
Abstract
Background
Diagnostic screening for pathogenic variants in breast cancer susceptibility genes, including BRCA1, BRCA2, PALB2, PTEN and TP53, may be offered to New Zealanders from suspected high-risk breast (and ovarian) cancer families. However, it is unknown how many high-risk pathogenic variant carriers in New Zealand are not offered genetic screening using existing triage tools and guidelines for breast (and ovarian) cancer patients.
Methods
Panel-gene sequencing of the coding and non-coding regions of the BRCA1 and BRCA2 genes, and the coding regions and splice sites of CDH1, PALB2, PTEN and TP53, was undertaken for an unselected cohort of 367 female breast cancer patients. A total of 1685 variants were evaluated using the ENIGMA and the ACMG/AMP variant classification guidelines.
Results
Our study identified that 13 (3.5%) breast cancer patients carried a pathogenic or likely pathogenic variant in BRCA1, BRCA2, PALB2, or PTEN. A significantly higher number of pathogenic variant carriers had grade 3 tumours (10/13) when compared to non-carriers; however, no other clinicopathological characteristics were found to be significantly different between (likely) pathogenic variant carriers and non-carriers, nor between variant of unknown significance carriers and non-carriers. Notably, 46% of the identified (likely) pathogenic variant carriers had not been referred for a genetic assessment and consideration of genetic testing.
Conclusion
Our study shows a potential under-ascertainment of women carrying a (likely) pathogenic variant in a high-risk breast cancer susceptibility gene. These results suggest that further research into testing pathways for New Zealand breast cancer patients may be required to reduce the impact of hereditary cancer syndromes for these individuals and their families.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In New Zealand, over 3000 people are diagnosed with breast cancer each year [1]. The risk of breast cancer is significantly enhanced when an individual carries a disease-predisposing variant in high-risk cancer susceptibility genes, such as BRCA1 and BRCA2, which, respectively, confer a 72% and 69% cumulative risk of breast cancer, and a 44% and 17% cumulative risk of ovarian cancer [2]. Identification of disease-associated genetic changes in breast cancer susceptibility genes, including BRCA1, BRCA2 and PALB2, not only has actionable implications for carriers, but also for the additional members of their family who are found to carry the high-risk variant. Breast cancer prevention strategies, including surgical interventions (bilateral mastectomy) and prophylactic hormone therapy (tamoxifen and aromatase inhibitors), can help to mitigate the risk of developing the disease, while increased surveillance measures can aid with earlier detection. Identification of high-risk variants can also influence clinical decisions around patient treatment, including surgical options (i.e. choosing to have a total mastectomy instead of a wide local excision), while PARP inhibitors and cisplatin are key chemotherapeutic options for the treatment of cancers in patients carrying BRCA1 and BRCA2 pathogenic variants [3, 4].
Powerful new high-throughput DNA sequencing technology is now being adopted by diagnostic laboratories worldwide, enabling cheaper genetic testing across the entire gene(s) of interest for a greater number of individuals. Targeted multigene panel sequencing enables testing of genes known to be associated with familial breast cancer in addition to other known hereditary cancer syndromes, of which breast cancer is a component, namely, TP53 (early onset breast cancer), CDH1 (invasive lobular, hereditary diffuse gastric cancer syndrome) and PTEN (Cowden syndrome) [5,6,7]. Interpreting genetic data from gene tests can be challenging for health care providers, requiring a multidisciplinary collaboration of clinicians and scientists worldwide. The international ENIGMA (Evidence-based Network for the Interpretation of Germline Mutant Alleles) consortium [8] is an important initiative that was established to share expert advice for classification of BRCA1 and BRCA2 variants in databases, including ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and BRCA Exchange (https://brcaexchange.org/). The ACMG/AMP (American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP)) standards and guidelines provide an alternative approach for the interpretation of genetic variants associated with genetic diseases [9]. These guidelines describe a framework for classifying variants using multiple categories and degrees of evidence defined as very strong, strong, moderate or supporting.
