
Abstract
Anti-estrogen and anti-HER2 treatments have been among the first and most successful examples of targeted therapy for breast cancer (BC). However, the treatment of triple-negative BC (TNBC) that lack estrogen receptor expression or HER2 amplification remains a major challenge. We previously discovered that approximately two-thirds of TNBCs express vitamin D receptor (VDR) and/or androgen receptor (AR) and hypothesized that TNBCs co-expressing AR and VDR (HR2-av TNBC) could be treated by targeting both of these hormone receptors. To evaluate the feasibility of VDR/AR-targeted therapy in TNBC, we characterized 15 different BC lines and identified 2 HR2-av TNBC lines and examined the changes in their phenotype, viability, and proliferation after VDR and AR-targeted treatment. Treatment of BC cell lines with VDR or AR agonists inhibited cell viability in a receptor-dependent manner, and their combination appeared to inhibit cell viability additively. Moreover, cell viability was further decreased when AR/VDR agonist hormones were combined with chemotherapeutic drugs. The mechanisms of inhibition by AR/VDR agonist hormones included cell cycle arrest and apoptosis in TNBC cell lines. In addition, AR/VDR agonist hormones induced differentiation and inhibited cancer stem cells (CSCs) measured by reduction in tumorsphere formation efficiency, high aldehyde dehydrogenase activity, and CSC markers. Surprisingly, we found that AR antagonists inhibited proliferation of most BC cell lines in an AR-independent manner, raising questions regarding their mechanism of action. In summary, AR/VDR-targeted agonist hormone therapy can inhibit HR2-av TNBC through multiple mechanisms in a receptor-dependent manner and can be combined with chemotherapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Breast cancers (BCs) are categorized in the clinic into three subtypes, including, estrogen receptor positive (ER+), epidermal growth factor receptor 2 positive (HER2+), and triple-negative BCs (TNBCs) [1]. Treatment of ER+ and HER2+ BCs has been successful through targeted therapy with anti-estrogen and anti-HER2 drugs. Due to the lack of these targets, neo-adjuvant chemotherapy is used for treatment of TNBC that are typically associated with poorer prognosis compared to other BC subtypes [2, 3]. In addition, up to half of the ER+ tumors eventually become resistant to anti-estrogens [4–6]. Therefore, there is an urgent need to identify and develop novel targeted therapy approaches for TNBC and hormone refractory ER+ BCs.
In an attempt to develop a normal cell lineage-based phylogenetic BC classification [7], we previously studied hormone receptors (HRs) in normal human breast tissues and compared them with human BCs [1]. To do so, we used multiplex immunofluorescent staining and analyzed simultaneous co-expression of the 14 lineage markers in ~15,000 normal breast cells and ~3000 BCs, and found that both normal luminal breast cells and BCs conform to four hormonal states (HR3, HR2, HR1, and HR0) based on co-expression of ER, androgen receptor (AR) and vitamin-D (Vit-D) receptor (VDR) [1]. We also found that there was 6.9-fold difference in overall survival between HR3 versus HR0 tumors [1]. Compared to the 1.7–2.1-fold differences typically reported in overall survival between ER+/LumA versus TNBC/basal-like BC [8, 9], these results suggest that the HR0–3 classification reveals BC sub-groups with highly significant outcome differences.
Utilizing the HR0–3 classification, we discovered that approximately two-thirds of TNBCs co-express AR and VDR (HR2-av TNBC) or express VDR alone (HR1-v TNBC); the remaining one-third of the TNBCs are triple negative for ER, AR, and VDR (HR0 TNBC) [1]. These findings raised the possibility of developing novel HR-targeted therapies for TNBC, for which the only existing option is chemotherapy at the moment. In the present study, we demonstrate that HR1-v and HR2-av TNBC cell lines can be targeted with AR and/or VDR agonist hormones alone and in combination. The combined effects of AR and VDR ligands not only reduce the viability of the cells, but also change their cancer stem cell (CSC) phenotype and differentiation. In addition, we found that AR and VDR agonist hormones can be combined with chemotherapeutic agents to successfully target TNBC cells.
Materials and methods
Cell culture and drug viability
All the BC and prostate cancer (PC) cell lines were purchased from ATCC and DSMZ and cultured in their respective media. The short tandem repeat profiling was used to validate the authenticity of the cell lines (Genetic Resources Core Facility, John Hopkins School of Medicine). All the drugs were prepared and used according to concentrations that were previously reported [10–14], detailed information for each drug is provided in Supplemental Table 2. Cell viability assays were carried out in 24 or 96 well plates with cell titer cell reagent as previously described [15]. The LD50 values of all the AR antagonists were calculated as described previously [16]. See supplemental methods for further details and analysis of additive or antagonistic activity determined by Bliss independent criterion.
Cell cycle and apoptosis assays
The cell cycle profile after AR or VDR agonist treatment was evaluated by bromodeoxy uridine (BrdU) pulse labeling followed by FACS analysis. Apoptosis was measured with FACS analysis of Annexin V and propidium iodide double staining following the manufacture’s protocol (Life Technologies). See supplemental methods for further details.
Tumorsphere and AldeFluor assays
Tumorsphere assays were performed as described previously [15]. AldeFluor assays were performed as described [17] using ALDEFLUOR kit (StemCell Technologies). The cells were treated with agonist(s) for 8 days with media changes every 3 days and analyzed according to manufacturer’s protocol by FACS. See supplemental methods for further details.
PCR-based human stem cell array
Human stem cell RT2 Profiler™ PCR array was used and data were analyzed according to manufacturer’s protocol and software (SA Biosciences). The genes with a 2-fold difference compared to the vehicle treated control group were shortlisted and plotted as a Heatmap generated using Microsoft Excel.
Statistical analysis
The statistical significance of the data was evaluating by performing Student’s T test using a cut-off of P-value <0.05.
Results
Majority of TNBC cell lines express AR and/or VDR
In order to identify BC cell lines that represent HR1-v, HR2-av, and HR0 phenotypes, we selected 15 lines based on their previously published profiles. Two well-established AR+ (LNCaP and LAPC-4) and two AR− (PC-3 and DU-145) PC cell lines were used to provide benchmark for relative scale of AR expression. Among the 15 BC cell lines, seven were TNBC (Fig. 1a) that included HR0 (BT-549 and SUM-1315), HR1-v (BT-20, MDA-MB-468 and SUM-159PT), and HR2-av (MFM-223 and CAL-148) cell lines (Fig. 1d). In addition to confirming these phenotypes with western blots, we tested the response of these cell lines to physiologic levels of AR and VDR agonists and determined that the cells we designate as HR1-v respond to VDR agonists but not AR agonists, HR2-av cell lines respond to both AR and VDR agonists, and HR0 cells did not respond to either AR or VDR agonists (Fig. 1b, c; Suppl. Fig. 1b). Therefore, the phenotypic HR0, HR1-v, and HR2-av designation of the cells in Fig. 1b are based on both biochemical AR and VDR expression and response to physiologic concentrations of their natural ligands.

