
Abstract
Purpose
A 3-biomarker homologous recombination deficiency (HRD) score is a key component of a currently FDA-approved companion diagnostic assay to identify HRD in patients with ovarian cancer using a threshold score of ≥ 42, though recent studies have explored the utility of a lower threshold (GIS ≥ 33). The present study evaluated whether the ovarian cancer thresholds may also be appropriate for major breast cancer subtypes by comparing the genomic instability score (GIS) distributions of BRCA1/2-deficient estrogen receptor–positive breast cancer (ER + BC) and triple-negative breast cancer (TNBC) to the GIS distribution of BRCA1/2-deficient ovarian cancer.
Methods
Ovarian cancer and breast cancer (ER + BC and TNBC) tumors from ten study cohorts were sequenced to identify pathogenic BRCA1/2 mutations, and GIS was calculated using a previously described algorithm. Pathologic complete response (pCR) to platinum therapy was evaluated in a subset of TNBC samples. For TNBC, a threshold was set and threshold validity was assessed relative to clinical outcomes.
Results
A total of 560 ovarian cancer, 805 ER + BC, and 443 TNBC tumors were included. Compared to ovarian cancer, the GIS distribution of BRCA1/2-deficient samples was shifted lower for ER + BC (p = 0.015), but not TNBC (p = 0.35). In the subset of TNBC samples, univariable logistic regression models revealed that GIS status using thresholds of ≥ 42 and ≥ 33 were significant predictors of response to platinum therapy.
Conclusions
This study demonstrated that the GIS thresholds used for ovarian cancer may also be appropriate for TNBC, but not ER + BC. GIS thresholds in TNBC were validated using clinical response data to platinum therapy.
Similar content being viewed by others


Introduction
Precision medicine can have important implications for management and treatment of individuals with cancer. Aggressive chemotherapy regimens can have intolerable side effects and carry the risk of weakening organ or immune functions without clinical benefit; even targeted anti-cancer drugs can damage healthy tissue. Identifying the presence of biomarkers that indicate whether tumors are likely to be sensitive to specific chemotherapies can optimize treatment selection, thus increasing the likelihood that patients will receive tolerable and effective treatment regimens. DNA-damaging agents are a targeted treatment which can exploit existing DNA deficiencies in tumors by inhibiting or overwhelming repair pathways [1]. Consistent with this concept, patients who have tumors with homologous recombination (HR) deficiency (HRD) may benefit from treatment with platinum and poly (ADP-ribose) polymerase (PARP) inhibitors [2,3,4,5].
Several markers of defects in DNA repair can be used to identify HRD, including the presence of germline or somatic pathogenic variants in BRCA1/2 and other genes involved in HR [6]. Individual measures of genomic instability, such as loss of heterozygosity (LOH), have also been utilized to determine tumor HRD status [7]. A 3-biomarker HRD signature assay was previously developed as a more robust way to measure HRD [8]. The test produces a combined genomic instability score (GIS) based on LOH, telomeric-allelic imbalance (TAI), and large-scale state transitions (LST) [8]. This test provides a comprehensive measure of tumor HRD, beyond what is captured by genetic deficiencies and/or a single measure of genomic instability (i.e., LOH) [7, 8]. Higher GIS is associated with treatment response to platinum-based therapies and PARP inhibitors, and GIS assessment is part of a United States Food and Drug Administration (FDA)–approved companion diagnostic for patients with ovarian cancer who may be eligible for PARP inhibitor treatment [9,10,11,12,13,14].
Currently, the 3-biomarker signature assay is FDA-approved to identify HRD in patients with ovarian cancer using a GIS threshold of ≥ 42. This threshold was determined using ovarian and breast cancer tumor samples, and was set as the 5th percentile of scores in BRCA-deficient tumors [11]. Tumors with mutations in BRCA1/2 are likely to have HRD. Therefore, the GIS distribution in known BRCA1/2-deficient samples can be used to set thresholds. Recently, a lower threshold of ≥ 33 set at the 1st percentile of scores in BRCA-deficient tumors in ovarian and breast cancer has been explored; this threshold was significantly associated with improved outcomes after platinum-based treatment in ovarian cancer [9, 10, 15]. However, the GIS distribution may vary between different cancers and even between different cancer subtypes due to differences in disease pathology. Therefore, determining an optimal GIS threshold for different types of HRD tumors is important. While current evidence suggests that a threshold of ≥ 33 may be the most appropriate cutoff for ovarian cancer, it is unclear whether this recommendation should be extended to breast cancer and to distinct breast cancer subtypes.
The present study evaluated whether a GIS threshold of ≥ 33 is also appropriate for two major breast cancer subtypes: estrogen receptor–positive (ER+) breast cancer (ER + BC) and triple-negative breast cancer (TNBC). To evaluate this, GIS distributions of BRCA1/2-deficient ER + BC and TNBC were assessed and compared to the GIS distribution of BRCA1/2-deficient ovarian cancer. Clinical outcomes were available for a subset of TNBC samples, allowing a potential GIS threshold to be set and to evaluate the ability of this potential GIS threshold to predict response to platinum therapy.