Publicly funded gene screening is offered to New Zealand breast cancer patients by Genetic Health Service New Zealand (GHSNZ) if they fulfil testing criteria provided by eviQ Cancer Genetics (Breast and Ovarian Referral Guidelines—https://www.eviq.org.au/). Based on these guidelines, individuals may meet criteria based on their tumour pathology (negative status for oestrogen receptor, progesterone receptor and ERBB2/HER2 expression), or where a BRCA1 and BRCA2 pathogenic variant probability of 10% or more is calculated using a validated pathogenic variant prediction tool, such as the BOADICEA assessment programme [10]. Currently, approximately 9% of eligible New Zealand patients who have undergone genetic screening were found to carry a clinically actionable genetic variant in BRCA1 or BRCA2. Many studies have shown that compared with multigene sequencing panels, testing of BRCA1 and BRCA2 alone ignores potentially actionable variants in a significant proportion of cases [11,12,13]. The panel of genes available for individuals who are offered gene screening through GHSNZ expanded in 2015 from BRCA1 and BRCA2, to also include PALB2 and TP53, and specific risk-associated variants in CHEK2 and ATM (screening of PTEN and CDH1 is offered if separate gene-specific criteria are met).
Early identification of carriers of high-risk variants in breast cancer susceptibility genes has the potential to reduce the number of breast and ovarian cancer cases and deaths in both the proband and their relatives. However, previous work has found that up to 50% of breast cancer patients who carry a pathogenic variant in a cancer-associated gene did not meet their national guidelines for genetic testing [14]. Recent studies from the United Kingdom and Australia have shown that multigene testing (BRCA1, BRCA2 and PALB2, and BRCA1 and BRCA2, respectively) for all breast cancer patients would be cost-effective when compared with current eligibility criteria based on personal and family-history testing [15, 16]. To date, the effectiveness of eligibility guidelines for genetic testing of breast cancer patients in New Zealand remains unclear.
This study used a gene panel sequencing approach to screen a consecutive series of unselected New Zealand female breast cancer patients for pathogenic variants in any of six high-risk breast cancer susceptibility genes (BRCA1, BRCA2, CDH1, PALB2, PTEN and TP53) and subsequently assessed the identified carriers for their eligibility for testing using national guidelines.
Methods
Patient cohort
The study cohort comprised 367 female breast cancer patients who underwent surgery at Christchurch Hospital (New Zealand) between May 2013 and April 2017. Informed, written consent was obtained for the tissue banking of pre-operative blood by the Christchurch Cancer Society Tissue Bank (CSTB) (HDEC 165TH92). Study-specific approval was obtained from the University of Otago Ethics Committee (H14/131) for this sequencing project. Extensively de-identified clinical and pathological characteristics were provided by the CSTB for all patients in the cohort, including additional family history and GHSNZ referral information for the patients identified to carry a (likely) pathogenic variant. Genetic screening results were ascertained for patients previously identified as eligible for publically funded gene screening by GHSNZ using the Breast and Ovarian Referral Guidelines eviQ (https://www.eviq.org.au/). This study was conducted in accordance with the CSTB guidelines, and under clinical guidance of Genetic Associates (JS, CL, SB) and a Senior Oncologist (BAR), thus providing an ethically approved process for actionable genomic variants to be returned to study participants.
Molecular analysis
Germline DNA was purified from three punches of 3 mm diameter from blood stored on Flinders Technology Associates (FTA) cards (Whatman) using QIAamp DNA Investigator kit (Qiagen), following the manufacturer’s instructions. Nineteen of the initial 367 samples obtained from the Cancer Society Tissue Bank returned a yield of less than 15 ng of genomic DNA and were excluded from further analysis. Library preparation (Ion AmpliSeq Library Kit 2.0 (ThermoFisher Scientific)) and sequencing (Ion Torrent) using a custom gene panel were completed by the Liggins Institute (Auckland, New Zealand). The custom panel was designed using Ion AmpliSeq designer software (Life Technologies) to target the exons and introns of BRCA1 and BRCA2, and the exonic regions of CDH1, PALB2, PTEN and TP53.
