
Abstract
X−linked adrenoleukodystrophy is a rare inherited demyelinating disorder characterized by an abnormal accumulation of very long chain fatty acids, mainly hexacosanoic acid (26:0), due to a mutation of the gene encoding for a peroxisomal membrane protein. The only available, and partially effective, therapeutic treatment consists of dietary intake of a 4:1 mixture of triolein and trierucin, called Lorenzo’s oil (LO), targeted to inhibit the elongation of docosanoic acid (22:0) to 26:0. In this study we tested whether, besides inhibiting elongation, an enhancement of peroxisomal beta oxidation induced by conjugated linoleic acid (CLA), will improve somatosensory evoked potentials and modify inflammatory markers in adrenoleukodystrophy females carriers. We enrolled five heterozygous women. They received a mixture of LO (40 g/day) with CLA (5 g/day) for 2 months. The therapeutic efficacy was evaluated by the means of plasma levels of 26:0, 26:0/22:0 ratio, modification of cerebrospinal fluid (CSF) inflammatory markers and somatosensory evoked potentials. Changes of fatty acid profile, and in particular CLA incorporation, were also evaluated in CSF and plasma. The results showed that CLA promptly passes the blood brain barrier and the mixture was able to lower both 26:0 and 26:0/22:0 ratio in plasma. The mixture improved somatosensory evoked potentials, which were previously found unchanged or worsened with dietary LO alone, and reduced IL-6 levels in CSF in three out of five patients. Our data suggest that the synergic activity of CLA and LO, by enhancing peroxisomal beta-oxidation and preventing 26:0 formation, improves the somatosensory evoked potentials and reduces neuroinflammation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
X−linked adrenoleukodystrophy (ALD) is an inherited demyelinating disorder. The disease is characterized by an abnormal accumulation of very long chain fatty acids (VLCFA), owing to a defect in VLCFA peroxisomal beta-oxidation. The gene responsible for ALD was cloned and originally termed adrenoleukodystrophy gene. This gene encodes a 745 amino acid peroxisomal transmembrane protein (ALDP) with the general structure of an ATP-binding cassette transporter (Mosser et al. 1993). The prevalence of the disease is 0.5 to 3.3 in 100,000 males (Heim et al. 1997; Kirk et al. 1998). There are seven distinct clinical phenotypes described in males, ranging from the more severe cerebral forms, to asymptomatic individuals or isolated adrenal insufficiency, without involvement of the central nervous system. Approximately 50% of women who are heterozygous for ALD develop an adrenomyeloneuropathy (AMN)-like syndrome, a higher proportion than what had been recognized in the past. The mean age at the onset of symptoms in heterozygous females (37 ± 14.6 years, range 32–73 years) is greater than in men with AMN (28 ± 9 years), clinical symptoms are milder and progression is slower. Cerebral and adrenal involvement are rare in females (Moser et al. 1991; Restuccia et al. 1997).
All the cerebral forms are inflammatory forms, associated with a rapidly progressive intensely inflammatory myelinopathy, that usually begins in the parietooccipital regions (Schaumburg et al. 1975; Ito et al. 2001) and may involve autoimmune mechanisms (Ito et al. 2001).
The accumulation of VLCFA in different tissues, including the brain, has been so far believed to be the major cause of ALD. This hypothesis stems from the evidence that an increase of 26:0 intercalated into the membrane (Ho et al. 1995) may account for the demyelination and may also increase immunoreactivity (Baes and Aubourg 2009). However, in a mouse model of ALD knock out for the ABC gene, while an accumulation of VLCFA has been described, only mild typical neurological signs of ALD were detected (Baes and Aubourg 2009). Therefore, it is likely that alongside the damage triggered by the accumulation of VLCFA, other factors, as a consequence and/or independently, contribute to the physiopathology of ALD.
Lorenzo's oil (LO) therapy is the only available and partially effective treatment for X−ALD patients.
