
Abstract
Lapatinib, an oral, reversible inhibitor of epidermal growth factor receptor and human epidermal growth factor receptor 2 (HER2) tyrosine kinase, has proven antitumor activity in HER2-positive metastatic breast cancer (MBC). Nanoparticle albumin-bound paclitaxel (nab-paclitaxel) is indicated for the treatment of breast cancer after failure of combination chemotherapy for metastatic disease or relapse within 6 months of adjuvant chemotherapy. This was an open-label, single-arm, multicenter, Phase II study to evaluate the efficacy and safety of nab-paclitaxel plus lapatinib in women with HER2 over-expressing MBC who had received no more than one prior chemotherapeutic regimen. The primary efficacy endpoint was the overall response rate (ORR). This was defined as the percentage of patients having either a complete response (CR) or partial response (PR). Secondary efficacy endpoints included progression-free survival (PFS), overall survival, duration of response (DoR), time to response (TTR), and time to progression (TTP). Investigator-assessed ORR was 53 % (n = 32, 95 % confidence interval (CI): 40.7–66.0) with the majority of patient responses demonstrating a PR (47 %). Four (7 %) patient responses demonstrated a CR, and ten (17 %) a stable disease. The median Kaplan–Meier estimate of investigator-assessed PFS, DoR, TTR, and TTP was 39.7 weeks (95 % CI 34.1–63.9), 48.7 weeks (95 % CI 31.7–57.1), 7.8 weeks (95 % CI 7.4–8.1), and 41 weeks (95 % CI 39.1–64.6), respectively. Lapatinib 1,000 mg with nab-paclitaxel 100 mg/m2 IV is feasible with manageable and predictable toxicity and an ORR of 53 % comparing favorably with other HER2-based combinations in this setting.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Breast cancer is the most commonly diagnosed tumor type in women in the United States with a significant proportion developing metastatic disease [1]. Metastatic breast cancer (MBC) is an incurable disease, and systemic treatment aims to prolong survival, control disease progression, alleviate symptoms, and enhance patient quality of life. Although early detection and improvements in therapy have augmented survival rates in recent years (5-year relative survival is 99 % for localized disease and 84 % for regional disease), the 5-year survival rates for patients with distant-stage disease remains low at 23 % [1].
Control of several cellular processes in breast cancer is dependent on the epidermal growth factor receptor (EGFR, also known as ErbB) family, and members of this family have been proposed as targets in cancer therapy. Amplification of human epidermal growth factor receptor 2 (HER2/ErbB2) leads to increased receptor homo- and hetero-dimerization and subsequent activation of downstream signaling pathways associated with cell proliferation, differentiation, survival, and angiogenesis [2]. Initial studies indicated that 20–25 % of tumors in women with early stage I–III breast cancers were HER2 positive [3, 4]. However, a larger and more recent study from Europe has reported HER2-positive breast cancer rates in the range of 13–19 % [5] which has both prognostic and predictive implications [3, 6].
HER2-positive tumors are associated with particularly aggressive disease and a poor prognosis with shorter overall survival (OS) and disease-free survival intervals [3]. Studies have shown that HER2-positive tumors are more likely to disseminate to major visceral sites as well as the central nervous system (CNS) [7, 8].
Trastuzumab, a monoclonal antibody directed against the HER2 receptor, has clinical activity in patients with HER2-positive breast carcinoma and is widely used in combination with cytotoxic agents [9–11]. A significant number of patients with HER2-positive MBC who are treated with trastuzumab experience symptomatic CNS metastases, potentially due to ineffective penetration of the CNS. Trastuzumab resistance may also develop, limiting trastuzumab’s ability to maintain disease control in the advanced as well as early stage disease settings [12]. As such, alternative therapeutic strategies and combinations need to be developed to target HER2 signaling.