Evaluation of androgen and vitamin D receptor agonists response in BC lines: a Western blot analysis of 15 breast cancer cell lines for ER, AR, VDR, PR, and Her2 expression. Two AR+ and two AR− prostate cancer cell lines, LNCaP, LAPC-4, PC-3, and DU-145, respectively, were used as controls for AR expression. 30 µg extract was loaded in each lane of a 4–12 % 20-well SDS-PAGE gel, which was transferred onto a nitrocellulose membrane and probed with the following antibodies: ERα (1:750, Santa Cruz sc-8002), progesterone receptor (1:1000, Thermofisher Scientific MA1-410), HER2 (1:2000, Abcam ab2428), androgen receptor (N-20) (1:1000, Abcam sc-816), vitamin D receptor (1:1000, Thermofisher Scientific MA5-14617). β-Actin is used as a loading control (1:5000, Sigma A2228). The results of VDR and AR agonist treatment experiments in panels b and c are plotted as percent viability of the cells in comparison to the vehicle control. Error bars represent mean ± standard error of mean (SEM). These experiments were repeated at least three times. The statistical significance of the drug treatments was determined using two-tailed Student’s T-test. *P < 0.05, **P < 0.01. Cal calcitriol, DHT dihydrotestosterone. b VDR agonist calcitriol treatment on cancer cell lines: breast cancer cell lines [black VDR(+) and hatched VDR(− or low)] were plated in their respective media with complete serum in triplicates. 24–48 h after plating, the cells were treated with 100 nM calcitriol for 6 days with media change containing fresh drug every 3 days. At the end of the experiment, the cells were trypsinized, stained with 0.1 % Trypan blue and viable cells were counted using Cellometer. c AR agonist dihydrotestosterone (DHT) treatment of cancer cell lines: all the cell lines were seeded at 30–40 % confluency into 24-well plates in triplicates in phenol red-free media containing charcoal stripped serum in order to exclude the interference from androgenic hormones in serum [black AR(+) and hatched AR(− or low expression)]. 48–72 h after plating, the cells were treated with 10 nM DHT for 8–10 days with media change every 3–4 days. At the end of the experiment, the cells were trypsinized, stained with 0.1 % Trypan blue and viable cells were counted using Cellometer. d The HR0–3 phenotype designation of selected TNBC cell lines based on AR/VDR expression and response to natural ligands
Inhibition of TNBC cell lines with calcitriol is VDR dependent
The role of VDR has been studied in cancers, showing that ligand bound VDR induces anti-proliferative, pro-apoptotic, and pro-differentiating effects both in vitro and in vivo [13, 18]. Here, we confirmed that natural VDR agonist 1α,25-dihyroxy vitamin D3 (calcitriol) inhibits cell viability in BC cell lines (Fig. 1b). No inhibition of cell viability was observed in VDR− breast cell line BT-549 demonstrating that the response to calcitriol is VDR dependent.
Inhibition of TNBC cell lines with dihydrotestosterone is AR dependent
While the notion of AR-targeted therapy for BC has been around since the early 1970s [19–21], whether AR agonists or AR antagonists should be used for this purpose has been contentious. Many studies show that AR agonists inhibit BC cell growth both in vivo and in vitro [22–30], and others indicated that AR antagonists can also inhibit breast tumor growth [31] and recently several clinical studies were initiated with AR antagonists in BC patients [32, 33]. Hence, based on the prior literature, it was not entirely clear whether AR agonists or antagonists should be used to treat AR+ TNBC. Thus, we started by testing the effects of both AR agonists and antagonists in a panel of AR+ and AR− BC cell lines including all three subtypes (ER+, HER2+, and TNBC). In addition, we used AR+ and AR− PCs as controls because PC cell lines have a well-established and specific response to AR ligands.
We found that AR agonists dihydrotestosterone (DHT) and R1881 stimulated proliferation of AR+ PC cell lines as expected. Importantly, there was no effect on AR− PC-3 cell line, which demonstrates that the effect of DHT and R1881 on cell proliferation is AR dependent in PC cell lines (Fig. 1c; Suppl. Fig. 1b).
Consistent with the opposing role of androgens in male versus female, DHT or R1881 treatment resulted in a decrease in cell proliferation and viability in AR+ BC cell lines (Fig. 1c; Suppl. Fig. 1b). The one exception was the ER− HER2+ BC cell lines in which AR agonists increase cell proliferation (data not shown), which was shown to be due to a cross-talk between HER2 and AR signaling pathways that is only observed in ER− background [10, 34, 35]. There was no response to DHT or R1881 in AR− BT-20, MDA-MB-468, and AR-low SUM-159PT cell lines indicating that at the effect of DHT and R1881 on cell proliferation is AR dependent in BC cell lines (Fig. 1c; Suppl. Fig. 1a, b).
In summary, AR agonists DHT and R1881 decrease proliferation of TNBC, ER+, and ER+/HER2+ BC cell lines and stimulate proliferation of ER−/HER2+/AR+ cell lines.
Inhibition of TNBC cell lines with AR antagonists is not dependent on AR
Next, we tested the effect of AR antagonists on BC cell line proliferation using three different drugs (flutamide, bicalutamide, and enzalutamide) on four AR+ and three AR−/low BC cell lines. In addition, two AR+ and two AR− PC cell lines were used as controls.
As expected, in PC cell lines, we observed a dose-dependent decrease in cell proliferation and viability in AR+ cell lines at low concentrations compared to AR− cell lines (Fig. 2a; Suppl. Fig. 2a). There was an approximately 5-fold difference in the LD50 values between AR+ versus AR− PC cell lines (Fig. 2b; Suppl. Fig. 2c), indicating that the response to AR antagonists in PC cell lines is AR dependent.

Evaluation of androgen receptor antagonists response in BC lines: a all the prostate cancer cell lines were seeded at 4000 cells/well on black-colored clear bottomed 96-well plates [LNCaP and LAPC-4, red lines AR(+) and PC-3 and DU-145, blue lines AR(− or low)]. 24 h after plating, the cells were treated with AR antagonists enzalutamide or bicalutamide for 4 days at concentrations ranging from 0.1 to 100 µM and cell viability was measured by cell titer blue reagent. The results were plotted as percent cell viability compared to vehicle control versus the concentration of the drug. Error bars represent mean ± SEM. b The IC50 values of enzalutamide and bicalutamide in prostate cancer cell lines were calculated using Prism software (version 6.05 GraphPad Software, Inc.) by non-linear regression analysis. The lethal dose LD50 was calculated as described in the “Materials and methods” section. Red bars AR(+) LNCaP and LAPC-4. Blue bars AR(−/low) PC-3 and DU-145. (*) DU-145 cell line did not reach 50 % inhibition, hence its LD50 value is >100 µM. c All the breast cancer cell lines were seeded at 4000 cells/well on black colored clear bottomed 96-well plates. Red lines AR(+) BC cell lines CAL-148, ZR-75-1, BT-474, and MFM-223. Blue lines AR(−/low) BC cell lines BT-20, MDA-MB-468, and SUM-159PT. 24 h after plating, the cells were treated with AR antagonists enzalutamide or bicalutamide for 4 days at concentrations from 0.1 to 100 µM and cell number and viability was measured by cell titer blue reagent. The results were plotted as percent cell viability compared to vehicle control versus the concentration of the drug. Error bars represent mean ± SEM. d The IC50 values of enzalutamide and bicalutamide in breast cancer cell lines were calculated using Prism software (version 6.05 GraphPad Software, Inc.) by non-linear regression analysis. The lethal dose LD50 was calculated as described in the “Materials and methods” section. Red bars AR(+), blue bars AR(− or low)]
Surprisingly, in BC cell lines, we did not observe a similar difference in the LD50 values of AR antagonists in AR+ versus AR− lines (Fig. 2d; Suppl. Fig. 2c). There was decrease in cell proliferation in BC cell lines with AR antagonists regardless of their AR protein expression (Fig. 2c; Suppl. Fig. 2b). These results suggest that the effect of AR antagonists in BC cell lines can be AR-independent. Therefore, we concentrated on AR agonists as drug of choice for further experiments in TNBC lines (Fig. 7a).
Treatment of TNBC with AR and VDR agonists in combination with chemotherapy
We previously showed that nearly two-thirds of TNBC have HR1-v (45 %) or HR2-av (18 %) phenotype, i.e., these tumors are negative for ER, PR, and HER2, but positive for VDR (HR1-v) or both AR and VDR (HR2-av) [1]. These TNBCs are currently treated with chemotherapeutic agents such as Taxol or cisplatin.