It consists of a 4:1 mixture of glycerol trioleate and glycerol trierucate. This mixture reduces plasma levels of VLCFA known to cause ALD, competitively inhibiting the elongase that forms VLCFA. Conversely, erucic acid while likely passes the blood–brain barrier, it may not be incorporated into brain lipids due to a very low desorption (Hamilton et al. 2007). As a net result, erucic acid may not accumulate sufficiently in the brain (Poulos et al. 1994), leaving open the question whether it is really effective in the central nervous system. We hypothesized that the addition of another fatty acid, that may induce peroxisomal beta-oxidation and accumulate in brain lipids, could act synergistically with LO. The possible candidate was conjugated linoleic acid (CLA), a group of isomers of linoleic acid, mainly found in foods derived from ruminants, which is characterized by a conjugated diene structure (Banni et al. 2004b). CLA is a high affinity ligand of the peroxisome proliferator activated receptor (PPAR) alpha (Moya-Camarena et al. 1999), a transcription factor for the genes involved in peroxisomal beta oxidation and thereby able to induce key enzymes for peroxisomal beta oxidation (Reddy and Hashimoto 2001).
The decrease of peroxisomal beta-oxidation may also lead to a decreased catabolism of proinflammatory eicosanoids (Diczfalusy and Alexson 1990; de Waart et al. 1994; Ferdinandusse et al. 2002) and lipid peroxidation products (Mayatepek and Lehmann 1996; Iannone et al. 2009). CLA, by increasing peroxisomal beta-oxidation, should also increase eicosanoid and oxidative stress product catabolism, acting as an antinflammatory and antioxidative factor. Given the prompt incorporation of CLA into the brain recently demonstrated in animal models (Alasnier et al. 2002; Fa et al. 2005; Sikorski et al. 2008) its activity in the central nervous system should also be ensured as recently suggested (Hunt et al. 2010).
A previous study showed that the long-term dietary supplementation of LO is not able to modify the progression of the neurophysiologic impairment of adult ALD patients repeatedly evaluated by evoked potentials (Restuccia et al. 1999).
The primary objective of our study was to determine whether CLA crosses the brain–blood barrier and thereby exerts a synergistic effect with LO in ameliorating biochemical, clinical and neurophysiological parameters in X-linked ALD patients. As conservative approach and because of the extreme rarity of the disease, we chose to recruit five female x-linked ALD carriers.
Materials and methods
Study design
Five ALD women (mildly symptomatic or asymptomatic) who were not treated with LO therapy at the study entry were recruited. A mixture of LO (40 g/day) with CLA (a mixture 1:1 c9,t11 and t10,c12 isomers) (5 g/day) was given for 2 months. No data on possible activity of CLA on neuroinflammation in humans are available, we therefore temptatively chose a dose sufficiently high to be incorporated in CNS lipids based on our previous study on rats (Fa et al. 2005).
No dietary lipid restriction was prescribed. The LO with CLA mixture was kindly provided by Sofedus srl, Milan, Italy.
Table 1 summarizes the clinical characteristics and the results of molecular analysis of the five patients.
After recruitment, during the 1st visit (t0), blood samples were collected from all the patients in order to analyze their baseline platelet count (Zinkham et al. 1993) and plasma lipid profile (including the fatty acid profile). At baseline, the somatosensory evoked potentials (SEPs) of upper and lower limbs were also performed in all the patients. The 2nd visit (t1) was scheduled after 4 weeks of treatment with LO + CLA to repeat blood sampling for platelet count and lipid analyses.
The 3rd visit (t2) was scheduled after 4 more weeks (8 weeks from baseline), to perform the second evaluation of the SEPs, and to repeat blood sampling for platelet count and lipid analyses.
One of the symptomatic patients with spastic paraparesis (patient 1) had been previously treated with LO and had stopped LO therapy one month before recruitment.
At baseline and during the 3rd visit, 25–30 drops of CSF (about 1.5 ml) of cerebrospinal fluid (CSF) were collected by lumbar puncture in all patients. Blood and CSF samples were centrifuged and stored at −80°C, until analyses.
The CSF samples were analysed for fatty acid composition, in order to verify whether CLA and erucic acid are able to pass, and to what extent, the blood–brain barrier.
The institutional review board approved the study, written informed consent was obtained from all patients at recruitment.
Lipid analyses
Total lipids were extracted from plasma or CSF using chloroform/methanol 2:1 (v/v) (Folch et al. 1957). Separation of phospholipids from total lipids was performed as previously reported (Banni et al. 2001). Aliquots were mildly saponified as previously described (Banni et al. 1996) in order to obtain free fatty acids for HPLC analysis. Separation of fatty acids was carried out by an Agilent 1100 HPLC system (Agilent, Palo Alto, Calif., USA) equipped with a diode array detector as previously reported (Angioni et al. 2002).
