherapy after breast-conserving surgery in elder women with early-stage breast cancer: a propensity-score matching analysis. Front Oncol 13:1012139. https://doi.org/10.3389/fonc.2023.1012139
Gupta GK, Collier AL, Lee D et al (2020) Perspectives on triple-negative breast cancer: current treatment strategies, unmet needs, and potential targets for future therapies. Cancers (Basel) 12(9):2392. https://doi.org/10.3390/cancers12092392
Schlosser S, Rabinovitch R, Shatz Z et al (2020) Radiation-associated secondary malignancies in BRCA mutation carriers treated with breast cancer. Int J Rad Oncol Biol Phys 107:353–350. https://doi.org/10.1016/j.ijrobp.2020.02.020
Lazzari G, Buono G, Zannino B, Silvano G (2021) Breast cancer adjuvant radiotherapy in BRCA1/2, TP53, ATM genes mutations: are there solved issues? Breast Cancer (Dove Med Press) 13:299–310. https://doi.org/10.2147/BCTT.S306075
Gonçalves D, Pires AS, Marques IA, Gomes I, Sousa G, Botelho MF, Abrantes AM (2022) An overview on radiation sensitivity in hereditary breast and ovarian cancer syndrome. Cancers 14(13):3254. https://doi.org/10.3390/cancers14133254
Thariat J, Chevalier F, Orbach D, Ollivier L, Marcy PY, Corradini N, Beddok A, Foray N, Bougeard G (2021) Avoidance or adaptation of radiotherapy in patients with cancer with Li-Fraumeni and heritable TP53-related cancer syndromes. Lancet Oncol 22(12):e562–e574. https://doi.org/10.1016/S1470-2045(21)00425-3
Tung NM, Boughey JC, Pierce LJ et al (2020) Management of hereditary breast cancer: American Society of Clinical Oncology, American Society for Radiation Oncology, and Society of Surgical Oncology Guideline. J Clin Oncol 38(18):2080–2106. https://doi.org/10.1200/JCO.20.00299
Okunola AO, Baatjes KJ, Zemlin AE et al (2023) Pathology-supported genetic testing for the application of breast cancer pharmacodiagnostics: family counselling, lifestyle adjustments and change of medication. Expert Rev Mol Diagn 23(5):431–443. https://doi.org/10.1080/14737159.2023.2203815
Acknowledgements
The authors thank each case for sharing their clinical and personal information and providing consent for publication purposes. Without them, this publication would not have been possible. We also acknowledge the Division of Human Genetics of the NHLS, Universitas Hospital, for providing the infrastructure needed for diagnostic testing.
Funding
Open access funding provided by University of the Free State. The data presented in this article represent diagnostic results. Patients were tested on the diagnostic platform of the NHLS, Division of Human Genetics, in Bloemfontein, South Africa.
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
NCV was responsible for the study conception, design and obtaining ethics approval. Material preparation, data analysis and reporting were performed by NCV, JO and KSN. MS, BR, MA, IB, KN and KV obtained informed consent from the patients for data-sharing and publication purposes and gathered and interpreted clinical data. MJK, IB, MA, BR and JO assisted with the compilation of the draft. All authors critically revised the manuscript and approved the final paper.
Corresponding author
Ethics declarations
Competing interests
No conflicts of Interest were reported.
Ethical approval
The study was performed in line with the principles of the Declaration of Helsinki. The translational research performed that resulted in the introduction of the panel onto the diagnostic platform of the NHLS Division of Human Genetics in Bloemfontein, South Africa, was reviewed and approved by the Health Sciences Research Ethics Committee of the University of the Free State (UFS-HSD2021/0704/2202), South Africa. Approval has also been granted for the publication of this case series (UFS-HSD2023/2194). Lastly, the NHLS permitted the use of the data (reference PR2235978).
Informed consent
The authors affirm that written informed consent for data sharing and the publication of patient details was gathered from each of the cases.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
van der Merwe, N.C., Buccimazza, I., Rossouw, B. et al. Clinical relevance of double heterozygosity revealed by next-generation sequencing of homologous recombination repair pathway genes in South African breast cancer patients. Breast Cancer Res Treat 207, 331–342 (2024). https://doi.org/10.1007/s10549-024-07362-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-024-07362-2