Lapatinib (Tykerb/Tyverb®, GlaxoSmithKline) is an orally active small molecule that inhibits the tyrosine kinase activity of HER2 and EGFR. Lapatinib has shown clinical activity in combination with capecitabine in HER2-positive tumors that progressed while on standard treatment, including trastuzumab [13]. Lapatinib, in combination with capecitabine was approved in 2007 by the U.S. Food and Drug Administration (US FDA) for the treatment of patients with advanced or MBC with HER2-positive tumors who have received prior therapy including an anthracycline, a taxane, and trastuzumab [14]. Lapatinib in combination with letrozole (an aromatase inhibitor) has also been shown to be effective in the treatment of post-menopausal women with hormone receptor-positive, HER2-positive MBC [15]. In 2010, the US FDA granted accelerated approval to lapatinib for use in combination with letrozole for the treatment of patients with HER2-positive MBC and for whom hormonal therapy is indicated [14].
Nanoparticle albumin-bound paclitaxel (nab-paclitaxel; Abraxane® for Injectable Suspension, Celgene Corporation) is a novel Cremophor® EL-free, non-crystalline, amorphous, albumin-bound nanoparticle formulation of paclitaxel suspended in normal saline. It is associated with a lower risk of hypersensitivity as well as an improved toxicity profile, and has no requirements for premedication compared with solvent-based paclitaxel [16]. Furthermore, the neuropathy that frequently accompanies paclitaxel administration was easily managed and resolved more rapidly in patients treated with nab-paclitaxel [16]. Nab-paclitaxel demonstrated significantly longer progression-free survival (PFS), compared with docetaxel, in patients with MBC [17, 18]. Nab-paclitaxel has been approved by the US FDA in 2005 for the treatment of breast cancer after failure of combination chemotherapy for metastatic disease or relapse within 6 months of adjuvant chemotherapy. Prior therapy should have included an anthracycline unless clinically contraindicated [19].
In early as well as advanced HER2-positive breast cancer, synergy between chemotherapy and HER2-directed therapy (such as trastuzumab) has significantly improved outcomes, including survival, compared with chemotherapy alone [10, 11]. However, despite treatment with taxane-based trastuzumab regimens in early stage disease, disease progressions are still evident. The proven activity of lapatinib in this setting (due to its differing mechanism of action) demonstrates that it is a suitable option in HER2-positive MBC. Unlike trastuzumab, it has been suggested that lapatinib can cross the blood–brain barrier, providing a rationale for testing lapatinib in patients with CNS metastases [20, 21]. Studies have also shown that HER3 can be transphosphorylated by p95 HER2 (a truncated version lacking the extracellular domain) in HER2-overexpressing breast cancer cell lines, and that this phosphorylation is inhibited by lapatinib, but not trastuzumab [22]. A more recent study demonstrated an OS benefit with lapatinib in combination with trastuzumab for patients with HER2-positive MBC [23]. Also, owing to the toxicity of solvent-based paclitaxel, evaluation of lapatinib in conjunction with nab-paclitaxel for the treatment of HER2-positive MBC is warranted.
Presented here are the results of a single-arm, multicenter Phase II study undertaken to determine the activity of lapatinib plus nab-paclitaxel in the first- and second-line setting in HER2-positive MBC.
Methods
Study design
This was an open-label, single-arm, multicenter, Phase II study (NCT00709761) to evaluate the efficacy and safety of nab-paclitaxel plus lapatinib in women with HER2-positive MBC who had received no more than one prior chemotherapeutic regimen in the metastatic setting. The planned enrollment was 60 patients with a lead in evaluation of the first 10 patients for safety of this combination. A safety analysis of the first ten enrolled patients treated for at least one cycle of the initial doses of nab-paclitaxel (125 mg/m2 IV on Day 1, 8, and 15 every 28 days) in combination with lapatinib (1,250 mg orally once daily on a continuous basis) in a 4-week cycle for a planned minimum of six cycles was performed. However, during the ongoing safety review of the first five patients, Grade 3 toxicities were observed in all five patients (four with neutropenia and one with neutropenic fever and diarrhea) and the decision was made to reduce the dose of both study drugs. All subsequent patients (n = 55) received nab-paclitaxel (100 mg/m2 IV on Day 1, 8, and 15 every 28 days) in combination with lapatinib (1,000 mg orally once daily on a continuous basis) in a 4-week cycle for a minimum of six cycles. If a complete response (CR) was obtained before six cycles, the patient had to receive two additional cycles of the combination regimen after which the CR was confirmed (a minimum of four cycles). After six cycles, patients continued to receive a daily dose of lapatinib 1,000 mg orally until disease progression or withdrawal from study due to unacceptable toxicity or consent withdrawal. Patients could also continue to receive nab-paclitaxel after six cycles if there was benefit without unacceptable toxicity.