Since saturated fatty acids are transparent to UV, they were measured, after methylation, by means of a gas chromatograph (Agilent, Model 6890, Palo Alto, CA) equipped with split ratio of 20:1 injection port, a flame ionization detector (FID), an autosampler (Agilent, Model 7673), a 100 m HP-88 fused capillary column (Agilent), and an Agilent ChemStation software system. The injector and detector temperatures were set at 250°C and 280°C respectively. H2 served as carrier gas (1 ml/min), and the FID gases were H2 (30 ml/min), N2 (30 ml/min), and purified air (300 ml/min). The temperature program was as follows: initial temperature was 120°C, programmed at 10°C/min to 210°C and 5°C/min to 230°C, then programmed at 25°C/min to 250°C and held for 2 min.
A further identification of fatty acids, and in particular of CLA has been carried out by LC-APCI-MS as previously described (Banni et al. 2004a).
Cytokine analyses
The total CSF samples were centrifuged at 1500 rpm for 10 min and the supernatants were immediately frozen at −80°C. IL-6 and IL-8 levels were quantified in CSF using sandwich immunoassays.
We decided to dose both these inflammatory markers in ALD, because IL-6 is one of the most important mediators of the acute phase response, it is capable of crossing the blood brain barrier (Banks et al. 1994) and initiating synthesis of PGE2 in the CNS. IL-8 is a chemokine produced by macrophages and epithelial cells, it is also synthesized by endothelial cells, which store IL-8 in their storage vesicles. The presence of IL-8 has been demonstrated in the CSF of patients with different diseases of the CNS (MacLullich et al. 2011; Vrethem et al. 2011).
All reagents were purchased from R&D System (R&D System Inc., Minneapolis, MN, USA). IL-6 was quantified using the Quantikine® High Sensitivity human IL-6 Immunoassay (Catalog Number: HS600B) according to the manufacturer’s instructions. The detection range of the kit was 0.15 – 10 pg/ml. IL-8 was quantified using a combination of a monoclonal anti-human CXCL8/IL-8 capture antibody (Catalog number: MAB208), a byotinilated human CXCL8 detection antibody (Catalog number: BAF208) and recombinant human CXCL8 (Catalog Number: 208-IL) as the standard. The detection range of this assay was 7.0 – 2000 pg/ml.
Somatosensory evoked potential (SEP) recording procedure
SEPs were recorded to both median and tibial nerve stimulation before and after 8 weeks’ treatment with a mixture of LO (40 g/day) with CLA (5 g/day).
For SEP recording we used the same procedure described in a previous study (Restuccia et al. 1997). Subjects lay on a bed in a quiet and semi-darkened room. Stimuli (0.3 ms duration, 5 Hz) were delivered by skin electrodes at the wrist for the median nerve, and at the ankle for the tibial nerve. Stimulus intensity was adjusted to slightly above the motor threshold. The stimulation rate was 1.5 Hz. An automatic artefact-rejection system excluded from the average all runs containing transients exceeding ±65 mV at any recording channel. In order to ensure baseline stabilization, SEPs were digitally filtered off-line by means of a digital filter with a bandpass of 30–3000 Hz. Two averages of 1000 trials each were obtained for each stimulated nerve. For median nerve SEP recording, electrodes were placed in the supraclavicular fossa (Erb’s point), over the sixth cervical vertebra (CV6), and in the parietal scalp regions contralateral and ipsilateral to the stimulated side. Erb’s point electrode was referred to an electrode placed in the frontal region (Fz, according to the 10–20 Intenational System), the CV6 electrode was referred to an electrode located immediately above the thyroid cartilage (AC), and the scalp electrodes were referred to the shoulder contralateral to the stimulated side (noncephalic reference). For tibial nerve SEP recording, electrodes were placed over the fourth lumbar (L4) vertebra [referred to the second lumbar (L2) vertebra], over the 12th dorsal (T12) vertebra (referred to an electrode located immediately above the umbilicus - Abd), at the Cz location of the 10–20 International System (referred to Fz) and in the temporal region (T3/T4) contralateral to the stimulation (referred to an electrode in the ipsilateral parietal region).