Variant classification
Variants were numbered in accordance to HGVS recommendations (https://varnomen.hgvs.org), where + 1 represents the first nucleotide of the ATG translation initiation codon. Reference transcripts for variant annotation were as follows: NM_007294.3 (BRCA1); NM_000059.3 (BRCA2); NM_004360 (CDH1), NM_024675 (PALB2), NM_000314.6 (PTEN) and NM_000546.5 (TP53). Variant calls were processed using Ion Reporter Software, then annotated with VEP (v92.3) [17], MaxEntScan splicing predictions [18], ClinVar annotation (2018-05 full release, processed using the MacArthur Lab ClinVar processing pipeline [19]) and REVEL pathogenicity predictions [20]. BRCA1 and BRCA2 variants were assessed using the ENIGMA classification guidelines (v2.5.1) (Supplementary Fig. 1) and ACMG/AMP guidelines [9]. CDH1, PALB2, PTEN and TP53 variants were classified using the ACMG/AMP guidelines, including the gene-specific guidelines for CDH1 and PTEN variants [9, 21, 22]. Variants were classified into one of five categories: Pathogenic; Likely Pathogenic; Variant of Unknown Significance; Likely Benign; or Benign. (Likely) pathogenic variants were confirmed by sequencing PCR fragments by capillary electrophoresis using a 3730xl DNA Analyzer (Applied Biosystems). PCR primer sequences are listed in Supplementary Table 1.
Results
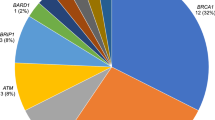
Results from gene screening (BRCA1, BRCA2, CDH1, PALB2, PTEN and TP53) germline variants in 367 New Zealand female breast cancer patients identified 1,685 single-nucleotide variants or indels. Sanger validated thirteen of the thirty variants that were classified as (likely) pathogenic, whereas the remaining seventeen variants were identified as false-positive calls (Supplementary Table 2). Of the validated variants, ten women carried a high-risk (likely) pathogenic variant in BRCA2, and three women carried one (likely) pathogenic variant in BRCA1, PTEN or PALB2, respectively (Table 1). Seven of these were frameshift variants, four were nonsense variants and two were donor site variants. Of the nineteen false calls, nine were located in homopolymer regions. The average minor allele frequency was 0.20 for all of the false calls and 0.49 for all of the true calls.
Referral to GHSNZ was recorded in the clinical notes of five of the ten patients confirmed to carry (likely) pathogenic variants in BRCA2 and for the two patients confirmed to carry a pathogenic variant in BRCA1 and PTEN, respectively (Table 1). Four of these seven referred patients had been tested for a known familial variant, whereas exploratory gene screening was undertaken for three patients.
The PALB2 carrier and five of the BRCA2 pathogenic variant carriers identified in this cohort had not been referred to GHSNZ. There was sufficient information in the hospital records for only one of these patients (ID 189) to establish that they would have met testing criteria. While this patient reported no known family cancer history, her age at diagnosis (36 y/o) alone meets eviQ criteria for referral (referral criteria: breast cancer < 40 years of age). The records for only one additional patient documented that family history was discussed at all during their treatment (ID 213), with one case of bowel cancer noted in their extended family.
Two of the non-referred patients (IDs 189 and 245) were from high-risk families known to carry a pathogenic variant. It is unknown whether these patients had chosen not to have predictive testing, or if they were unaware of their respective family histories. However, as noted above, patient ID 189 met criteria for referral based on her age at diagnosis alone. There was no indication that family history had been discussed for one of these patients (ID 245), whose age alone (74 years at diagnosis) may have influenced the clinician’s decision around GHSNZ referral.
A novel likely pathogenic variant in PTEN was identified in one patient, who was 31 years old at diagnosis and met eviQ criteria for referral (referral criteria: breast cancer < 40 years of age). This patient has a family history of breast cancer, but it was unknown whether she had clinical features or a family history indicative of PTEN hamartoma tumour syndrome. The PTEN variant (c.406dup) is predicted to cause a termination codon 44 amino acids downstream of the variant site, completely removing the crucial C2 and C-Terminal Tail domains of the 403-amino acid protein (Supplementary Fig. 2).
PALB2 and PTEN screening was not routinely offered at the time each of the two patients found to carry a variant in one of these genes (IDs 77 and 225) presented with breast cancer. The PTEN variant carrier was eligible for BRCA1 and BRCA2 screening and was identified to carry the BRCA1[c.594-2A > C_c.641A > G] haplotype, which is now known to be benign [23].
Forty-one variants of unknown significance were identified in 35 (9.8%) women (Supplementary Table 3). Twenty of these variants were identified in BRCA2, nine in BRCA1, two in CDH1, eight in PALB2 and two in TP53. The majority were missense (n = 34), while the remainder included four intronic variants and three variants located near a splice site (within 6 nt). Within our study cohort, 319 (86.9%) women were not found to carry any VUS or (likely) pathogenic variants (Table 2).