Efficacy assessments were performed every 8 weeks and at the end of investigational treatment. All patients are being followed for survival. Lapatinib treatment delays of up to 2 weeks were permitted for resolution of toxicities, other than in the event of protocol defined liver abnormalities, left ventricular cardiac dysfunction, or interstitial pneumonitis. Only one dose reduction of both lapatinib (to 750 mg) and nab-paclitaxel (to 80 mg/m2) was permitted for related toxicity.
Safety assessments were performed throughout the study and included physical examinations, Eastern Cooperative Oncology Group (ECOG) performance status (PS), vital signs, clinical laboratory evaluations, cardiac monitoring (left ventricular ejection fraction function at 12-week intervals for patients while receiving lapatinib), and recording of adverse events (AEs) and serious AEs (SAEs).
Patient population
Female patients ≥18 years of age with histologically confirmed HER2-positive invasive breast cancer (defined as HER2 positive score [>2.2] by fluorescence in situ hybridization or 3+ amplification by immunohistochemistry) who presented with de novo stage IV disease or had stage IV disease at a relapse after curative-intent surgery were enrolled in the study.
Patients were required to have received no more than one prior chemotherapeutic regimen in the metastatic setting. Prior endocrine therapy or trastuzumab treatment in the neoadjuvant, adjuvant, or metastatic setting was permitted. If a taxane had been previously administered, progression must have occurred ≥12 months after completion of this treatment. Patients were also required to have an ECOG PS of 0 or 1, and measurable disease, according to Response Evaluation Criteria in Solid Tumors version 1.0 guidelines [24]. A cardiac ejection fraction of at least 50 %, as measured by echocardiogram or multigated acquisition scan, within the institutional range of normal was also essential. Patients with stable CNS metastases (stable for at least 3 months) were eligible if they were not taking steroids or enzyme-inducing anticonvulsants. Bisphosphonate therapy for bone metastases was allowed. However, treatment must have been initiated prior to the first dose of study medication. Key exclusion criteria included active cardiac, hepatic, or biliary diseases, concurrent treatment with other anticancer or investigational agents, peripheral neuropathy of Common Terminology Criteria for Adverse Events Grade 2 or greater, and diseases or surgeries affecting gastrointestinal function including malabsorption syndrome, gastric resection, and uncontrolled inflammatory bowel disease.
Study endpoints
The primary efficacy endpoint was overall response rate (ORR), based on confirmed responses from investigator assessment of best overall response. ORR was defined as the percentage of patients having either a CR or partial response (PR). Secondary efficacy endpoints included PFS (defined as time from the start of treatment until the earliest date of disease progression or death due to any cause, if sooner), OS (defined as time from the start of treatment until death due to any cause), duration of response (DoR) (defined for the subset of patients who show a CR or PR—time from first documented evidence of CR or PR until the first documented sign of disease progression or death due to any cause), and time to response (TTR) (defined for the subset of patients who show a CR or PR as time from the start of treatment until first documented evidence of PR or CR, whichever is recorded first). When tumor response was confirmed at a repeat assessment, the TTR was taken to be the first time the response was observed. Time to progression (TTP) (defined as time from the start of treatment until the earliest date of disease progression, or death due to breast cancer, if sooner) was also included as another secondary efficacy endpoint.