For the median nerve SEPs, we evaluated the peak latencies of: 1) the peripheral N9 potential, generated by the brachial plexus volley and recorded by the Erb’s electrode, 2) the spinal N13 response, generated within the cervical grey matter and identifiable in the CV6-AC trace, 3) and 4) the scalp far-field P9 and P14 responses, recorded from the scalp electrodes and generated in the brachial plexus and in the lower brainstem respectively, and 5) the contralateral N20 and P25 responses, recorded from the parietal electrode and generated by neurons in the primary somatosensory (SI) area. We also calculated the P9-N20 interpeak interval to assess the conduction time in central somatosensory pathways, and the P9-P14 and P14-N20 interpeak intervals, to evaluate the somatosensory conduction along the cervical dorsal columns and the intracranial lemniscal pathways respectively. Lastly, we evaluated the peak-to peak amplitudes of the spinal N13 response, the P14 far-field scalp potential and the N20 scalp response (Desmedt and Cheron 1980; Yamada et al. 1986).
For the tibial nerve SEPs, we evaluated the peak latencies of: 1) the cauda equina response (CE), recorded at L4 and generated by the peripheral volley along the L5-S1 roots, 2) the spinal N24 response, identifiable in the T12-Abd trace and generated by the lumbo-sacral dorsal horn neurons, 3) and 4) the scalp N37 and P40 responses, recorded from the contralateral temporal electrode and the Cz lead respectively and probably generated in the SI area. Moreover, the peak-to-peak amplitudes of the N24 potential and of both the scalp N37 and P40 SEP components were measured. Finally, we calculated the N24-P40 interpeak interval to assess the conduction time in central somatosensory pathways.
The SEP values obtained in our patients were referred to the normative data for upper and lower limb SEPs reported in our previous studies (Restuccia et al. 1993; Restuccia et al. 1994; Valeriani et al. 2000).
Statistic analysis
SEP latencies and amplitudes were compared between both sides and between before and after treatment. Paired Student’s t-test was used to compare the SEP latencies and the interpeak intervals, while Wilcoxon-test was used to compare the SEP amplitudes. Statistic significance was fixed at p < 0.05.
Wilcoxon-test was used to compare lipid profile findings at different time points.
Due to the small number and the wide scatter of the data, cytokine analysis results are shown as raw data without statistic analysis.
Results
Clinical data
No significant adverse effects, potentially related to the administration of the mixture of LO + CLA, were observed during the study period. One patient presented with severe headache after the lumbar puncture, and requested hospitalization for two days.
In three out of five subjects, neurological examination was normal before and after treatment. One patient had a spastic paraparesis (patient 1) and another patient showed a mild tactile and thermic sensory loss in lower limbs (patient 4) before treatment. Their clinical examination did not change after treatment.
Lipid metabolism findings
LO + CLA did not significantly affect platelet count. Total cholesterol was reduced during therapy in four out of five individuals, LDL decreased while HDL tended to increase, and triglycerides were relatively unchanged (Table 2). The changes in cholesterolemia could be ascribed to the particularly high intake of oleic acid, which is widely known to decrease LDL cholesterol (Hu et al. 1997).
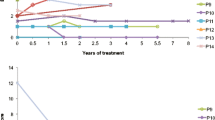
Figures 1 and 2 clearly show that in all patients the mixture LO + CLA treatment decreased the two parameters taken into consideration to evaluate very long chain saturated fatty acid elongation activity: plasma levels of 26:0 (Fig. 1) and 26:0/22:0 ratio (Fig. 2). In CSF, VLCFAs as well as erucic acid were under the detectable levels.

Plasma levels of 26:0 at different time points (* indicates p < 0.05 versus t0)

Plasma 26.0/22:0 ratio at different time points (* indicates p < 0.05 versus t0)
Treatment with the mixture LO + CLA significantly increased CLA levels in plasma (Fig. 3a) and in CSF (Fig. 3c). The 22:6/20:5 ratio also increased in plasma and CSF but not significantly (Fig. 3b and d, respectively). This ratio is an indirect marker of peroxisomal beta oxidation, because to form 22:6 from 20:5 a step in peroxisome is obligatory (Ferdinandusse et al. 2001). In fact, 20:5 after two steps of elongation forms 24:5, with one step of delta 6 desaturase forms 24:6 and with a final and limiting step in the peroxisome is beta-oxidized to 22:6. The intermediates fatty acids (22:5, 24:5 and 24:6) do not accumulate in sufficient amount to be detected in CSF and in plasma.

CLA and 22:6/20:5 levels in plasma and CSF at different time points (* indicates p < 0.05 versus t0)
The two components of LO, oleic and erucic acids significantly increased in plasma, even though the rise of oleic acid became significant only after two months (data not shown). No changes were detected in CSF (data not shown).