The majority of the (likely) pathogenic variant carriers had grade 3 tumours (10/13), which was significantly higher compared to non-carriers (P < 0.05). No other clinical characteristics were significantly different between (likely) pathogenic carriers and non-carriers, or between VUS carriers and non-carriers (Table 2). Of the patients in our cohort who specified their ethnicity, 7.1% (23/322) identified as Māori or as having Māori ancestry (Table 2). On average, Māori were found to be slightly younger at diagnosis compared to non-Māori (57.6 vs 63.1 years), and a higher proportion were diagnosed with grade 3 tumours compared to non-Māori (60.9% vs 47.8%) (Supplementary Table 4), but these clinical differences did not reach statistical significance (P < 0.05).
Discussion
This study screened 367 female breast cancer patients in New Zealand for risk-associated variants in the familial breast cancer susceptibility genes BRCA1, BRCA2, CDH1, PALB2, PTEN and TP53. Thirteen patients (3.5%) were identified to carry (likely) pathogenic variants: ten in BRCA2 and one in BRCA1, PALB2 and PTEN, respectively.
BRCA1/2 (likely) pathogenic variants were identified in 3.0% (11/367) of the cohort, which is within the range of observations in comparable international studies (1.8–5.4%) [24,25,26,27]. An additional two likely pathogenic variants (0.5%) were identified in PALB2 and PTEN. (Likely) pathogenic variants in non-BRCA1/2 breast cancer susceptibility genes (CDH1, PALB2, PTEN and TP53) have typically been observed in small numbers in other studies. PALB2 and PTEN pathogenic variants were detected in 0.2% of a US breast cancer cohort analysed by Tung et al. [28]. In a German cohort of 581 breast cancer patients with a strong family history of breast/ovarian cancers, pathogenic variants in PALB2, TP53 and CDH1 were only found in 1%, 0.3% and 0% of the patients, respectively [29]. These results indicate that pathogenic variants in these genes are rare, even in very select high-risk populations. Assessing variants across all genes (BRCA1, BRCA2, CDH1, PALB2, PTEN and TP53) in our study also found 11.7% of New Zealand patients carrying a variant of unknown significance, which was comparable to similar international breast cancer studies (6.4% [30]; 9.9% [27]; 14.7% [31]).
Six of the 13 (likely) pathogenic variant carriers identified in our cohort were not referred to GHSNZ and there was sufficient information in the clinical notes for only one of these patients to suggest that they did meet the criteria for referral. Extrapolating this result nationwide suggests that around 2% of the approximately 3000 breast cancer cases diagnosed annually may carry a (likely) pathogenic variant in a susceptibility gene that predisposes to cancer, but would fail to be referred to GHSNZ. The issues that may prevent referral include that patients may not always be aware of their family cancer history, and/or of genetic testing in their extended families, as evidenced by two of the high-risk variant carriers identified in our study. Furthermore, one high-risk pathogenic variant carrier identified in our study (ID 67) refused all treatment options at the time of diagnosis, which may have affected referral to GHSNZ, while acceptable treatment options were explored. Due to this, clinicians may not be able to obtain the information required to establish if a patient is eligible for genetic referral, while in some cases the patient may not have been asked for this information [32]. These issues would each have direct implications for risk management advice offered to patients and their families.
The gene panel offered by GHSNZ since 2015 was not available when the PALB2 and PTEN variant carriers identified in this work presented with breast cancer. While the more recent implementation of panel screening includes PALB2 (in addition to TP53 and specific variants in ATM and CHEK2), separate gene-specific criteria need to be met to also include PTEN. In addition, re-testing would need to have been offered, or exploratory testing in a close relative undertaken, to identify non-BRCA1/2 disease-predisposing variants in high-risk patients tested prior to 2015.
Universal gene screening of breast cancer patients has been previously reported as a cost-effective option to overcome the under-referral of breast cancer patients who do not meet the current referral guidelines [16, 33]. Implementation of such a regime in New Zealand would overcome the factors identified in this study that limited patient referral, while simultaneously capturing many of the families that carry (likely) pathogenic variants in non-BRCA1/2 genes that were not included in screening at the time of their referral. This strategy would improve identification of high-risk families in New Zealand, while the information gained would also help to guide the treatment plans of affected patients and aid with decisions around prevention for the other identified carriers in each patient’s family.