We found that single agent VDR agonist (calcitriol) can reduce proliferation of both HR1-v and HR2-av TNBC cell lines (Fig. 1c), and combination of calcitriol with Taxol resulted in an additive or synergistic decrease in cell proliferation and viability in two different HR1-v TNBC lines (Fig. 3a) using Bliss independence criterion approach [36–38]. Additionally, similar results were observed when the combination of calcitriol and Taxol was tested in two TNBC lines, MFM-223, and CAL-148 (Suppl. Fig. 3).

Evaluation of AR- and VDR-targeted therapy in combination with chemotherapy in TNBC: the effect on cell viability was measured after 8 days of treatment with various drugs listed in a–d by counting viable cells after 0.1 % Trypan blue staining using Cellometer in all experiments. Whether the combination of drugs resulted in additive, synergistic, or antagonistic interactions was determined by Bliss independent criterion approach, explained in detail in the supplemental methods. Y-axis represents percentage viability of the cells after treatment compared to vehicle control. Error bars represent mean ± SEM. Cal calcitriol, DHT dihydrotestosterone, Cis cisplatin, Tax paclitaxel. ‡ represents additive effect on the combination of drugs and § represents additive or synergistic effect. a VDR-targeted therapy in combination with paclitaxel (Taxol) in TNBC HR1-v cell lines. Two VDR(+) TNBC lines BT-20 and SUM-159PT were treated with vehicle (gray bar), calcitriol (black bars 10 nM) or Taxol (white bar BT-20: 1 nM, SUM-159PT: 0.5 nM) or a combination of calcitriol and Taxol (hatched bar) for 6 days with media change with fresh drug every 3 days. b Combination of AR- and VDR-targeted therapy in TNBC HR2-av cell lines. MFM-223 and CAL-148 cell lines were treated with DHT (black bar 10 nM), calcitriol (white bar MFM-223: 25 nM and CAL-148: 10 nM), or combination of DHT and calcitriol (hatched bars). c Combination of AR- and VDR-targeted therapy with paclitaxel in TNBC HR2-av cell lines. MFM-223 and CAL-148 cell lines were treated with DHT (black bar 10 nM), Taxol (white bar 0.5 nM), calcitriol (single hatched bar MFM-223: 25 nM and CAL-148: 10 nM), or combination of DHT + calcitriol + Taxol (double hatched black bar). d Combination of AR- and VDR-targeted therapy with cisplatin in TNBC HR2-av cell lines. MFM-223 and CAL-148 cell lines were treated with DHT (black bar 10 nM), cisplatin (white bar 10 nM), calcitriol (single hatched bar MFM-223: 25 nM and CAL-148: 10 nM), or combination of DHT + calcitriol + cisplatin (double hatched black bar)
In HR2-av TNBC cell lines, we found that combination of AR and VDR agonists had an additive inhibitory effect in two different HR2-av TNBC cell lines (Fig. 3b). Next, we examined combining AR- and VDR-targeted therapy with chemotherapy, and found that combination of AR (DHT)- and VDR (calcitriol)-targeted therapy with Taxol or cisplatin has an additive effect in reducing cell viability (Fig. 3c, d).
It is worth pointing out that in order to demonstrate additivity in these experiments, we used doses lower than IC50 for each drug (Suppl. Fig. 4). Therefore, it is possible to achieve a greater reduction in cell numbers and viability, close to 100 % cell death, when these drugs are combined between IC50 and IC90 dose range.
The mechanism of tumor cell inhibition by AR- and VDR-targeted hormones
The VDR and AR agonists have been shown to have cell-context dependent pleiotropic effects on cell cycle, apoptosis, autophagy, or differentiation depending on the cell type and dose [13, 14, 25, 39–42]. Therefore, we examined these potential mechanisms in HR2-av cell lines MFM-223 and CAL-148.
There was an increase in G1 phase of the cell cycle in MFM-223 with both DHT and calcitriol individually, and their combination resulted in additive G0/G1 cell cycle arrest (Fig. 4a). In contrast, we did not observe a change in the cell cycle profile of CAL-148 with VDR or AR agonists (Fig. 4b).

Analysis of cell cycle and apoptosis after AR and/or VDR therapy: a, b the effect of DHT or calcitriol treatment on cell cycle was analyzed in TNBC cell lines MFM-223 (a) and CAL-148 (b) with DHT (10 nM, black bars) or calcitriol (10 or 25 nM, white bars) or DHT + calcitriol (hatched bars) after 4 days. After BrdU incorporation, cells were fixed and stained with 2 µg anti-BrdU FITC antibody and 50 µg/ml propidium iodide solution and evaluated by flow cytometric analysis. Y-axis represents percentage population of cells in a given phase (G1, S or G2/M) of cell cycle. Error bars represent mean ± SEM. Statistical analysis was carried out compared to the control group. *P < 0.05, **P < 0.01. c The effect of DHT or calcitriol treatment on apoptosis was analyzed in TNBC cell lines MFM-223 and CAL-148 that were treated with DHT (10 nM, black bars) and calcitriol (10 or 25 nM, white bars) alone or in combination (hatched bars) for 4 days. A positive control with 10 µM etoposide treatment was included in the experiments (dotted bar). All the cells (attached and floating) were collected, double stained with Annexin V and propidium iodide to identify apoptotic cells. Y-axis represents the combined early and late apoptotic cell population. The error bars represent mean ± SEM. Statistical analysis was carried out compared to the control group. *P < 0.05, **P < 0.01. d PARP cleavage as a measure of apoptosis was measured with Western blots in HR2-av TNBC cell lines treated with DHT or calcitriol. MFM-223 and CAL-148 treated as described in panel c and were harvested with RIPA buffer. 30 µg Protein was loaded in each well 7.5 % SDS-PAGE gel; after gel transfer on to nitrocellulose membrane, they were probed with anti-PARP antibody (1:1000, Cell Signaling 9542). β-Tubulin was used as a loading control
An increase in apoptosis was observed in CAL-148 with combination of DHT and calcitriol evaluated by Annexin V/PI co-staining (Fig. 4c) as well as PARP cleavage (Fig. 4d). Importantly, the apoptotic effects of DHT and calcitriol were minimal when they were used alone, consistent with an additive effect. In contrast, there was no change in apoptosis of MFM-223 cells with either hormone alone or in combination (Fig. 4c, d). Since many chemotherapeutic agents induce G2/M arrest followed by apoptosis [43, 44], we examined whether combining DHT and calcitriol with cisplatin in MFM-223 and CAL-148 would interfere with chemotherapy-induced G2/M arrest and apoptosis. We also found that co-treatment with AR/VDR agonists plus chemotherapy resulted in either no change or additive increase in G2/M arrest and apoptosis in MFM-223 and CAL-148 (Suppl. Fig. 5).
Lastly, we found that autophagy did not change with co-treatment of MFM-223 and CAL-148 cell lines with AR and VDR (DHT and Cal), measured by FACS using Cyto-ID® fluorescent dye or by western blots for autophagy marker LC3B (Suppl. Fig. 6).
Cumulatively, these results indicate that AR and VDR stimulation can additively inhibit proliferation of BC cells through cell cycle arrest or apoptosis depending on the cellular context, suggesting a rationale for their combined use. Furthermore, combining both hormones with chemotherapy can additively increase apoptosis in cancer cell lines.
AR- and VDR-targeted hormones can inhibit cancer stem cell phenotype
Both androgens and Vit-D are well-known regulators of cellular differentiation [14, 40–42, 45–49]. Therefore, we examined whether they also change the differentiation state, CSC marker expression and tumorsphere formation efficiency (TFE) of BC cell lines. We found that calcitriol significantly inhibited TFE in both MFM-223 and CAL-148 cell lines and DHT inhibited TFE in CAL-148 (Fig. 5a; Suppl. Fig. 7). No further decrease in TFE was observed with co-treatment with both hormones at the doses we tested.