Methods
Tumor samples
This retrospective study assessed ovarian and breast cancer tumors from ten individual study cohorts: Hennessy et al. [16], The Cancer Genome Atlas (TCGA) Research Network, [17, 18] NCT01372579, [19] NCT00148694/NCT00580333, [11] PrECOG 0105, [11] Timms et al., [8] TBCRC008, [20] TBCRC030, [21] and the OlympiAD trial [22]. All tumors with a known GIS from patients with ovarian cancer, ER + BC, or TNBC were selected for inclusion in the current analysis. Tumors with a known GIS from patients with ER-negative breast cancer were excluded from the analysis. Additional details on patient and specimen characteristics, inclusion and exclusion criteria, any treatments received, patient follow-up, and the time period of case collection are described, as applicable, in previous publications for the individual study cohorts. All included samples were obtained under protocols approved by an Institutional Review Board [8, 11, 16,17,18,19,20,21,22]. REMARK reporting guidelines have been followed as applicable [23].
MyChoice testing
MyChoice testing (Myriad Genetics) was performed to determine somatic BRCA1/2 status and GIS. Public TCGA data were downloaded from the Cancer Genomics Hub and run through MyChoice software, as previously described [24]. For all other specimens, MyChoice CDx testing was performed at the central Myriad Genetics reference laboratory at the time of the initial investigation [8, 11, 16, 19,20,21,22] following previously published methods [11, 25]. Details of the test, including test kit contents, requirements for biological specimens, test results and interpretation, and performance characteristics are provided in the technical specifications document [26].
BRCA1/2 sequencing
Gene mutation detection for BRCA1/2 and single-nucleotide polymorphism whole-genome analysis were performed using a custom hybridization capture method, as described previously [25]. Pathogenic BRCA mutation status was defined as a deleterious or suspected deleterious mutation in BRCA1 or BRCA2, regardless of heterozygosity. BRCA wildtype (BRCAwt) refers to a sample with no deleterious or suspected deleterious mutation in BRCA1 or BRCA2. BRCA deficiency was defined as loss of function resulting from a germline or somatic deleterious or suspected deleterious variant in BRCA1 or BRCA2 with LOH in the affected gene, or by multiple deleterious or suspected deleterious mutations in the same BRCA gene. BRCA-intact refers to a sample that is not BRCA1/2 deficient, regardless of BRCA mutation status.
Genomic instability score
GIS was calculated using an algorithm that combines measures of LOH, TAI, and LST, as previously described [25]. Binary GIS status was determined based on whether GIS scores were above or below a threshold of ≥ 33 or ≥ 42.
Pathologic complete response
Pathologic complete response (pCR) to preoperative chemotherapy was available for TNBC samples from five cohorts (NCT01372579, [19] NCT00148694/NCT00580333, [11] PrECOG 0105, [11] TBCRC008, [20] and TBCRC030 [21]). pCR status was not available for ER + samples. In some studies, residual cancer burden (RCB) [27] was used and pCR status was not available. Patients with data on RCB [11, 19,20,21] after treatment with platinum therapy were dichotomized into those with complete response (RCB-0) and those with incomplete response (RCB-I/II/III). Patients with RCB-0 who did not receive crossover treatment prior to surgery and who did not exit treatment due to progression or toxicity were considered to have achieved pCR.
Statistics
Two-sided Kolmogorov-Smirnov tests were used to compare GIS distributions in BRCA1/2-deficient ER + BC samples by human epidermal growth factor receptor 2 (HER2) status. Additionally, the GIS distributions of BRCA1/2-deficient ER + BC and TNBC samples were compared to that of BRCA1/2-deficient ovarian cancer samples.
Binomial logistic regression was used to measure the ability of binary GIS status (i.e., scores above or below the threshold) to predict pCR status in TNBC tumors. Odds ratios (ORs) with 95% profile likelihood confidence intervals (CIs) and partial likelihood ratio test p-values were reported. Sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV), were calculated by comparing binary GIS status and binary pCR status, where a pCR event with a GIS above the threshold was considered a true positive. Univariable three-parameter logistic regression models optimized for the upper bound, slope, and midpoint were used to estimate the probability of pCR for each GIS value.
All p-values were considered significant at the α = 0.05 level.
Results
Ovarian cancer tumors
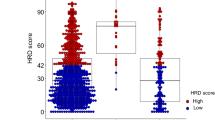
A total of 560 ovarian cancer tumors from two cohorts (Hennessy et al. [16], and The Cancer Genome Atlas Network– Ovarian [18]) were included, 20.5% of which were known to be BRCA1/2-deficient (N = 115/560; Table 1). Among BRCA1/2-deficient samples, 67.8% (N = 78/115) had a pathogenic mutation in BRCA1, 31.3% (N = 36/115) had a pathogenic mutation in BRCA2, and 0.9% (N = 1/115) had a pathogenic mutation in both BRCA1 and BRCA2. The GIS distributions are shown in Fig. 1a for BRCA1/2-deficient and BRCA-intact tumors and Supplemental Fig. 1 for BRCA1-deficient and BRCA2-deficient tumors. The median GIS was 62 in BRCA1/2-deficient ovarian cancer tumors and 31 in BRCA-intact tumors (Table 1, Supplemental Fig. 2). In this analysis, the GIS distribution of BRCA1/2-deficient ovarian cancer samples was used as a comparator to evaluate GIS distributions in BRCA1/2-deficient ER + BC and TNBC samples.