Statistical analyses
All efficacy analyses were conducted on the intent-to-treat population, which comprised all patients who received at least one dose of study drug. Exact binomial 95 % confidence interval (CI) for the overall response rate was used. The Kaplan–Meier estimate for the median PFS, OS, DoR, TTR, and TTP were summarized, along with approximate 95 % CI, if there were a sufficient number of events. The estimate of the standard error was computed by Greenwood’s formula [25]. The study was not designed for inference testing. Sample-size considerations were based entirely on feasibility. No statistical hypotheses were tested, and the focus was to estimate the ORR who received no more than 1 prior treatment for HER2-positive MBC. A sample size of 60 was targeted to insure that the lower limit of the 95 % CI exceeded 50 % for the overall response rate.
Results
Patient population
From July 2, 2008 to January 5, 2011, 60 patients were enrolled and treated at 14 centers in the United States. Patient demographics and prior anticancer treatments are summarized in Table 1. The median time from initial diagnosis (any stage) was 28.9 months; 25 % of the patients had previously received one prior regimen in the metastatic setting and 57 % of patients had previously received neo/adjuvant therapy. Disease burden at baseline is shown in Table 2. The most common sites of metastatic disease were lymph nodes, followed by the lung, liver, bone, and breast.
Primary efficacy results: overall tumor response rate
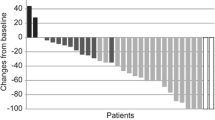
Lapatinib + nab-paclitaxel was efficacious with respect to investigator-assessed ORR (53 % [n = 32, 95 % CI 40.7–66.0]) with the majority of patients having a PR (28; 47 %), four patients (7 %) a CR and ten patients (17 %) showing stable disease (SD) (Table 3). A total of ten patients had unknown responses. These were patients who withdrew on or before the first response assessment time point of 8 weeks with no tumor assessments available with the exception of one patient who withdrew at week 9 due to diarrhea and refused to have the bone scans.
Secondary efficacy results
The median Kaplan–Meier estimate of investigator-assessed PFS was 39.7 weeks (95 % CI 34.1–63.9) (Table 3). As of the data cut-off, 13 patients (22 %) had died. Therefore, the median OS was not reached as the data were not mature (i.e., >75 % of the patients were censored for the endpoint). The median Kaplan–Meier estimate of investigator-assessed TTR and DoR was 7.8 weeks (95 % CI 7.4–8.1) and 48.7 weeks (95 % CI 31.7–57.1) for patients who responded (CR or PR). The Kaplan–Meier estimate for median TTP was 41 weeks (95 % CI 39.1–64.6). TTP was similar to PFS in this study as the majority of PFS events (either progression or death due to any cause) were disease progressions (28 out of 30). Two PFS events were deaths due to causes other than breast cancer and were considered a competing risk for the TTP analysis.
Lapatinib and nab-paclitaxel exposure
The median duration of exposure to lapatinib was 5.6 months (range, 0–19 months). The mean daily lapatinib dose was 893.1 mg. Approximately half of the study population (31 patients; 52 %) completed six cycles of treatment with nab-paclitaxel, which was the duration of treatment specified in the protocol. A total of 13 patients (22 %) continued to participate in the study for further cycles of treatment. In total, 73 lapatinib dose interruptions occurred in 34 patients (57 %) were of short duration (51/73 interruptions were ≤7 days in duration), and were mainly due to non-hematologic toxicities (52 %). Twenty-six reductions in the dose of lapatinib were made in 24 patients (40 %) and were also mainly due to non-hematologic toxicities (85 %). Twenty-four doses of nab-paclitaxel were delayed in 15 patients (25 %), and were most commonly due to non-hematologic toxicity (42 %). Sixteen dose reductions of nab-paclitaxel occurred in 13 patients (22 %) mainly due to hematologic toxicities (56 %). Doses of nab-paclitaxel were missed by 33 patients (55 %) and the majority of these patients missed one or two doses (53 missed doses in total). The most common reason for missing a dose was non-hematologic toxicity (47 %).