The clinical characteristics of all breast cancer cases reported by two northern New Zealand regional registries over a 13-year period have been summarized previously [34]. When comparing the clinical features of the two cohorts, our cohort reported a higher proportion of women presenting with a grade 3 tumour (48.8% vs 29.1%), fewer women under 50 years of age (21.0% vs 28.2%) and a lower number of women of Māori ethnicity (6.4% vs 9.5%). The latter finding reflects the population demographic differences between the North Island and the South Island of New Zealand, with higher proportion of people reporting Māori descent in the North Island. Four VUS carriers, but none of the (likely) pathogenic variant carriers, identified in our cohort reported to be of Māori decent. A novel pathogenic variant was identified in one patient in our study suggesting that there may be other unique high-risk variants in the New Zealand population. Future work to expand the cohort to include patients from the North Island would help us better understand the genetic predisposition to cancer in Māori, while also further capturing the genetic diversity of the New Zealand population.
This study is the first to demonstrate the frequency of (likely) pathogenic genetic variants in BRCA1, BRCA2, TP53, CDH1, PTEN and PALB2 in a cohort of unselected breast cancer patients in New Zealand. Furthermore, we provide evidence that a significant proportion of (likely) pathogenic variant carriers are not being referred to a clinical genetic service, even when some of these patients were eligible under the current referral guidelines. Variant screening in all women diagnosed with breast cancer has been shown to be cost-effective, while reducing the risk that variant carriers remain unidentified [16, 33]. Gene panel testing of all breast cancer patients in New Zealand would improve the identification of high-risk individuals, allowing for timely predictive testing and appropriate risk management, while also reducing the impact of hereditary cancer syndromes for these individuals and their families.
References
Ministry of Health (2016) Cancer: new registrations and deaths 2013. Wellington
Kuchenbaecker KB, Hopper JL, Barnes DR, Phillips KA, Mooij TM, Roos-Blom MJ, Jervis S, van Leeuwen FE, Milne RL, Andrieu N, Goldgar DE, Terry MB, Rookus MA, Easton DF, Antoniou AC, Brca, Consortium BC, McGuffog L, Evans DG, Barrowdale D, Frost D, Adlard J, Ong KR, Izatt L, Tischkowitz M, Eeles R, Davidson R, Hodgson S, Ellis S, Nogues C, Lasset C, Stoppa-Lyonnet D, Fricker JP, Faivre L, Berthet P, Hooning MJ, van der Kolk LE, Kets CM, Adank MA, John EM, Chung WK, Andrulis IL, Southey M, Daly MB, Buys SS, Osorio A, Engel C, Kast K, Schmutzler RK, Caldes T, Jakubowska A, Simard J, Friedlander ML, McLachlan SA, Machackova E, Foretova L, Tan YY, Singer CF, Olah E, Gerdes AM, Arver B, Olsson H (2017) Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA 317(23):2402–2416. https://doi.org/10.1001/jama.2017.7112
Byrski T, Huzarski T, Dent R, Marczyk E, Jasiowka M, Gronwald J, Jakubowicz J, Cybulski C, Wisniowski R, Godlewski D, Lubinski J, Narod SA (2014) Pathologic complete response to neoadjuvant cisplatin in BRCA1-positive breast cancer patients. Breast Cancer Res Treat 147(2):401–405. https://doi.org/10.1007/s10549-014-3100-x
Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, Friedlander M, Arun B, Loman N, Schmutzler RK, Wardley A, Mitchell G, Earl H, Wickens M, Carmichael J (2010) Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. The Lancet 376(9737):235–244. https://doi.org/10.1016/s0140-6736(10)60892-6