BRCAwt, BRCA wildtype; ER+, estrogen receptor positive; GIS, genomic instability score; IQR, interquartile range; pCR, pathologic complete response; TNBC, triple negative breast cancer.
Age was not readily accessible for a total of 16 patients: 11/560 (2.0%) ovarian samples; 3/805 (0.4%) ER + samples; and 2/443 (0.5%) TNBC samples, including none of the TNBC clinical validation tumors.

Distribution of GIS by cancer type and BRCA status. (a) The distribution of GIS for BRCA1/2-deficient and BRCA-intact tumors in ovarian cancer, TNBC, and ER + BC. (b) The distribution of GIS for BRCA1/2-deficient tumors fit to a normal distribution for ovarian cancer, TNBC, and ER + BC
ER + BC tumors
A total of 805 ER + BC tumors were included from five cohorts (The Cancer Genome Atlas Network – Breast, [17] PrECOG 0105, [11] Timms et al., [8] TBCRC008, [20] and the OlympiAD trial [22]; Table 1). Of these, 579 were ER + HER2-, 174 were ER + HER2+, and 52 were ER + with unknown HER2 status. To determine whether it would be appropriate to combine all ER + BC tumors, the GIS distributions of BRCA1/2-deficient tumors for ER + HER2- (N = 60) and ER + HER2+ (N = 10) were compared. No significant differences were observed between GIS distributions of ER + HER2- and ER + HER2 + BRCA1/2-deficient tumors (p = 0.80; Supplemental Fig. 3). However, with only ten BRCA1/2-deficient ER + HER2 + samples, this comparison is underpowered. In the future, when more ER + HER2 + samples are available, ER + HER2- and ER + HER2 + samples may be compared more rigorously. In these analyses, all ER + samples were analyzed together to increase statistical power.
Among all ER + BC tumors, 8.8% (N = 71/805; N = 60 ER + HER2-, N = 10 ER + HER+, and N = 1 ER + HER2 status unknown) were BRCA1/2-deficient; of those, 40.8% (N = 29/71) had a pathogenic mutation in BRCA1, and 59.2% (N = 42/71) had a pathogenic mutation in BRCA2 (see Supplemental Fig. 1 for GIS distributions of BRCA1-deficient and BRCA2-deficient samples). The GIS distributions of BRCA1/2-deficient and BRCA-intact tumors are shown in Fig. 1a. The median GIS was 56 in BRCA1/2-deficient ER + BC tumors and 14 in BRCA-intact tumors (Table 1, Supplemental Fig. 2). A significant difference was observed between the GIS distributions for BRCA1/2-deficient ER + BC tumors and ovarian cancer tumors (p = 0.015; Fig. 1b), indicating that a separate threshold should be established for ER + BC tumors. A potential GIS threshold will be established in a future study when clinical outcomes for ER + BC tumors treated with platinum or other DNA-damaging agents are available.
TNBC tumors
A total of 443 TNBC tumors were included from seven cohorts (The Cancer Genome Atlas Network – Breast, [17] NCT01372579, [19] NCT00148694/NCT00580333, [11] PrECOG 0105, [11] Timms et al., [8] TBCRC008, [20] and TBCRC030 [21]; Table 1). Among the 56 (12.6%) BRCA1/2-deficient TNBC tumors, 47 (83.9%) had a pathogenic mutation in BRCA1, 8 (14.3%) had a pathogenic mutation in BRCA2, and 1 (1.8%) had pathogenic mutations in both BRCA1 and BRCA2 (see Supplemental Fig. 1 for GIS distributions of BRCA1-deficient and BRCA2-deficient samples). The GIS distributions of BRCA1/2-deficient and BRCA-intact tumors are shown in Fig. 1a. The median GIS was 64 in BRCA1/2-deficient TNBC tumors and 40 in BRCA-intact tumors (Table 1, Supplemental Fig. 2). When comparing the GIS distributions of BRCA1/2-deficient samples, TNBC tumors were significantly different from ER + BC tumors (p < 0.001; Fig. 1b), but not significantly different from ovarian cancer tumors (p = 0.35; Fig. 1b). This indicates that the same thresholds used for ovarian cancer tumors may also be appropriate for TNBC tumors.
Clinical validation of thresholds in TNBC
GIS thresholds of ≥ 42 and ≥ 33 have been previously validated in patients with ovarian cancer [9,10,11, 15]. Because the GIS distributions in BRCA1/2-deficient ovarian and TNBC samples were similar, the thresholds used for ovarian cancer were applied to the TNBC samples in this study. The TNBC clinical validation cohort (samples from the following preoperative trials: NCT01372579, [19] NCT00148694/NCT00580333, [11] PrECOG 0105, [11] TBCRC008, [20] and TBCRC030 [21]) included 211 platinum-treated samples (N = 55 with pCR), 171 of which were BRCAwt tumors (N = 39 with pCR)) (Table 1). GIS distributions for all TNBC clinical validation samples (full clinical validation cohort) and for the subset of BRCAwt samples (BRCAwt clinical validation cohort) are summarized by binary pCR status (i.e., pCR vs. no pCR) in Fig. 2.