Safety
The most commonly reported AEs were diarrhea, fatigue, nausea, and rash (Table 4). The maximum toxicity grade of the majority of frequently reported AEs was Grade 1 or 2. Grade 3 diarrhea was reported at a frequency of 20 %, contributing to a high overall frequency (62 %) of AEs with a maximum toxicity Grade of 3. AEs of neutropenia also reached a maximum toxicity Grade of 3 for 22 % of patients. A total of five patients (8 %) experienced Grade 4 events (diarrhea, nausea, fatigue, febrile neutropenia, and hypokalemia) and all these AEs were considered treatment-related.
Eighteen patients (30 %) reported SAEs (Table 5). The most frequently reported SAEs were dehydration (n = 3; 5 %) and diarrhea (n = 3; 5 %).
Forty-nine patients (82 %) discontinued study treatment, predominantly due to disease progression (28 patients), and 10/49 patients (17 %) experienced an AE that led to the permanent discontinuation of study treatment (Table 6). Overall, 13 deaths were reported in this study with the primary cause of death due to breast cancer (n = 10/13; 17 %). Two patients (3 %) died due to SAEs (sudden death, acute renal failure) which were considered to be related to treatment. There were no clinically significant changes in vital signs, body weight, and ECOG PS during the study.
Discussion
The primary objective of this study was to determine the ORR in patients with HER2-positive MBC who received first-line or second-line treatment with lapatinib in combination with weekly nab-paclitaxel. This study was not designed for inference testing thus definitive conclusions cannot be derived from the results. A clinical benefit was observed for treatment with lapatinib plus nab-paclitaxel with respect to investigator-assessed ORR in 32 patients (53 %). The highest number of responses was PR (28 patients; 47 %). Four patients (7 %) had a CR and ten patients (17 %) showed SD.
ORR in this study was lower compared with other studies conducted to investigate paclitaxel and lapatinib combination therapy. In a Phase III, multicenter, double-blind, placebo-controlled study in which patients with HER2-positive MBC (Stage IV) received treatment with paclitaxel and either lapatinib or placebo (NCT00281658), ORR was 69 % [26].
In a Phase II study in which 57 female patients with HER2-positive MBC (Stage IV) were randomized to receive paclitaxel and lapatinib (NCT00356811) ORR was 77 % [27]. These differences could be due in part to the small sample size, confounded by the inclusion of both first-line and second-line patients and the fact that ten patients in this study had an unknown response (no tumor assessments available).
The clinically meaningful activity observed in the primary endpoint was supported by investigator-assessed PFS, where the median Kaplan–Meier estimate of PFS was 39.7 weeks. These results are consistent with those of other trials evaluating paclitaxel and lapatinib combination therapy. In two similar studies, the median PFS were 9.7 months (41.7 weeks) (NCT00281658) [26] and 47.9 weeks (NCT00356811) [27].
The median duration of lapatinib and nab-paclitaxel treatment was 24.3 and 24 weeks, respectively. These data are comparable to results from a randomized, multicenter, double-blind, placebo-controlled, 2-Arm, Phase III Study of lapatinib in combination with paclitaxel in subjects previously untreated for advanced or MBC (NCT00075270) [28]. In this study, the median duration of lapatinib treatment was 19.9 weeks, and the median duration of paclitaxel treatment was 15.1 and 16.1 weeks in the paclitaxel-lapatinib and paclitaxel-placebo arms, respectively.
The maximum toxicity grade of the majority of AEs was Grade 3 or less, and most resolved without sequelae. For hematologic toxicities, Grade 3 neutropenia and anemia was observed in 22 and 2 % of patients, respectively, which was lower compared with 35 and 4 % observed in the lapatinib plus paclitaxel study (NCT00281658) [26]. However, the non-hematologic toxicities observed in this study were consistent with the NCT00281658 study [26]. Grade 3 diarrhea, rash, and fatigue were reported by 20, 5, and 8 % of the patients, respectively, in this study compared with 20, 4, and 2 % in the NCT00281658 study. Although Grade 3 AEs of diarrhea and neutropenia were reported by 20 and 22 % of patients, respectively, low numbers of patients withdrew due to these AEs (7 % due to diarrhea, none due to neutropenia). SAEs were reported by 30 % of patients. The most frequently reported SAEs were dehydration (5 %) and diarrhea (5 %). Two patients died during the study due to SAEs (sudden death in a patient with a medical history of cardiac arrhythmias, and acute renal failure in a patient with a medical history of uncontrolled diabetes).