Effect of AR and/or VDR therapy on cancer stem cell and differentiation phenotype: a the effect of DHT and calcitriol on tumorsphere formation in TNBC HR2-av cell lines MFM-223 and CAL-148. The cells were trypsinized and single cell suspensions were plated into six-well low adhesion plates in 4-ml sphere media (seeding: MFM-223 10,000 and CAL-148 6000) containing DHT (10 nM, black bars) or calcitriol (MFM-223 25 nM and CAL-148 10 nM, white bars), or DHT + calcitriol (hatched bars). After 2 weeks during which spheres are formed from single cells, the plates were stained with iodo-nitrotetrazolium chloride overnight and the spheres were counted using GelCount. Y-axis represents the TFE, tumorsphere formation efficiency (number of spheres formed per the number of cells seeded in a well) represented as fold change in comparison to control group (no treatment, vehicle only). The statistical significance of the drug treatments was determined using two-tailed Student’s T-test. Error bars represent mean ± SEM. *P < 0.05, **P < 0.01. b AldeFluor assay on HR2-av cell lines MFM-223 and CAL-148 after AR and VDR treatment. The cells were treated as described in panel a for 8 days with media change containing fresh drug every 3 days and ALDH+ population was examined by AldeFluor assay kit using FACS profiles representing ALDH+ population before (top) and after (bottom) co-treatment. The graph shows ALDH+ cells plotted as percentage in each treatment group. Gray bar vehicle treatment, black bar DHT, white bar Cal, hatched DHT and Cal combination. Error bars represent mean ± SEM. The statistical significance of the drug treatments was determined using two-tailed Student’s T-test. *P < 0.05, **P < 0.01. c, d Western blot analysis of differentiation and cancer stem cell markers in HR2-av cell lines MFM-223 and CAL-148 after DHT and calcitriol treatment. The cells were treated with DHT (10 nM) and/or calcitriol (MFM-223: 25 nM and CAL-148: 10 nM) for 8 days with media including drug every 3 days. The cell lysates with 30 µg protein were loaded in each lane of a 4–15 % SDS-PAGE gel and after transfer to nitrocellulose membrane, it was probed with the following antibodies at appropriate dilutions: E-cadherin (BD Biosciences 61081), vimentin (Sigma 5255), cytokeratin 5 (Abcam ab75869), cytokeratin 18 (Thermofisher Scientific MS-142-P), Claudin-4 (Invitrogen 32-9400), ALDH1A1 (Cell Signalling mAb12035), CD326 (Abcam ab32392), CD49f (ABD Serotec MCA1457), CD133 (Sigma C9493), CD24 (Santa Cruz SC-53660), CD166 (Abcam ab49496), Nanog (Abcam ab109250), and Musashi (Abcam ab52865). β-Actin or β-tubulin were used as loading controls. e mRNA expression of stem cell markers. MFM-223 cells were treated with 10 nM DHT and/or 25 nM calcitriol for 96 h. The mRNA was quantified using a PCR-based array (Qiagen). The expression level of each gene was determined by comparing the treatment to vehicle control, and genes with more than 2-fold change are presented here in a Heatmap generated using conditional formatting in Microsoft Excel. Scale bar red represents high expression, green represents low expression
High aldehyde dehydrogenase (ALDH) activity is one of the features of CSCs, which is measured by AldeFluor assay [17, 50]. Consistent with the TFE results, we found that treatment with AR or VDR agonists decreased ALDH+ cells additively resulting in 5–100-fold decrease in the frequency of ALDH+ cells with co-treatment of HR2-av TNBC MFM-223 and CAL-148 cell lines (Fig. 5b; Suppl. Fig. 8).
Next, we examined other CSC-associated markers after treatment with AR or VDR agonists alone or in combination; we found that CD49f, Musashi, CD133, and CD326 are down-regulated with combination treatment (Fig. 5c). In addition, we observed an increase in differentiation markers Claudin-4 and cytokeratin 18 and down-regulation of cytokeratin 5 (MFM-223) as well as reciprocal down-regulation of vimentin and up-regulation of cytokeratin 18 (CAL-148) consistent with more differentiated epithelial phenotype (Fig. 5d; Suppl. Fig. 9).
Lastly, we used a PCR-based CSC pathway array that includes 84 common CSC-associated genes and found a 2–5-fold decrease in the mRNA expression of CD44, SOX2, MME (CD10), ALDH1A1, and PPAR-gamma with AR and VDR hormone co-treatment compared to control (Fig. 5e).
Cumulatively, these results indicate that in addition to increasing apoptosis and cell cycle arrest, AR and VDR agonists also inhibit CSC phenotype and induce differentiation in HR2-av TNBC cell lines.
Synthetic AR and VDR ligands can be used to treat HR2-av TNBC cell lines
One concern with using natural AR agonists in the clinic is the potential virilizing side effects and aromatization to estrogens, which can be overcome by a new class of drugs, known as non-steroidal selective AR modulators (SARMs). We selected to test SARM compound enobosarm (GTx-024), since it is currently being tested in clinical trials [51]. We found that similar to AR agonists DHT and R1881, GTx-024 increased proliferation of AR+ PC cell line LNCaP, and inhibited proliferation of AR+ BC cell lines. This effect was specific to AR because GTx-024 had no effect on AR− PC and BC cell lines (Fig. 6a).

Selective androgen receptor modulators and VDR analogs alone or in combination results in anti-proliferative effects: for all the panels a–e, the Y-axis represents percentage viability of the cells after drug treatment compared to vehicle control. Error bars represent mean ± SEM. The statistical significance was validated by two-tailed Students T-test compared to vehicle control. *P < 0.05, **P < 0.01. The combination of drugs resulted in additive, synergistic or antagonistic was determined by Bliss independent criterion approach, explained in detail in the supplemental methods. ‡ Represents additive effect on the combination of drugs and § represents additive or synergistic effect. a Selective androgen receptor modulator (SARM) treatment in breast and prostate cancer cell lines. All the cell lines [black AR(+) and hatched AR(− or low)] were treated with SARM compound GTx-024 at 100 nM concentration for 8–10 days with media change every 3 days. The cells were then trypsinized, stained with 0.1 % Trypan blue and viable cells were counted using Cellometer. b Comparison of VDR agonist analog EB1089 and calcitriol treatment in TNBC cell lines. Selected TNBC cell lines that express VDR (BT-20, SUM-159PT, MFM-223 and CAL-148) were treated with either 5 nM calcitriol (gray bar) or 5 nM EB1089 (black bar) for 6 days with media change every 3 days. The viable cells were counted after staining with 0.1 % Trypan blue. c Combination of VDR agonist analog EB1089 with chemotherapy drug paclitaxel in TNBC HR1-v cells lines. BT-20 and SUM-159PT cell lines were treated with EB1089 and/or paclitaxel for 6 days and viable cells were counted using Cellometer. EB1089 (black bars BT-20 1 nM, SUM-159PT 2.5 nM), Taxol (white bars BT-20 1 nM, SUM-159PT 0.5 nM), combination of EB1089 + Taxol (hatched bars). d Combination of SARM compound GTx-024 with VDR analog EB1089 in HR2-av TNBC cell line MFM-223. Gray bar vehicle, black bar GTx-024 (100 nM), white bar EB1089 (0.5 nM), hatched bar combination of GTx-024 and EB1089. e Triple combination of SARM GTx-024, VDR analog EB1089 and Taxol in HR2-av TNBC cell line MFM-223. Gray bar vehicle, black bar GTx-024 (100 nM), white bar Taxol (0.75 nM), single hatched bar EB1089 (0.5 nM), double hatched black bar combination of all the three drugs
The natural Vit-D ligands can cause hypercalcemia in patients, which prevents achieving clinically effective anti-tumor activity. However, over 1500 Vit-D analogs have been synthesized which may potentially have low calcemic effect [52–54]. We selected seocalcitol (EB1089), since its affinity to VDR is similar to natural ligand calcitriol, yet it has a 50–200-fold higher anti-proliferative activity and reduced hypercalcemic effect [55, 56]. We found that treatment of HR1-v and HR2-av cell lines with EB1089 resulted in a dose-dependent decrease in cell viability. Moreover, as reported before, EB1089 treatment was more potent than calcitriol at low doses (Fig. 6b; Suppl. Fig. 10a).