Distribution of GIS by pCR status for TNBC in (a) the full clinical validation cohort, and (b) the BRCAwt clinical validation cohort. Samples are stratified based on whether pCR was achieved (“pCR” vs. “No pCR”)
Univariable logistic regression models were used to evaluate the ability of GIS status, using thresholds of ≥ 33 and ≥ 42, to predict binary pCR status in both the full clinical validation cohort and in the BRCAwt clinical validation cohort. In both cohorts, GIS status using thresholds of ≥ 33 and ≥ 42 were significant predictors of pCR. Compared to the GIS threshold status of ≥ 42, the GIS threshold status of ≥ 33 resulted in a larger effect size in both the full clinical validation cohort (GIS ≥ 33: OR 11.1, 95% CI 3.9–47.1, p = 2.2 × 10− 7; GIS ≥ 42: OR 8.2, 95% CI 3.5–22.3, p = 5.6 × 10− 8) and the BRCAwt clinical validation cohort (GIS ≥ 33: OR 9.4, 95% CI 3.2–40.4, p = 5.6 × 10− 6; GIS ≥ 42: OR 7.0, 95% CI 2.9–19.6, p = 3.0 × 10− 6). A comparison of ORs from univariable logistic regression models evaluating the ability of GIS status set at a range of thresholds to predict pCR is shown in Supplemental Fig. 4.
To evaluate the ability of the GIS status using thresholds of ≥ 33 and ≥ 42 to predict pCR, a bivariable logistic regression model was run with both GIS threshold statuses as binary variables. In the full clinical validation cohort, the GIS threshold status of ≥ 42 was significant (OR 3.6, 95% CI 1.1–15.8, p = 0.03), while the GIS threshold status of ≥ 33 was not (OR 3.6, 95% CI 0.6–21.0, p = 0.15). In the same model fit in the BRCAwt clinical validation cohort, neither of the GIS threshold statuses were significant (GIS ≥ 33: OR 3.6, 95% CI 0.6–21.3, p = 0.15; GIS ≥ 42: OR 3.0, 95% CI 0.9–13.7, p = 0.07). These results demonstrate that the GIS threshold status of ≥ 42 adds significant information to the GIS threshold status of ≥ 33 in the full clinical validation cohort.
Sensitivity, specificity, PPV, and NPV for the pre-specified thresholds are reported in Table 2 for GIS thresholds of ≥ 33 and ≥ 42.
A high proportion of samples with pCR events had a GIS ≥ 33 in both the full clinical validation cohort (94.5%, N = 52/55) and the BRCAwt clinical validation cohort (92.3%, N = 36/39). The proportion of pCR events captured by the threshold decreased at the higher GIS threshold of ≥ 42 (full clinical validation cohort: 89.1%, N = 49/55; BRCAwt clinical validation cohort: 84.6%, N = 33/39); a GIS between 33 and 42 captured pCR events in an additional 5.5% of the full clinical validation cohort and 7.7% of the BRCAwt subset.
The difference in utility between a threshold of ≥ 33 and ≥ 42 can also be characterized by the difference in probability of pCR as calculated by a three-parameter logistic regression with continuous GIS predicting binary pCR status (Fig. 3). In both the full clinical validation cohort and the BRCAwt clinical validation cohort, patients with GIS between 33 and 42 had an intermediate probability of pCR; a GIS threshold of ≥ 33 separated patients with a low probability of response from patients with a moderate to high probability of response. The opposite was true for the GIS threshold of ≥ 42, which would only identify patients with the highest likelihood of response.

The probability of pCR in TNBC for a range of GIS from 3-parameter logistic regression models fit for the full clinical validation cohort (N = 211, solid line) and the BRCAwt clinical validation cohort (N = 171, dashed line). The vertical grey dashed lines represent potential thresholds of ≥ 33 and ≥ 42
Discussion
In the present study, the GIS distributions of BRCA1/2-deficient tumors were evaluated for two different major breast cancer subtypes. The GIS distribution of BRCA1/2-deficient tumors for ER + BC was significantly different from the distribution for ovarian cancer, indicating that the GIS threshold used for ovarian cancer may not be appropriate for ER + BC. The GIS distribution for BRCA1/2-deficient TNBC tumors in this study was not statistically significantly different from ovarian cancer but was significantly different from ER + BC tumors. Additionally, the clinical validation analysis demonstrated the ability of the GIS ≥ 33 and ≥ 42 thresholds to predict platinum-based therapy pCR in a subset of the TNBC samples. Together, these findings highlight the importance of determining individual thresholds for different cancer lineages and for different cancer subtypes.