Lalloo F, Varley J, Moran A, Ellis D, O’Dair L, Pharoah P, Antoniou A, Hartley R, Shenton A, Seal S, Bulman B, Howell A, Evans DG (2006) BRCA1, BRCA2 and TP53 mutations in very early-onset breast cancer with associated risks to relatives. Eur J Cancer 42(8):1143–1150. https://doi.org/10.1016/j.ejca.2005.11.032
Liaw D, Marsh DJ, Li J, Dahia PL, Wang SI, Zheng Z, Bose S, Call KM, Tsou HC, Peacoke M (1997) Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet 16(1):64–67
Benusiglio PR, Malka D, Rouleau E, De Pauw A, Buecher B, Nogues C, Fourme E, Colas C, Coulet F, Warcoin M, Grandjouan S, Sezeur A, Laurent-Puig P, Moliere D, Tlemsani C, Di Maria M, Byrde V, Delaloge S, Blayau M, Caron O (2013) CDH1 germline mutations and the hereditary diffuse gastric and lobular breast cancer syndrome: a multicentre study. J Med Genet 50(7):486–489. https://doi.org/10.1136/jmedgenet-2012-101472
Spurdle AB, Healey S, Devereau A, Hogervorst FB, Monteiro AN, Nathanson KL, Radice P, Stoppa-Lyonnet D, Tavtigian S, Wappenschmidt B, Couch FJ, Goldgar DE (2012) ENIGMA-evidence-based network for the interpretation of germline mutant alleles: an international initiative to evaluate risk and clinical significance associated with sequence variation in BRCA1 and BRCA2 genes. Hum Mutat 33(1):2–7. https://doi.org/10.1002/humu.21628
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, Committee ALQA (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med 17(5):405–424. https://doi.org/10.1038/gim.2015.30
Lee A, Mavaddat N, Wilcox AN, Cunningham AP, Carver T, Hartley S, Babb de Villiers C, Izquierdo A, Simard J, Schmidt MK, Walter FM, Chatterjee N, Garcia-Closas M, Tischkowitz M, Pharoah P, Easton DF, Antoniou AC (2019) BOADICEA: a comprehensive breast cancer risk prediction model incorporating genetic and nongenetic risk factors. Genet Med. https://doi.org/10.1038/s41436-018-0406-9
Desmond A, Kurian AW, Gabree M, Mills MA, Anderson MJ, Kobayashi Y, Horick N, Yang S, Shannon KM, Tung N, Ford JM, Lincoln SE, Ellisen LW (2015) Clinical actionability of multigene panel testing for hereditary breast and ovarian cancer risk assessment. JAMA Oncol 1(7):943–951. https://doi.org/10.1001/jamaoncol.2015.2690
Kurian AW, Hare EE, Mills MA, Kingham KE, McPherson L, Whittemore AS, McGuire V, Ladabaum U, Kobayashi Y, Lincoln SE, Cargill M, Ford JM (2014) Clinical evaluation of a multiple-gene sequencing panel for hereditary cancer risk assessment. J Clin Oncol 32(19):2001–2009. https://doi.org/10.1200/JCO.2013.53.6607
Couch FJ, Shimelis H, Hu C, Hart SN, Polley EC, Na J, Hallberg E, Moore R, Thomas A, Lilyquist J, Feng B, McFarland R, Pesaran T, Huether R, LaDuca H, Chao EC, Goldgar DE, Dolinsky JS (2017) Associations between cancer predisposition testing panel genes and breast cancer. JAMA Oncol 3(9):1190–1196. https://doi.org/10.1001/jamaoncol.2017.0424
Beitsch PD, Whitworth PW, Hughes K, Patel R, Rosen B, Compagnoni G, Baron P, Simmons R, Smith LA, Grady I (2019) Underdiagnosis of hereditary breast cancer: are genetic testing guidelines a tool or an obstacle? J Clin Oncol 37(6):453–460
Sun L, Brentnall A, Patel S, Buist DSM, Bowles EJA, Evans DGR, Eccles D, Hopper J, Li S, Southey M, Duffy S, Cuzick J, Dos Santos SI, Miners A, Sadique Z, Yang L, Legood R, Manchanda R (2019) A cost-effectiveness analysis of multigene testing for all patients with breast cancer. JAMA Oncol. https://doi.org/10.1001/jamaoncol.2019.3323
Tuffaha HW, Mitchell A, Ward RL, Connelly L, Butler JRG, Norris S, Scuffham PA (2018) Cost-effectiveness analysis of germ-line BRCA testing in women with breast cancer and cascade testing in family members of mutation carriers. Genet Med 20(9):985–994. https://doi.org/10.1038/gim.2017.231
McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, Flicek P, Cunningham F (2016) The ensembl variant effect predictor. Genome Biol 17(1):122. https://doi.org/10.1186/s13059-016-0974-4
Shamsani J, Kazakoff SH, Armean IM, McLaren W, Parsons MT, Thompson BA, O’Mara TA, Hunt SE, Waddell N, Spurdle AB (2019) A plugin for the ensembl variant effect predictor that uses MaxEntScan to predict variant spliceogenicity. Bioinformatics 35(13):2315–2317. https://doi.org/10.1093/bioinformatics/bty960
Zhang X, Minikel EV, O’Donnell-Luria AH, MacArthur DG, Ware JS, Weisburd B (2017) ClinVar data parsing Wellcome. Open Res 2:33. https://doi.org/10.12688/wellcomeopenres.11640.1
Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, Musolf A, Li Q, Holzinger E, Karyadi D, Cannon-Albright LA, Teerlink CC, Stanford JL, Isaacs WB, Xu J, Cooney KA, Lange EM, Schleutker J, Carpten JD, Powell IJ, Cussenot O, Cancel-Tassin G, Giles GG, MacInnis RJ, Maier C, Hsieh CL, Wiklund F, Catalona WJ, Foulkes WD, Mandal D, Eeles RA, Kote-Jarai Z, Bustamante CD, Schaid DJ, Hastie T, Ostrander EA, Bailey-Wilson JE, Radivojac P, Thibodeau SN, Whittemore AS, Sieh W (2016) REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet 99(4):877–885. https://doi.org/10.1016/j.ajhg.2016.08.016
Mester JL, Ghosh R, Pesaran T, Huether R, Karam R, Hruska KS, Costa HA, Lachlan K, Ngeow J, Barnholtz-Sloan J, Sesock K, Hernandez F, Zhang L, Milko L, Plon SE, Hegde M, Eng C (2018) Gene-specific criteria for PTEN variant curation: recommendations from the ClinGen PTEN Expert Panel. Hum Mutat 39(11):1581–1592. https://doi.org/10.1002/humu.23636
Lee K, Krempely K, Roberts ME, Anderson MJ, Carneiro F, Chao E, Dixon K, Figueiredo J, Ghosh R, Huntsman D, Kaurah P, Kesserwan C, Landrith T, Li S, Mensenkamp AR, Oliveira C, Pardo C, Pesaran T, Richardson M, Slavin TP, Spurdle AB, Trapp M, Witkowski L, Yi CS, Zhang L, Plon SE, Schrader KA, Karam R (2018) Specifications of the ACMG/AMP variant curation guidelines for the analysis of germline CDH1 sequence variants. Hum Mutat 39(11):1553–1568. https://doi.org/10.1002/humu.23650
de la Hoya M, Soukarieh O, Iea L-P, Vega A, Walker LC, van Ierland Y, Baralle D, Santamariña M, Lattimore V, Wijnen J, Whiley P et al (2016) Combined genetic and splicing analysis of BRCA1 c.[594–2A>C; 641A>G] highlights the relevance of naturally occurring in-frame transcripts for developing disease gene variant classification algorithms. Hum Mol Genet 25(11):2256–2268
Li J, Wen WX, Eklund M, Kvist A, Eriksson M, Christensen HN, Torstensson A, Bajalica-Lagercrantz S, Dunning AM, Decker B, Allen J, Luccarini C, Pooley K, Simard J, Dorling L, Easton DF, Teo SH, Hall P, Borg A, Gronberg H, Czene K (2019) Prevalence of BRCA1 and BRCA2 pathogenic variants in a large, unselected breast cancer cohort. Int J Cancer 144(5):1195–1204. https://doi.org/10.1002/ijc.31841
Gomes MC, Costa MM, Borojevic R, Monteiro AN, Vieira R, Koifman S, Koifman RJ, Li S, Royer R, Zhang S, Narod SA (2007) Prevalence of BRCA1 and BRCA2 mutations in breast cancer patients from Brazil. Breast Cancer Res Treat 103(3):349–353. https://doi.org/10.1007/s10549-006-9378-6
Rodriguez RC, Esperon AA, Ropero R, Rubio MC, Rodriguez R, Ortiz RM, Anta JJ, de los Rios M, Carnesolta D, del Olivera MC, Vansam SS, Royer R, Akbari MR, Donenberg T, Narod SA (2008) Prevalence of BRCA1 and BRCA2 mutations in breast cancer patients from Cuba. Fam Cancer 7(3):275–279. https://doi.org/10.1007/s10689-008-9187-7
Li G, Guo X, Tang L, Chen M, Luo X, Peng L, Xu X, Wang S, Xiao Z, Yi W, Dai L, Wang J (2017) Analysis of BRCA1/2 mutation spectrum and prevalence in unselected Chinese breast cancer patients by next-generation sequencing. J Cancer Res Clin Oncol 143(10):2011–2024. https://doi.org/10.1007/s00432-017-2465-8