This study established a dose regimen of nab-paclitaxel (100 mg/m2 IV on Day 1, 8, and 15 every 28 days) in combination with lapatinib (1,000 mg orally once daily on a continuous basis) in a 4-week cycle as a feasible treatment for HER2-positive MBC with manageable and predictable toxicity. The incidence and severity of AEs for the combination treatment was considered to be consistent with the known safety profiles of lapatinib and nab-paclitaxel. Overall, the data in this study are consistent with those reported for other studies of lapatinib in combination with paclitaxel. Although this study was not designed for inference testing, the results suggest a promising signal of efficacy and no new safety signals.
References
Siegel R, Naishadham D, Jemal A (2012) Cancer statistics, 2012. CA Cancer J Clin 62:10–29
Yarden Y, Sliwkowski MX (2001) Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2:127–137
Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL (1987) Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235:177–182
Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE et al (1989) Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 244:707–712
Blows FM, Driver KE, Schmidt MK, Broeks A, van Leeuwen FE, Wesseling J et al (2010) Subtyping of breast cancer by immunohistochemistry to investigate a relationship between subtype and short and long term survival: a collaborative analysis of data for 10,159 cases from 12 studies. PLoS Med 7:e1000279
Mass RD, Press MF, Anderson S, Cobleigh MA, Vogel CL, Dybdal N et al (2005) Evaluation of clinical outcomes according to HER2 detection by fluorescence in situ hybridization in women with metastatic breast cancer treated with trastuzumab. Clin Breast Cancer 6:240–246
Gabos Z, Sinha R, Hanson J, Chauhan N, Hugh J, Mackey JR et al (2006) Prognostic significance of human epidermal growth factor receptor positivity for the development of brain metastasis after newly diagnosed breast cancer. J Clin Oncol 24:5658–5663
Kallioniemi OP, Holli K, Visakorpi T, Koivula T, Helin HH, Isola JJ (1991) Association of c-erbB-2 protein over-expression with high rate of cell proliferation, increased risk of visceral metastasis and poor long-term survival in breast cancer. Int J Cancer 49:650–655
Piccart-Gebhart MJ, Procter M, Leyland-Jones B, Goldhirsch A, Untch M, Smith I et al (2005) Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N Engl J Med 353:1659–1672
Romond EH, Perez EA, Bryant J, Suman VJ, Geyer CE Jr, Davidson NE et al (2005) Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med 353:1673–1684
Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A et al (2001) Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 344:783–792
Montemurro F, Donadio M, Clavarezza M, Redana S, Jacomuzzi ME, Valabrega G et al (2006) Outcome of patients with HER2-positive advanced breast cancer progressing during trastuzumab-based therapy. Oncologist 11:318–324
Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, Pienkowski T et al (2006) Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med 355:2733–2743
http://www.cancer.gov/cancertopics/druginfo/fda-lapatinib. Accessed 2 Oct 2012
Schwartzberg LS, Franco SX, Florance A, O’Rourke L, Maltzman J, Johnston S (2010) Lapatinib plus letrozole as first-line therapy for HER-2+ hormone receptor-positive metastatic breast cancer. Oncologist 15:122–129
Gradishar WJ, Tjulandin S, Davidson N, Shaw H, Desai N, Bhar P et al (2005) Phase III trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil-based paclitaxel in women with breast cancer. J Clin Oncol 23:7794–7803
Gradishar WJ, Krasnojon D, Cheporov S, Makhson AN, Manikhas GM, Clawson A et al (2009) Significantly longer progression-free survival with nab-paclitaxel compared with docetaxel as first-line therapy for metastatic breast cancer. J Clin Oncol 27:3611–3619