Compared to BRCA1/2-deficient ovarian cancer tumors, the GIS distribution was significantly different for BRCA1/2-deficient ER + BC tumors, but not TNBC tumors. This may not be surprising, given that there are known similarities in the molecular signatures of ovarian cancer and TNBC. For example, messenger RNA expression is similarly up- or down-regulated in some genes (e.g., AKT3, CCNE1, MYC, RB1) and high mutation rates in specific genes (e.g., BRCA1, RB1, TP53) are observed in both TNBC and ovarian cancer [28, 29]. Further, both TNBC and ovarian cancer are considered copy number-driven cancers [17, 18]. Patients with ovarian cancer and TNBC are also more likely to have mutations in BRCA1 than BRCA2, [30, 31] while, the opposite is true for patients with ER + BC [32]. Differences in the underlying biology, and thus GIS, between pathogenic BRCA1-mutated and BRCA2-mutated tumors may at least partially explain the observed differences between GIS distributions for TNBC and ER + BC.
GIS thresholds of ≥ 33 and ≥ 42, set at the 1st and 5th percentile of BRCA-deficient tumors, respectively, have been validated previously in ovarian cancer [10, 12, 13, 15]. Therefore, both thresholds were evaluated in the TNBC clinical validation cohort. When evaluated in independent analyses, both the GIS threshold statuses of ≥ 33 and ≥ 42 were found to significantly predict pCR to platinum therapy, although a non-significantly larger effect size was observed for the GIS threshold status of ≥ 33 compared to ≥ 42 (OR 11.1 vs. 8.2). In a bivariable model that assessed the relationship between the two threshold statuses (i.e., evaluated whether one threshold added significant information to the other) in the full clinical validation cohort, the GIS threshold status of ≥ 42 was significant, while the GIS threshold status of ≥ 33 was not. In the BRCAwt clinical validation cohort, neither of the GIS threshold statuses in the bivariable model were found to be significant. While the analysis in the full clinical validation cohort indicated that the GIS threshold status of ≥ 42 added significant predictive information to the GIS threshold status of ≥ 33, the null findings in the BRCAwt analysis suggested that the two GIS threshold statuses had similar predictive value for pCR. The clinical significance of these inconsistent findings was unclear; therefore, sensitivity and specificity were evaluated to assess the clinical validity of the two thresholds.
In both the full clinical validation cohort and the BRCAwt clinical validation cohort, the GIS threshold of ≥ 42 had lower sensitivity, but higher specificity than the ≥ 33 threshold. When selecting a GIS threshold to identify patients who will benefit from DNA-damaging agents (e.g., platinum, PARP inhibitors), it is important to consider the appropriate balance of sensitivity and specificity. The GIS threshold of ≥ 42 will result in fewer false positives (i.e., fewer patients who will not benefit from treatment being categorized HRD-positive), but also will result in fewer true positives (i.e., fewer patients who will benefit from treatment being categorized as HRD-positive). Among the patients who achieved pCR to platinum therapy, 5.5% of patients in the full clinical validation cohort and 7.7% of patients in the BRCAwt cohort would not be identified as eligible for treatment using the threshold of ≥ 42. In clinical settings, it may be beneficial to utilize a lower threshold of ≥ 33 in order to maximize the identification of eligible patients given a paucity of alternative treatment choices. The decision to pursue treatment with DNA-damaging agents can then be considered on an individual basis, which may be dependent upon several clinical factors.
The balance of sensitivity and specificity should also be considered when selecting a GIS threshold for clinical trials. This is particularly relevant in cases where study eligibility criteria may influence the GIS distribution. For example, clinical trials that have enrollment criteria that enrich for patients with HR-deficient tumors (e.g., BRCA1/2-mutated tumors, high-grade and/or serous subtypes, platinum-sensitive tumors) will shift the distribution toward a higher GIS, as patients with pathogenic BRCA1/2–mutated tumors have higher GIS. A higher GIS threshold may appear appropriate based on high specificity alone, which may mean that fewer patients who will benefit from treatment will be categorized as HRD-positive. However, whether it may be appropriate to prioritize specificity or sensitivity could depend on the study population, or other clinical factors (e.g., first-line treatment, metastatic disease).
To date, most of the studies evaluating HRD status and clinical outcomes in breast cancer have used a threshold of ≥ 42 to identify HRD-positive tumors. Several single-arm studies have demonstrated that HRD-positive status is associated with improved clinical outcomes after platinum-based therapy in ovarian cancer [10, 15, 33] and TNBC [11, 19, 34]. However, in randomized trials, no association between HRD and chemotherapy benefit was observed [21, 35]. One study that evaluated platinum-based treatment in metastatic TNBC reported no association between HRD status and clinical outcomes [35]. In that study, HRD testing was performed on treatment-naïve tumor samples, and therefore it is possible that reversion of BRCA mutations could have precluded an association between HRD status and outcomes. However, it is also possible that an association between HRD status and outcomes would have been observed if the GIS threshold of ≥ 33 had been used.
One limitation of this study was the absence of clinical outcomes data for ER + BC; future studies will be needed to identify and validate potential thresholds. Additionally, the clinical outcomes evaluated for TNBC were limited to platinum-based therapy response. The thresholds discussed here should also be validated using other DNA-damaging agents (e.g., PARP inhibitors) in future studies. Further, the availability of data on receptor/molecular sub-types was limited in this study. It would be beneficial to compare additional tumor characteristics (e.g., Luminal A, Luminal B, HER2, etc.) in future studies to determine whether these thresholds should be broadly applied to all breast cancer sub-types.