Next we examined combining synthetic VDR agonist EB1089 with chemotherapy in HR1-v BC cell lines, and found that there is an additive decrease in cell viability when EB1089 is combined with cisplatin or Taxol (Fig. 6c; Suppl. Fig. 10b). Furthermore, combining GTx-024 and EB1089 with or without Taxol produced additive inhibition of cell viability of the TNBC cell lines (Fig. 6d, e; Suppl. Fig. 10c, d). Cumulatively, these results suggest that AR- and VDR-targeted hormone therapy should be considered in the clinic using synthetic AR and VDR ligands with lower virilizing and calcemic side effects.
Discussion
In brief, our study demonstrates that two different hormones that activate AR and VDR can be used alone, in combination with each other as well as with chemotherapy to inhibit proliferation of TNBC cell lines by increased apoptosis and G1/S arrest. In addition, these hormones inhibit TNBC CSC phenotype and induce differentiation. These observations raise the possibility of targeting approximately two-thirds of the TNBCs [ 1] with AR and/or VDR HR-targeted therapies.
Vitamin D receptor: its role in breast cancer treatment
VDR is expressed in around 90 % of the breast tumors [57]. Of the six natural Vit-D compounds, calcitriol is the most active and stable form of Vit-D [52]. Several clinical trials have been conducted with calcitriol as a single agent in breast tumors, with the conclusion that the therapeutic dose required to induce anti-tumor activity is difficult to achieve due to hypercalcemia, renal stones [52, 58, 59], and vascular calcification [60]. Therefore, VDR agonists with low calcemic effects are needed to translate our findings to the clinic. Only a few of the more than 1500 Vit-D analogs have been tested in vivo so far [52–54]. Seocalcitol (EB1089) which we used in our study was tested in a Phase-I clinical trial and was found to be well tolerated and had some activity in BC patients [61].
The role of androgen agonists in breast cancer treatment
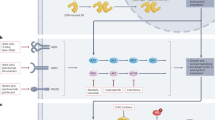
In early 1970s, post-menopausal women with breast cancer were treated with testosterone with favorable responses [62–65]. However, conversion of testosterone into estrogens in tissues [66, 67], virilizing side effects, and development of effective anti-ER therapies as an alternative have slowed further development of AR-targeted therapy for BC [68]. An additional challenge was the seemingly discrepant response to AR agonists in different cell lines [22, 25–27, 69, 70]. However, a recent review of ER+ BC cell lines suggest that while experimental variations in cell density, serum, drugs, and cell counting methods contributed to conflicting results, in the studies that reported a response to AR agonists (n = 33), the majority (78 %) showed that AR activation results in inhibition of cell proliferation in ER+ BC lines [68]. In ER−/HER2+ BC, it was shown that the growth inhibitory effect of AR agonists is switched into growth stimulation by the cross-talk between HER2 and AR [10, 34, 35]. But, this switch only occurs in ER− cells, i.e., AR agonists stimulate proliferation in ER− HER2+ cell lines, but inhibit proliferation in ER+ HER2+ cell lines. In addition to these preclinical data, it was found that high AR expression correlates with low grade and better outcome in BC [71] and we previously reported that AR expression is mutually exclusive with proliferation in human BC sections [1]. We also found that SARMs such as GTx-024 are effective in inhibiting proliferation of BC cell lines and showed clinical activity in a Phase II trial with few virilizing side effects [51, 72–74]. Cumulatively these observations indicate that AR agonists can inhibit proliferation of AR+ BC, except in ER− HER2+ tumors (Fig. 7).

a Summary of androgen receptor response to AR agonists and AR antagonists in TNBC and prostate cancer cell lines. Green arrows represent activation of the receptor, whereas red arrow represents de-activation of the receptor. b Proposed hormone receptor-targeted therapy strategy for triple-negative breast cancers
The role of androgen antagonists in breast cancer treatment
In our study, AR antagonists inhibited proliferation of all BC cell lines regardless of their high, low or negative AR expression status. This is very different than the observations in PC in which there was a >5-fold difference between in the LD50 of AR antagonists in AR+ versus AR− PC cell lines. Importantly, AR-independent inhibition of TNBC cell line proliferation was observed with all three antagonists indicating that this is not an isolated finding. Furthermore, the response of the same TNBC cell lines to the AR agonists DHT and R1881 was AR-specific, which indicates that these TNBC cell lines have a functional AR receptor. At the same time, the response of PC cell lines to AR antagonists was AR dependent ruling out methodological errors. These results indicate that the anti-proliferative effect of AR antagonists in BC cell lines may possibly be due to an off-target effect. Thus, while these drugs have some activity in the clinic, whether this is through AR or some other target remains to be determined. It is worth pointing out that it is already known that some AR antagonists can have non-AR activities; for example, enzalutamide inhibits GABA-A receptors [75], bicalutamide inhibits CYP27A1 [76], and both flutamide and bicalutamide bind to PR [77]. Hence it is possible that these drugs may have activity in cells that are ostensibly negative for AR activity through other genes that are yet to be discovered. Therefore, genetic background of these individual cell lines may play a role in the AR antagonist response in BC cell lines. However, one might have expected the genetic background could affect agonist response equally, which was not the case. The seemingly off-target AR-antagonist effect appears to emerge only in breast cells and not in prostate cells, which may be due to tissue-specific regulation of signaling pathways. For example, at the gene expression level, it was found that AR binds to the PTEN promoter as a repressor, inhibiting its transcription in PC cells. In contrast, AR stimulates PTEN gene expression as an activator in BC cells [78]. Therefore, while surprising, the tissue-specific drug response may not be completely unexpected. Many drugs have unexpected activities due to binding to unknown targets or unknown interactions between the known drug target and other biochemical pathways [79].
Role of hormones in inhibiting CSC population
There is growing evidence that BCs consist of a heterogeneous population of different subtypes of cells including non-CSCs and CSCs that possess the ability of self-renewal [80, 81] and thought to be associated with resistance to the standard therapies and metastasis [82, 83]. Hence, targeting CSCs may be important for complete tumor remission.
We found that hormonal co-stimulation of AR and VDR simultaneously leads to reduction of CSC population, evidenced by a decrease in TFE, decrease in ALDH activity and down-regulation of markers associated with the CSC phenotype. High expression of CD49f regulates pluripotency factors such as OCT4 and SOX2 [84] and Musashi regulates Notch which is one of the key pathways that regulate self-renewal potency of the cells [85, 86]. Therefore, inactivation of CD49f, SOX2, and Notch signaling may provide a mechanism through which AR and VDR treatment can decrease CSC population.
In summary, we show that co-targeting AR and VDR with agonist hormones can be an effective strategy to target CSCs. Our results also suggest that the selection of AR agonists in the treatment of BC will depend on ER and HER2 status, and combination of AR and VDR agonists can be additive with chemotherapy.