Tung N, Lin NU, Kidd J, Allen BA, Singh N, Wenstrup RJ, Hartman AR, Winer EP, Garber JE (2016) Frequency of germline mutations in 25 cancer susceptibility genes in a sequential series of patients with breast cancer. J Clin Oncol 34(13):1460–1468. https://doi.org/10.1200/JCO.2015.65.0747
Kraus C, Hoyer J, Vasileiou G, Wunderle M, Lux MP, Fasching PA, Krumbiegel M, Uebe S, Reuter M, Beckmann MW, Reis A (2017) Gene panel sequencing in familial breast/ovarian cancer patients identifies multiple novel mutations also in genes others than BRCA1/2. Int J Cancer 140(1):95–102. https://doi.org/10.1002/ijc.30428
Yang XR, Devi BCR, Sung H, Guida J, Mucaki EJ, Xiao Y, Best A, Garland L, Xie Y, Hu N, Rodriguez-Herrera M, Wang C, Jones K, Luo W, Hicks B, Tang TS, Moitra K, Rogan PK, Dean M (2017) Prevalence and spectrum of germline rare variants in BRCA1/2 and PALB2 among breast cancer cases in Sarawak Malaysia. . Breast Cancer Res Treat. https://doi.org/10.1007/s10549-017-4356-8
Singh J, Thota N, Singh S, Padhi S, Mohan P, Deshwal S, Sur S, Ghosh M, Agarwal A, Sarin R, Ahmed R, Almel S, Chakraborti B, Raina V, DadiReddy PK, Smruti BK, Rajappa S, Dodagoudar C, Aggarwal S, Singhal M, Joshi A, Kumar R, Kumar A, Mishra DK, Arora N, Karaba A, Sankaran S, Katragadda S, Ghosh A, Veeramachaneni V, Hariharan R, Mannan AU (2018) Screening of over 1000 Indian patients with breast and/or ovarian cancer with a multi-gene panel: prevalence of BRCA1/2 and non-BRCA mutations. Breast Cancer Res Treat 170(1):189–196. https://doi.org/10.1007/s10549-018-4726-x
Tan YY (2013) Referral of patients with suspected hereditary breast-ovarian cancer or lynch syndrome for genetic services: a systematic review. J Commun Med Health Educ. https://doi.org/10.4172/2161-0711.1000255
Manchanda R, Patel S, Gordeev VS, Antoniou AC, Smith S, Lee A, Hopper JL, MacInnis RJ, Turnbull C, Ramus SJ, Gayther SA, Pharoah PDP, Menon U, Jacobs I, Legood R (2018) Cost-effectiveness of population-based BRCA1, BRCA2, RAD51C, RAD51D, BRIP1, PALB2 mutation testing in unselected general population women. J Natl Cancer Inst 110(7):714–725. https://doi.org/10.1093/jnci/djx265
Seneviratne S, Lawrenson R, Harvey V, Ramsaroop R, Elwood M, Scott N, Sarfati D, Campbell I (2016) Stage of breast cancer at diagnosis in New Zealand: impacts of socio-demographic factors, breast cancer screening and biology. BMC Cancer 16:129. https://doi.org/10.1186/s12885-016-2177-5
Funding
This study was supported by grants from the Royal Society of New Zealand (Rutherford Discovery Fellowship, L.W.), the Breast Cancer Foundation New Zealand (Belinda Scott Clinical Fellowship, V.L.) and the National Health and Medical Research Council (Investigator Fellowship, A.S.). V.L., L.W., H.M. and B.R. also acknowledge continued support by the Mackenzie Charitable Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. The Christchurch Cancer Society Tissue Bank received ethical approval from the Health and Disability Ethics Committee (New Zealand) (HDEC 165TH92), while study specific approval was obtained from the University of Otago Ethics Committee (H14/131). This article does not contain any studies with animals performed by any of the authors.
Informed consent
Signed informed consent was obtained from all individual participants included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lattimore, V., Parsons, M.T., Spurdle, A.B. et al. Under-ascertainment of breast cancer susceptibility gene carriers in a cohort of New Zealand female breast cancer patients. Breast Cancer Res Treat 185, 583–590 (2021). https://doi.org/10.1007/s10549-020-05986-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-020-05986-8