The present study demonstrated that the optimal GIS threshold of ≥ 33 for ovarian cancer is also appropriate to predict platinum-therapy response for TNBC but may not be appropriate for ER + BC. Future studies evaluating the association between these thresholds and clinical outcomes will be required to demonstrate expanded clinical validity in response to other treatments, and in other breast cancer subtypes. The different GIS distributions observed in this study highlight the need for cancer-specific and cancer subtype–specific GIS thresholds. This will be especially important as evaluations of HRD to identify candidates for treatment with DNA-damaging agents become more commonly used in clinical practice and expand to different cancers.
Data availability
The datasets analyzed during the current study are not publicly available due to patient privacy but are available from the corresponding author on reasonable request.
References
Konstantinopoulos PA, Ceccaldi R, Shapiro GI, D’Andrea AD (2015) Homologous recombination Deficiency: exploiting the fundamental vulnerability of Ovarian Cancer. Cancer Discov 11:1137–1154. https://doi.org/10.1158/2159-8290.CD-15-0714
Ledermann JA, Drew Y, Kristeleit RS (2016) Homologous recombination deficiency and ovarian cancer. Eur J Cancer 49–58. https://doi.org/10.1016/j.ejca.2016.03.005
Foo T, George A, Banerjee S (2020) PARP inhibitors in ovarian cancer: an overview of the practice-changing trials. Genes Chromosomes Cancer
Chan CY, Tan KV, Cornelissen B (2021) PARP inhibitors in Cancer diagnosis and therapy. Clin Cancer Res 6:1585–1594. https://doi.org/10.1158/1078-0432.CCR-20-2766
O’Kane GM, Connor AA, Gallinger S (2017) Characterization, detection, and treatment approaches for homologous recombination Deficiency in Cancer. Trends Mol Med 12:1121–1137. https://doi.org/10.1016/j.molmed.2017.10.007
Hoppe MM, Sundar R, Tan DSP, Jeyasekharan AD (2018) Biomarkers for homologous recombination Deficiency in Cancer. J Natl Cancer Inst 7:704–713. https://doi.org/10.1093/jnci/djy085
Abkevich V, Timms KM, Hennessy BT, Potter J, Carey MS, Meyer LA, Lanchbury JS (2012) Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br J Cancer 10:1776–1782. https://doi.org/10.1038/bjc.2012.451
Timms KM, Abkevich V, Hughes E, Neff C, Reid J, Morris B, Chen J (2014) Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res 6:1–9
Stronach EA, Paul J, Timms KM, Hughes E, Brown K, Neff C, Brown R (2018) Biomarker Assessment of HR Deficiency, Tumor BRCA1/2 mutations, and CCNE1 Copy Number in Ovarian Cancer: Associations with clinical outcome following platinum Monotherapy. Mol Cancer Res 7:1103–1111. https://doi.org/10.1158/1541-7786.MCR-18-0034
Hodgson DR, Dougherty BA, Lai Z, Fielding A, Grinsted L, Spencer S, Barrett JC (2018) Candidate biomarkers of PARP inhibitor sensitivity in ovarian cancer beyond the BRCA genes. Br J Cancer 11:1401–1409. https://doi.org/10.1038/s41416-018-0274-8
Telli ML, Timms KM, Reid J, Hennessy B, Mills GB, Jensen AL (2016) Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin Cancer Res 15:3764-73. DOI: https://doi.org/10.1158/1078-0432.CCR-15-2477
Ray-Coquard I, Pautier P, Pignata S, Pérol D, González-Martín A, Berger R, Mäenpää J (2019) Olaparib plus bevacizumab as first-line maintenance in ovarian cancer. N Engl J Med 25:2416–2428
Moore KN, Secord AA, Geller MA, Miller DS, Cloven N, Fleming GF, Monk BJ (2019) Niraparib monotherapy for late-line treatment of ovarian cancer (QUADRA): a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol 5:636–648. https://doi.org/10.1016/S1470-2045(19)30029-4
U.S. Food & Drug Administration. Premarket Approval (PMA) (2019) September 22, 2022]; Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpma/pma.cfm?id=P190014
How JA, Jazaeri AA, Fellman B, Daniels MS, Penn S, Solimeno C, Yates MS (2021) Modification of homologous recombination Deficiency score threshold and association with long-term survival in epithelial ovarian Cancer. Cancers (Basel) 5:946. https://doi.org/10.3390/cancers13050946
Hennessy BT, Timms KM, Carey MS, Gutin A, Meyer LA, Flake DD 2nd,., Mills GB (2010) Somatic mutations in BRCA1 and BRCA2 could expand the number of patients that benefit from poly (ADP ribose) polymerase inhibitors in ovarian cancer. J Clin Oncol 22:3570–3576. https://doi.org/10.1200/JCO.2009.27.2997
The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 7418:61–70. DOI: https://doi.org/10.1038/nature11412