References
Santagata S, Thakkar A, Ergonul A, Wang B, Woo T, Hu R, Harrell JC, McNamara G, Schwede M, Culhane AC, Kindelberger D, Rodig S, Richardson A, Schnitt SJ, Tamimi RM, Ince TA (2014) Taxonomy of breast cancer based on normal cell phenotype predicts outcome. J Clin Investig 124(2):859–870. doi:10.1172/JCI70941
Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P, Narod SA (2007) Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res Off J Am Assoc Cancer Res 13(15 Pt 1):4429–4434. doi:10.1158/1078-0432.CCR-06-3045
Haffty BG, Yang Q, Reiss M, Kearney T, Higgins SA, Weidhaas J, Harris L, Hait W, Toppmeyer D (2006) Locoregional relapse and distant metastasis in conservatively managed triple negative early-stage breast cancer. J Clin Oncol Off J Am Soc Clin Oncol 24(36):5652–5657. doi:10.1200/JCO.2006.06.5664
Ma CX, Sanchez CG, Ellis MJ (2009) Predicting endocrine therapy responsiveness in breast cancer. Oncology 23(2):133–142
Dowsett M, Cuzick J, Ingle J, Coates A, Forbes J, Bliss J, Buyse M, Baum M, Buzdar A, Colleoni M, Coombes C, Snowdon C, Gnant M, Jakesz R, Kaufmann M, Boccardo F, Godwin J, Davies C, Peto R (2010) Meta-analysis of breast cancer outcomes in adjuvant trials of aromatase inhibitors versus tamoxifen. J Clin Oncol Off J Am Soc Clin Oncol 28(3):509–518. doi:10.1200/JCO.2009.23.1274
Early Breast Cancer Trialists’ Collaborative G (2005) Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet 365(9472):1687–1717. doi:10.1016/S0140-6736(05)66544-0
Dontu G, Ince TA (2015) Of mice and women: a comparative tissue biology perspective of breast stem cells and differentiation. J Mammary Gland Biol Neoplasia 20(1–2):51–62. doi:10.1007/s10911-015-9341-4
Harrell JC, Prat A, Parker JS, Fan C, He X, Carey L, Anders C, Ewend M, Perou CM (2012) Genomic analysis identifies unique signatures predictive of brain, lung, and liver relapse. Breast Cancer Res Treat 132(2):523–535. doi:10.1007/s10549-011-1619-7
Parise CA, Caggiano V (2014) Breast cancer survival defined by the ER/PR/HER2 subtypes and a surrogate classification according to tumor grade and immunohistochemical biomarkers. J Cancer Epidemiol 2014:469251. doi:10.1155/2014/469251
Ni M, Chen Y, Lim E, Wimberly H, Bailey ST, Imai Y, Rimm DL, Liu XS, Brown M (2011) Targeting androgen receptor in estrogen receptor-negative breast cancer. Cancer Cell 20(1):119–131. doi:10.1016/j.ccr.2011.05.026
Garay JP, Karakas B, Abukhdeir AM, Cosgrove DP, Gustin JP, Higgins MJ, Konishi H, Konishi Y, Lauring J, Mohseni M, Wang GM, Jelovac D, Weeraratna A, Sherman Baust CA, Morin PJ, Toubaji A, Meeker A, De Marzo AM, Lewis G, Subhawong A, Argani P, Park BH (2012) The growth response to androgen receptor signaling in ERalpha-negative human breast cells is dependent on p21 and mediated by MAPK activation. Breast Cancer Res 14(1):R27. doi:10.1186/bcr3112
Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA (2011) Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Investig 121(7):2750–2767. doi:10.1172/JCI45014
Flanagan L, Packman K, Juba B, O’Neill S, Tenniswood M, Welsh J (2003) Efficacy of Vitamin D compounds to modulate estrogen receptor negative breast cancer growth and invasion. J Steroid Biochem Mol Biol 84(2–3):181–192
Pendas-Franco N, Gonzalez-Sancho JM, Suarez Y, Aguilera O, Steinmeyer A, Gamallo C, Berciano MT, Lafarga M, Munoz A (2007) Vitamin D regulates the phenotype of human breast cancer cells. Differ Res Biol Divers 75(3):193–207. doi:10.1111/j.1432-0436.2006.00131.x
Wang B, Lee CW, Witt A, Thakkar A, Ince TA (2015) Heat shock factor 1 induces cancer stem cell phenotype in breast cancer cell lines. Breast Cancer Res Treat 153(1):57–66. doi:10.1007/s10549-015-3521-1
Ince TA, Sousa AD, Jones MA, Harrell JC, Agoston ES, Krohn M, Selfors LM, Liu W, Chen K, Yong M, Buchwald P, Wang B, Hale KS, Cohick E, Sergent P, Witt A, Kozhekbaeva Z, Gao S, Agoston AT, Merritt MA, Foster R, Rueda BR, Crum CP, Brugge JS, Mills GB (2015) Characterization of twenty-five ovarian tumour cell lines that phenocopy primary tumours. Nat Commun 6:7419. doi:10.1038/ncomms8419
Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, Schott A, Hayes D, Birnbaum D, Wicha MS, Dontu G (2007) ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 1(5):555–567. doi:10.1016/j.stem.2007.08.014
Freedman DM, Chang SC, Falk RT, Purdue MP, Huang WY, McCarty CA, Hollis BW, Graubard BI, Berg CD, Ziegler RG (2008) Serum levels of vitamin D metabolites and breast cancer risk in the prostate, lung, colorectal, and ovarian cancer screening trial. Cancer Epidemiol Biomark Prev Publ Am Assoc Cancer Res Cospons Am Soc Prev Oncol 17(4):889–894. doi:10.1158/1055-9965.EPI-07-2594
Goldenberg IS (1964) Testosterone propionate therapy in breast cancer. JAMA 188:1069–1072
Gordan GS, Graham WP III, Goldman L, Papac R, Sheline GE, Vaeth J, Witt J (1963) Hormonal treatment of disseminated cancer of the female breast. Calif Med 98:189–194
Allegra JC, Lippman ME, Thompson EB, Simon R, Barlock A, Green L, Huff KK, Do HM, Aitken SC (1979) Distribution, frequency, and quantitative analysis of estrogen, progesterone, androgen, and glucocorticoid receptors in human breast cancer. Cancer Res 39(5):1447–1454
Cops EJ, Bianco-Miotto T, Moore NL, Clarke CL, Birrell SN, Butler LM, Tilley WD (2008) Antiproliferative actions of the synthetic androgen, mibolerone, in breast cancer cells are mediated by both androgen and progesterone receptors. J Steroid Biochem Mol Biol 110(3–5):236–243. doi:10.1016/j.jsbmb.2007.10.014
Dauvois S, Geng CS, Levesque C, Merand Y, Labrie F (1991) Additive inhibitory effects of an androgen and the antiestrogen EM-170 on estradiol-stimulated growth of human ZR-75-1 breast tumors in athymic mice. Cancer Res 51(12):3131–3135
de Launoit Y, Dauvois S, Dufour M, Simard J, Labrie F (1991) Inhibition of cell cycle kinetics and proliferation by the androgen 5 alpha-dihydrotestosterone and antiestrogen N, n-butyl-N-methyl-11-[16′ alpha-chloro-3′,17 beta-dihydroxy-estra-1′,3′,5′-(10′)triene-7′ alpha-yl] undecanamide in human breast cancer ZR-75-1 cells. Cancer Res 51(11):2797–2802
Greeve MA, Allan RK, Harvey JM, Bentel JM (2004) Inhibition of MCF-7 breast cancer cell proliferation by 5alpha-dihydrotestosterone; a role for p21(Cip1/Waf1). J Mol Endocrinol 32(3):793–810
Macedo LF, Guo Z, Tilghman SL, Sabnis GJ, Qiu Y, Brodie A (2006) Role of androgens on MCF-7 breast cancer cell growth and on the inhibitory effect of letrozole. Cancer Res 66(15):7775–7782. doi:10.1158/0008-5472.CAN-05-3984