Gradishar WJ, Krasnojon D, Cheporov S, Makhson AN, Manikhas GM, Clawson A et al (2012) Phase II trial of nab-paclitaxel compared with docetaxel as first-line chemotherapy in patients with metastatic breast cancer: final analysis of overall survival. Clin Breast Cancer 12:313–321
http://www.cancer.gov/cancertopics/druginfo/fda-nanoparticle-paclitaxel. Accessed 2 Oct 2012
Gril B, Palmieri D, Bronder JL, Herring JM, Vega-Valle E, Feigenbaum L et al (2008) Effect of lapatinib on the outgrowth of metastatic breast cancer cells to the brain. J Natl Cancer Inst 100:1092–1103
Pestalozzi BC, Brignoli S (2000) Trastuzumab in CSF. J Clin Oncol 18:2349–2351
Xia W, Liu LH, Ho P, Spector NL (2004) Truncated ErbB2 receptor (p95ErbB2) is regulated by heregulin through heterodimer formation with ErbB3 yet remains sensitive to the dual EGFR/ErbB2 kinase inhibitor GW572016. Oncogene 23:646–653
Blackwell KL, Burstein HJ, Storniolo AM, Rugo HS, Sledge G, Aktan G et al (2012) Overall survival benefit with lapatinib in combination with trastuzumab for patients with human epidermal growth factor receptor 2-positive metastatic breast cancer: final results from the EGF104900 study. J Clin Oncol 30:2585–2592
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L et al (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92(205):16
Greenwood M (1926) The natural duration of cancer. In: Reports on public health and medical subjects. London, England: His Majesty's Stationery Office, pp 1–26
Guan ZZ, Xu BH, DeSilvio M, Shen ZZ, Arponwirat W, Tong ZS et al (in press) Randomized trial of lapatinib vs placebo added to paclitaxel in the treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol
Jagiello-Gruszfeld A, Tjulandin S, Dobrovolskaya N, Manikhas A, Pienkowski T, DeSilvio M et al (2010) A single-arm phase II trial of first-line paclitaxel in combination with lapatinib in HER2-overexpressing metastatic breast cancer. Oncology 79:129–135
Di Leo A, Gomez HL, Aziz Z, Zvirbule Z, Bines J, Arbushites MC, et al (2008) Phase III, double-blind, randomized study comparing lapatinib plus paclitaxel with placebo plus paclitaxel as first-line treatment for metastatic breast cancer. J Clin Oncol 26:5544–5552. Erratum in: J Clin Oncol 2009;27:1923
Acknowledgments
We would like to thank all of our patients and their family members for participating in this clinical trial and Celgene/Abraxis BioScience for providing the nab-paclitaxel for this clinical trial. LTP111111 investigators: Robert F. Asbury, Evergreen Hematology & Oncology, Spokane, Washington; Min-Chun Chen, Multicare Health System, Tacoma, Washington; Ellen Chuang, Weill Medical College of Cornell University New York, New York; James Congdon, Providence Everett Medical Center, Everett, Washington; Kiem D. Liem, Pacific Shores Medical Group, Long Beach, California; Paula Silverman, University Hospitals of Cleveland, Cleveland, Ohio; Jacqueline Vuky, Virginia Mason Medical Center, Seattle, Washington. Editorial and medical writing support was provided by Sarah White and Karen Yee at Fishawack Scientific Communications Ltd., funded by GlaxoSmithKline.
Conflict of interest
Denise A. Yardley, Lowell Hart, Linda Bosserman, Mansoor N. Salleh, David M. Waterhouse, Maura K. Hagan, and Paul Richards have no conflict of interest to declare. Michelle L. DeSilvio and Janine M. Mahoney are employees of, and own stock in, GlaxoSmithKline. Yasir Nagarwala is a former employee of GlaxoSmithKline and an employee of Celgene; he owns stock in GlaxoSmithKline and Celgene.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Yardley, D.A., Hart, L., Bosserman, L. et al. Phase II study evaluating lapatinib in combination with nab-paclitaxel in HER2-overexpressing metastatic breast cancer patients who have received no more than one prior chemotherapeutic regimen. Breast Cancer Res Treat 137, 457–464 (2013). https://doi.org/10.1007/s10549-012-2341-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-012-2341-9