The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 7353:609–15. DOI: https://doi.org/10.1038/nature10166
Kaklamani VG, Jeruss JS, Hughes E, Siziopikou K, Timms KM, Gutin A, Gradishar W (2015) Phase II neoadjuvant clinical trial of carboplatin and eribulin in women with triple negative early-stage breast cancer (NCT01372579). Breast Cancer Res Treat 3:629–638. https://doi.org/10.1007/s10549-015-3435-y
Connolly RM, Leal JP, Goetz MP, Zhang Z, Zhou XC, Jacobs V (2015) TBCRC 008: early change in 18F-FDG uptake on PET predicts response to preoperative systemic therapy in human epidermal growth factor receptor 2-negative primary operable breast cancer. J Nucl Med 1:31 – 7. DOI: https://doi.org/10.2967/jnumed.114.144741
Mayer EL, Abramson V, Jankowitz R, Falkson C, Marcom PK, Traina T, Winer EP (2020) TBCRC 030: a phase II study of preoperative cisplatin versus paclitaxel in triple-negative breast cancer: evaluating the homologous recombination deficiency (HRD) biomarker. Ann Oncol 11:1518–1525. https://doi.org/10.1016/j.annonc.2020.08.2064
Robson M, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, Conte P (2017) Olaparib for metastatic breast Cancer in patients with a germline BRCA mutation. N Engl J Med 6:523–533. https://doi.org/10.1056/NEJMoa1706450
McShane LM, Altman DG, Sauerbrei W, Taube SE, Gion M, Clark GM (2006) REporting recommendations for tumor MARKer prognostic studies (REMARK). Breast Cancer Res Treat 2:229–235. https://doi.org/10.1007/s10549-006-9242-8
Siedel JH, Ring KL, Hu W, Dood RL, Wang Y, Baggerly K, Sood AK (2021) Clinical significance of homologous recombination deficiency score testing in endometrial Cancer. Gynecol Oncol 3:777–785. https://doi.org/10.1016/j.ygyno.2020.12.010
Patel JN, Braicu I, Timms KM, Solimeno C, Tshiaba P, Reid J, Ganapathi RN (2018) Characterisation of homologous recombination deficiency in paired primary and recurrent high-grade serous ovarian cancer. Br J Cancer 9:1060–1066. https://doi.org/10.1038/s41416-018-0268-6
Myriad Genetic Laboratories, Inc. myChoice CDx® Technical Information. FDA Premarket Application Number: P190014/S003. Accessed: 25 Jan 2023. https://www.accessdata.fda.gov/cdrh_docs/pdf19/P190014S003C.pdf
Symmans WF, Peintinger F, Hatzis C, Rajan R, Kuerer H, Valero V, Pusztai L (2007) Measurement of residual breast cancer burden to predict survival after neoadjuvant chemotherapy. J Clin Oncol 28:4414–4422. https://doi.org/10.1200/JCO.2007.10.6823
Longacre M, Snyder NA, Housman G, Leary M, Lapinska K, Heerboth S, Sarkar S (2016) A comparative analysis of genetic and epigenetic events of breast and ovarian Cancer related to Tumorigenesis. Int J Mol Sci 5:759. https://doi.org/10.3390/ijms17050759
Koboldt D, Fulton R, McLellan M, Schmidt H, Kalicki-Veizer J, McMichael J, Mardis E (2012) Comprehensive molecular portraits of human breast tumours. Nature 7418:61–70
Ramus SJ, Gayther SA (2009) The contribution of BRCA1 and BRCA2 to ovarian cancer. Mol Oncol 2:138–150. https://doi.org/10.1016/j.molonc.2009.02.001
Peshkin BN, Alabek ML, Isaacs C (2011) BRCA1/2 mutations and triple negative breast cancers. Breast Dis 1–2:25–33
Bane AL, Beck JC, Bleiweiss I, Buys SS, Catalano E, Daly MB, O’Malley FP (2007) BRCA2 mutation-associated breast cancers exhibit a distinguishing phenotype based on morphology and molecular profiles from tissue microarrays. Am J Surg Pathol 1:121–128. https://doi.org/10.1097/01.pas.0000213351.49767.0f
Stronach EA, Paul J, Timms KM, Hughes E, Brown K, Neff C, Steel JH (2018) Biomarker Assessment of HR Deficiency, Tumor BRCA1/2 mutations, and CCNE1 Copy Number in Ovarian Cancer: Associations with clinical outcome following platinum MonotherapyBiomarkers of Ovarian Cancer and Survival Outcome. Mol Cancer Res 7:1103–1111
Loibl S, Weber K, Timms K, Elkin E, Hahnen E, Fasching P, Braun S (2018) Survival analysis of carboplatin added to an anthracycline/taxane-based neoadjuvant chemotherapy and HRD score as predictor of response—final results from GeparSixto. Ann Oncol 12:2341–2347
Tutt A, Tovey H, Cheang MCU, Kernaghan S, Kilburn L, Gazinska P, Bliss JM (2018) Carboplatin in BRCA1/2-mutated and triple-negative breast cancer BRCAness subgroups: the TNT Trial. Nat Med 5:628–637. https://doi.org/10.1038/s41591-018-0009-7
Acknowledgements
The authors are grateful for the funding support to the Translational Breast Cancer Research Consortium (TBCRC) from The Breast Cancer Research Foundation and Susan G. Komen, as well as funding support from AstraZeneca.