Ortmann J, Prifti S, Bohlmann MK, Rehberger-Schneider S, Strowitzki T, Rabe T (2002) Testosterone and 5 alpha-dihydrotestosterone inhibit in vitro growth of human breast cancer cell lines. Gynecol Endocrinol Off J Int Soc Gynecol Endocrinol 16(2):113–120
Poulin R, Baker D, Labrie F (1988) Androgens inhibit basal and estrogen-induced cell proliferation in the ZR-75-1 human breast cancer cell line. Breast Cancer Res Treat 12(2):213–225
Reese CC, Warshaw ML, Murai JT, Siiteri PK (1988) Alternative models for estrogen and androgen regulation of human breast cancer cell (T47D) growth. Ann NY Acad Sci 538:112–121
Sutherland RL, Hall RE, Pang GY, Musgrove EA, Clarke CL (1988) Effect of medroxyprogesterone acetate on proliferation and cell cycle kinetics of human mammary carcinoma cells. Cancer Res 48(18):5084–5091
Anestis A, Karamouzis MV, Dalagiorgou G, Papavassiliou AG (2015) Is androgen receptor targeting an emerging treatment strategy for triple negative breast cancer? Cancer Treat Rev 41(6):547–553. doi:10.1016/j.ctrv.2015.04.009
Barton VN, D’Amato NC, Gordon MA, Lind HT, Spoelstra NS, Babbs BL, Heinz RE, Elias A, Jedlicka P, Jacobsen BM, Richer JK (2015) Multiple molecular subtypes of triple-negative breast cancer critically rely on androgen receptor and respond to enzalutamide in vivo. Mol Cancer Ther 14(3):769–778. doi:10.1158/1535-7163.MCT-14-0926
Cochrane DR, Bernales S, Jacobsen BM, Cittelly DM, Howe EN, D’Amato NC, Spoelstra NS, Edgerton SM, Jean A, Guerrero J, Gomez F, Medicherla S, Alfaro IE, McCullagh E, Jedlicka P, Torkko KC, Thor AD, Elias AD, Protter AA, Richer JK (2014) Role of the androgen receptor in breast cancer and preclinical analysis of enzalutamide. Breast Cancer Res 16(1):R7. doi:10.1186/bcr3599
Ni M, Chen Y, Fei T, Li D, Lim E, Liu XS, Brown M (2013) Amplitude modulation of androgen signaling by c-MYC. Genes Dev 27(7):734–748. doi:10.1101/gad.209569.112
Robinson JL, Macarthur S, Ross-Innes CS, Tilley WD, Neal DE, Mills IG, Carroll JS (2011) Androgen receptor driven transcription in molecular apocrine breast cancer is mediated by FoxA1. EMBO J 30(15):3019–3027. doi:10.1038/emboj.2011.216
Bliss CI (1939) The toxicity of poisons applied jointly. Ann Appl Biol 26:585–615
Greco WR, Bravo G, Parsons JC (1995) The search for synergy: a critical review from a response surface perspective. Pharmacol Rev 47(2):331–385
Goldoni M, Johansson C (2007) A mathematical approach to study combined effects of toxicants in vitro: evaluation of the Bliss independence criterion and the Loewe additivity model. Toxicol In Vitro 21(5):759–769
Kandouz M, Lombet A, Perrot JY, Jacob D, Carvajal S, Kazem A, Rostene W, Therwath A, Gompel A (1999) Proapoptotic effects of antiestrogens, progestins and androgen in breast cancer cells. J Steroid Biochem Mol Biol 69(1–6):463–471
Wahler J, So JY, Cheng LC, Maehr H, Uskokovic M, Suh N (2015) Vitamin D compounds reduce mammosphere formation and decrease expression of putative stem cell markers in breast cancer. J Steroid Biochem Mol Biol 148:148–155. doi:10.1016/j.jsbmb.2014.10.016
Baratta M, Grolli S, Poletti A, Ramoni R, Motta M, Tamanini C (2000) Role of androgens in proliferation and differentiation of mouse mammary epithelial cell line HC11. J Endocrinol 167(1):53–60
Buras RR, Schumaker LM, Davoodi F, Brenner RV, Shabahang M, Nauta RJ, Evans SR (1994) Vitamin D receptors in breast cancer cells. Breast Cancer Res Treat 31(2–3):191–202
Siddik ZH (2003) Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene 22(47):7265–7279. doi:10.1038/sj.onc.1206933
Yeung TK, Germond C, Chen X, Wang Z (1999) The mode of action of Taxol: apoptosis at low concentration and necrosis at high concentration. Biochem Biophys Res Commun 263(2):398–404. doi:10.1006/bbrc.1999.1375
Heer R, Robson CN, Shenton BK, Leung HY (2007) The role of androgen in determining differentiation and regulation of androgen receptor expression in the human prostatic epithelium transient amplifying population. J Cell Physiol 212(3):572–578. doi:10.1002/jcp.21154
Welsh J, Wietzke JA, Zinser GM, Byrne B, Smith K, Narvaez CJ (2003) Vitamin D-3 receptor as a target for breast cancer prevention. J Nutr 133(7 Suppl):2425S–2433S
Jeong Y, Swami S, Krishnan AV, Williams JD, Martin S, Horst RL, Albertelli MA, Feldman BJ, Feldman D, Diehn M (2015) Inhibition of mouse breast tumor-initiating cells by calcitriol and dietary vitamin D. Mol Cancer Ther 14(8):1951–1961. doi:10.1158/1535-7163.MCT-15-0066
Pervin S, Hewison M, Braga M, Tran L, Chun R, Karam A, Chaudhuri G, Norris K, Singh R (2013) Down-regulation of vitamin D receptor in mammospheres: implications for vitamin D resistance in breast cancer and potential for combination therapy. PLoS One 8(1):e53287. doi:10.1371/journal.pone.0053287
So JY, Lee HJ, Smolarek AK, Paul S, Wang CX, Maehr H, Uskokovic M, Zheng X, Conney AH, Cai L, Liu F, Suh N (2011) A novel Gemini vitamin D analog represses the expression of a stem cell marker CD44 in breast cancer. Mol Pharmacol 79(3):360–367. doi:10.1124/mol.110.068403
Marcato P, Dean CA, Giacomantonio CA, Lee PW (2011) Aldehyde dehydrogenase: its role as a cancer stem cell marker comes down to the specific isoform. Cell Cycle 10(9):1378–1384
Coss CC, Jones A, Dalton JT (2014) Selective androgen receptor modulators as improved androgen therapy for advanced breast cancer. Steroids 90:94–100. doi:10.1016/j.steroids.2014.06.010
Mehta RG, Peng X, Alimirah F, Murillo G, Mehta R (2012) Vitamin D and breast cancer: emerging concepts. Cancer Lett. doi:10.1016/j.canlet.2012.10.034
Jones G (2010) Vitamin D analogs. Endocrinol Metab Clin N Am 39(2):447–472, table of contents. doi:10.1016/j.ecl.2010.02.003
Chiellini G, DeLuca HF (2011) The importance of stereochemistry on the actions of vitamin D. Curr Top Med Chem 11(7):840–859
Hansen CM, Hamberg KJ, Binderup E, Binderup L (2000) Seocalcitol (EB 1089): a vitamin D analogue of anti-cancer potential. Background, design, synthesis, pre-clinical and clinical evaluation. Curr Pharm Des 6(7):803–828
Hansen CM, Maenpaa PH (1997) EB 1089, a novel vitamin D analog with strong antiproliferative and differentiation-inducing effects on target cells. Biochem Pharmacol 54(11):1173–1179
Ditsch N, Toth B, Mayr D, Lenhard M, Gallwas J, Weissenbacher T, Dannecker C, Friese K, Jeschke U (2012) The association between vitamin D receptor expression and prolonged overall survival in breast cancer. J Histochem Cytochem Off J Histochem Soc 60(2):121–129. doi:10.1369/0022155411429155
Gross C, Krishnan AV, Malloy PJ, Eccleshall TR, Zhao XY, Feldman D (1998) The vitamin D receptor gene start codon polymorphism: a functional analysis of FokI variants. J Bone Miner Res Off J Am Soc Bone Miner Res 13(11):1691–1699. doi:10.1359/jbmr.1998.13.11.1691
Krishnan AV, Trump DL, Johnson CS, Feldman D (2010) The role of vitamin D in cancer prevention and treatment. Endocrinol Metab Clin N Am 39(2):401–418, table of contents. doi:10.1016/j.ecl.2010.02.011
Mizobuchi M, Towler D, Slatopolsky E (2009) Vascular calcification: the killer of patients with chronic kidney disease. J Am Soc Nephrol 20(7):1453–1464. doi:10.1681/ASN.2008070692
Gulliford T, English J, Colston KW, Menday P, Moller S, Coombes RC (1998) A phase I study of the vitamin D analogue EB 1089 in patients with advanced breast and colorectal cancer. Br J Cancer 78(1):6–13
Adair FE, Herrmann JB (1946) The use of testosterone propionate in the treatment of advanced carcinoma of the breast. Ann Surg 123(6):1023–1035
Fels E (1944) Treatment of breast cancer with testosterone propionate. J Clin Endocrinol Metab 4(3):121–125. doi:10.1210/jcem-4-3-121
Raynaud A, Lacassagne A (1939) Sur le Mecanisme d’une Action Preventive de la Testosterone sur le Carcinome Mammaire de la Souris. C R Acad Soc Biol Paris 131(586):1939
Ulrich P (1939) Testosterone (hormone male) et son R6le possible dans le Traitment de Certains Cancers du Sein. Unio Int Contra Cancrum 4(4):377
Farrow JH, Woodard HQ (1942) The influence of androgenic and estrogenic substances on the serum calcium: in cases of skeletal metastases from mammary cancer. J Am Med Assoc 118(5):339–343. doi:10.1001/jama.1942.02830050001001
West CD, Damast BL, Sarro SD, Pearson OH (1956) Conversion of testosterone to estrogens in castrated, adrenalectomized human females. J Biol Chem 218(1):409–418
Garay JP, Park BH (2012) Androgen receptor as a targeted therapy for breast cancer. Am J Cancer Res 2(4):434–445
Hackenberg R, Hofmann J, Holzel F, Schulz KD (1988) Stimulatory effects of androgen and antiandrogen on the in vitro proliferation of human mammary carcinoma cells. J Cancer Res Clin Oncol 114(6):593–601
Lippman M, Bolan G, Huff K (1976) The effects of androgens and antiandrogens on hormone-responsive human breast cancer in long-term tissue culture. Cancer Res 36(12):4610–4618
Vera-Badillo FE, Templeton AJ, de Gouveia P, Diaz-Padilla I, Bedard PL, Al-Mubarak M, Seruga B, Tannock IF, Ocana A, Amir E (2014) Androgen receptor expression and outcomes in early breast cancer: a systematic review and meta-analysis. J Natl Cancer Inst 106(1):djt319. doi:10.1093/jnci/djt319
Dobs AS, Boccia RV, Croot CC, Gabrail NY, Dalton JT, Hancock ML, Johnston MA, Steiner MS (2013) Effects of enobosarm on muscle wasting and physical function in patients with cancer: a double-blind, randomised controlled phase 2 trial. Lancet Oncol 14(4):335–345. doi:10.1016/S1470-2045(13)70055-X
Ferriman D, Gallwey JD (1961) Clinical assessment of body hair growth in women. J Clin Endocrinol Metab 21(11):1440–1447. doi:10.1210/jcem-21-11-1440
Narayanan R, Ahn S, Cheney MD, Yepuru M, Miller DD, Steiner MS, Dalton JT (2014) Selective androgen receptor modulators (SARMs) negatively regulate triple-negative breast cancer growth and epithelial:mesenchymal stem cell signaling. PLoS One 9(7):e103202. doi:10.1371/journal.pone.0103202
Foster WR, Car BD, Shi H, Levesque PC, Obermeier MT, Gan J, Arezzo JC, Powlin SS, Dinchuk JE, Balog A, Salvati ME, Attar RM, Gottardis MM (2011) Drug safety is a barrier to the discovery and development of new androgen receptor antagonists. Prostate 71(5):480–488. doi:10.1002/pros.21263
Mast N, Lin JB, Pikuleva IA (2015) Marketed drugs can inhibit cytochrome P450 27A1, a potential new target for breast cancer adjuvant therapy. Mol Pharmacol 88(3):428–436. doi:10.1124/mol.115.099598
Fan F, Hu R, Munzli A, Chen Y, Dunn RT II, Weikl K, Strauch S, Schwandner R, Afshari CA, Hamadeh H, Nioi P (2015) Utilization of human nuclear receptors as an early counter screen for off-target activity: a case study with a compendium of 615 known drugs. Toxicol Sci Off J Soc Toxicol 145(2):283–295. doi:10.1093/toxsci/kfv052
Wang Y, Romigh T, He X, Tan MH, Orloff MS, Silverman RH, Heston WD, Eng C (2011) Differential regulation of PTEN expression by androgen receptor in prostate and breast cancers. Oncogene 30(42):4327–4338. doi:10.1038/onc.2011.144
MacDonald ML, Lamerdin J, Owens S, Keon BH, Bilter GK, Shang Z, Huang Z, Yu H, Dias J, Minami T, Michnick SW, Westwick JK (2006) Identifying off-target effects and hidden phenotypes of drugs in human cells. Nat Chem Biol 2(6):329–337. doi:10.1038/nchembio790
Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, Visvader J, Weissman IL, Wahl GM (2006) Cancer stem cells—perspectives on current status and future directions: AACR workshop on cancer stem cells. Cancer Res 66(19):9339–9344. doi:10.1158/0008-5472.CAN-06-3126
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF (2003) Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA 100(7):3983–3988. doi:10.1073/pnas.0530291100
Alison MR, Lim SM, Nicholson LJ (2011) Cancer stem cells: problems for therapy? J Pathol 223(2):147–161. doi:10.1002/path.2793
Dean M, Fojo T, Bates S (2005) Tumour stem cells and drug resistance. Nat Rev Cancer 5(4):275–284. doi:10.1038/nrc1590
Yu KR, Yang SR, Jung JW, Kim H, Ko K, Han DW, Park SB, Choi SW, Kang SK, Scholer H, Kang KS (2012) CD49f enhances multipotency and maintains stemness through the direct regulation of OCT4 and SOX2. Stem Cells 30(5):876–887. doi:10.1002/stem.1052
Weinmaster G (2000) Notch signal transduction: a real rip and more. Curr Opin Genet Dev 10(4):363–369
Lagadec C, Vlashi E, Frohnen P, Alhiyari Y, Chan M, Pajonk F (2014) The RNA-binding protein Musashi-1 regulates proteasome subunit expression in breast cancer- and glioma-initiating cells. Stem Cells 32(1):135–144. doi:10.1002/stem.1537
Acknowledgments
Research for this article was supported in part by Funding to T. A. Ince from Breast Cancer Research Foundation and Play for P.I.N.K., NIEHS R01-ES024991 and NCI R01-CA146445 from NIH Roadmap Epigenomics Project, as well as Funds from Women’s Cancer Association of UM and Sylvester Comprehensive Cancer Center. We thank Dr. Aurea D. Sousa, Dr. Kerry Burnstein, Dr. Dorraya El-Ashry, and Dr. Joyce Slingerland for helpful discussion and their suggestions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no conflicts of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Thakkar, A., Wang, B., Picon-Ruiz, M. et al. Vitamin D and androgen receptor-targeted therapy for triple-negative breast cancer. Breast Cancer Res Treat 157, 77–90 (2016). https://doi.org/10.1007/s10549-016-3807-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-016-3807-y