Funding
This analysis was funded by Myriad Genetics, Inc.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval Statement
The analysis described in this manuscript was performed using de-identified data obtained from previously Institutional Review Board–approved protocols. No protected health information is reported; only aggregate data are presented in the manuscript. Therefore, this analysis did not meet the U.S. Health and Human Services definition of research on human subjects (HHS 46.102) and did not require Institutional Review Board approval.
Consent to participate and Publish
Patient consent for this manuscript was not required given that this was a retrospective analysis of aggregate data.
Competing Interests/Disclosures
Ms. Lenz, Mr. Neff, Ms. Solimeno, Dr. Cogan, Dr. Slavin, Dr. Gutin, Dr. Lanchbury and Dr. Timms were all employed by Myriad Genetics at the time of this study and received salary and stock options. Dr. Abramson has received consulting fees from AstraZeneca, Eisai, Daiichi Sankyo, Macrogenics, and Seagen. Dr. Boughey has received research funding from Lilly and participated in a Data Safety Monitoring Board with Cairns Surgical. Dr. Falkson has received honoraria from Exact Sciences, Curio Sciences, Agendia, and Biotheranostics for advisory boards; has received honoraria from OncLive/MJH Life Sciences for Speaker’s Bureau activity; and has received research funding to institution from Oncolytics Biotech, Quantumleap Health, and Eli Lilly. Dr. Goetz has received personal fees for CME activities from Research to Practice and Clinical Education Alliance; has received consulting fees to the Mayo Clinic from AstraZeneca, Biovica, Biotheranostics, Blueprint Medicines, Eagles Pharmaceuticals, Lilly, Novartis, Pfizer, and Sermonix; and has received grant funding to the Mayo Clinic from Lilly, Pfizer, and Sermonix. Dr. Ford has received research funding from Pfizer, Genentech, Merus, PUMA, and AstraZeneca. Dr. Kaklamani has received speaker fees from Pfizer, Gilead, Genentech, Genomic Health, Puma, Eisai, Novartis, Daiichi Sankyo, and Seagen; has received consultant fees from Puma, AstraZeneca, Athenex, and Gilead; and has received research funding from Eisai. Dr. Marcom is employed by Veracyte, Inc. Dr. Richardson is an inventor on an IP owned by Partners Healthcare and licensed to Myriad Genetics. Dr. Storniolo has received consulting fees from AstraZeneca. Dr. Tung has received research funding from AstraZeneca and has consulted for GSK. Dr. Hodgson, Dr. Lai, and Mr. Dearden were employed by AstraZeneca at the time of the study and received salary and restricted stock shares. Dr. Hennessy has received research funding from Bayer, AstraZeneca, and Daiichi Sankyo and has received royalties from Myriad Genetics. Dr. Mayer has received consulting fees from Lilly, Novartis, AstraZeneca, and Gilead. Dr. Mills is on the scientific advisory board for Amphista, Astex, AstraZeneca, BlueDot, Chrysalis Biotechnology, Ellipses Pharma, ImmunoMet, Infinity, Ionis, Leapfrog Bio, Lilly, Medacorp, Nanostring, Nuvectis, PDX Pharmaceuticals, Quereator, Roche, SignalChem Lifesciences, Tarveda, Turbine, and Zentalis Pharmaceuticals; has stock options with BlueDot, Catena Pharmaceuticals, ImmunoMet, Nuvectis, SignalChem, Tarveda, and Turbine; has licensed the HRD assay to Myriad Genetics and DSP patents to Nanostring; and has sponsored research support from AstraZeneca. Dr. Connolly has received research funding from Pfizer; has received research funding to institution for clinical trials from MSD Ireland, Pfizer, Daiichi Sankyo, and AstraZeneca; and has consulted for Seagen and AstraZeneca/Daiichi without renumeration. Dr. Telli reports research support (to her institution) from AbbVie, Arvinas, Bayer, Biothera, Calithera Biosciences, EMD Serono, Genentech, GlaxoSmithKline, Hummingbird Biosciences, Medivation, Merck, Novartis, OncoSec, Pfizer, PharmaMar, Tesaro and Vertex and has received consulting/advisory fees from AbbVie, Aduro Biotech, AstraZeneca, Blueprint Medicines, Celgene, Daiichi Sankyo, Genentech/Roche, Gilead, GlaxoSmithKline, G1 Therapeutics, Guardant, Immunomedics, Lilly, Merck, Natera, Novartis, OncoSec, Pfizer, RefleXion and Sanofi. Dr. Stearns has received research grants (to his institution) from AbbVie, Biocept, Pfizer, Novartis, QUE Oncology, and Puma Biotechnology; is a member of the advisory board for Novartis; is the chair of the Data Safety Monitoring Board for AstraZeneca; and has received nonfinancial support from Foundation Medicine. All other authors have no disclosures to report.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lenz, L., Neff, C., Solimeno, C. et al. Identifying homologous recombination deficiency in breast cancer: genomic instability score distributions differ among breast cancer subtypes. Breast Cancer Res Treat 202, 191–201 (2023). https://doi.org/10.1007/s10549-023-07046-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-023-07046-3