Cytokine assessment
IL-8 was not detectable in the CSF (data not shown). On the contrary, at study entry IL-6 was clearly detectable in CSF of all patients. After LO + CLA administration, IL-6 levels were decreased in three out of five patients (patients 1–3), while remained substantially unchanged in two patients (patients 4 and 5). The results are shown in Table 3.
Neurophysiologic findings before treatment
Table 4 summarizes SEP results. SEP amplitudes and latencies were not different between right and left side stimulation (p > 0.05). Median nerve SEPs were normal in all patients. After tibial nerve stimulation, the CE response and spinal N24 potential were within normal limits in all patients. The scalp P40 potentials resulted abnormally delayed bilaterally in four out of five subjects (patients 1–4). The central somatosensory conduction after both the right and left tibial nerve stimulation (N24-P40 interval) was prolonged in four out of five patients (patients 1–4).
Neurophysiologic findings after treatment
SEP amplitudes and latencies were not different between right and left side stimulation (p > 0.05).
Median nerve SEPs remained normal in all patients and did not differ from those recorded before treatment (p > 0.05). After tibial nerve stimulation, the scalp P40 potential resulted was abnormally delayed only in one out of five subjects (patient 1). Conversely, in three other patients showing a delayed P40 latency before treatment (patients 2, 3 and 4), the P40 potential was recorded with a normal latency after treatment. After treatment, the average P40 latency in our patients decreased significantly from 47.16 ± 4.59 ms to 40.83 ± 4.47 ms (p: 0.003) and the N37 latency decreased from 45.33 ± 4,03 ms to 40.49 ± 4.05 ms (p: 0.019). Moreover, the N24-P40 interpeak interval decreased from 24.40 ± 5.59 ms to 18.79 ± 3.67 ms (p: 0.016). Lastly, treatment led to a slight increase of both N37 and the P40 amplitudes. Overall, in four out of five patients (patients 2–5) tibial nerve SEPs were significantly improved after treatment.
Discussion
The accumulation of VLCFA in different tissues, including the brain, has been considered not only a marker but also a major determinant of ALD pathogenesis. At present, LO remains the sole therapeutic approach available for ALD targeted to contrast VLCFA accumulation. One of the two components of LO, oleic acid, aims at lowering dietary saturated fatty acids, which are also banned from the ALD patient’s diet. The other component, erucic acid, aims at reducing the elongation of docosanoic (22:0) to 26:0, by interfering with microsomal elongases. However, while LO is very effective in decreasing plasma VLCFAs, it does not seem sufficient to prevent ALD (Di Biase et al. 1998). The efficacy of treatment with LO is questionable, even when the treatment begins at disease onset. This may be explained by the hypothesis that VLCFA accumulation is only one of the factors contributing to the disease. ALD is due to defects of the ALDP, an integral peroxisomal membrane protein (Douar et al. 1994). Furthermore, a VLCFA acyl-CoA synthetase, has been found reduced in normal white matter from ALD patients and it has been associated with the early pathogenesis and severity of disease progression (Asheuer et al. 2005). Thus, mutations in different genes account for the tissue accumulation of 26:0, through the impaired peroxisomal beta oxidation of peculiar fatty acids which are, almost exclusively, beta oxidized in peroxisomes. The non enzymatic free radical products eicosanoids (including isoprostanes) are beta-oxidized in peroxisomes as a mechanism of their catabolism (Jedlitschky et al. 1993; Leung and Glauert 1996; Mayatepek and Tiepelmann 1996; Ferdinandusse et al. 2002; Iannone et al. 2009; Banni et al. 2010), thereby a lower eicosanoid peroxisomal beta oxidation may increase inflammation and oxidative stress contributing to ALD pathogenesis.
CLA is preferentially beta oxidized in peroxisomes (Banni et al. 2004a). It has been observed that the fatty acids that are a good substrate of peroxisomal beta oxidation are biological ligands for PPAR alpha (Latruffe and Vamecq 1997). PPAR alpha regulates the gene expression of membrane transporter proteins, VLCFA acyl-CoA synthetase and all the enzymes involved in peroxisomal beta oxidation (Latruffe and Vamecq 1997). Our study demonstrates for the first time the prompt entry of CLA through the blood–brain barrier in humans. Furthermore, because the ratio in percentage between CLA concentrations in CSF and plasma was not significantly different at T0 and T1, (3.8 ± 1.6 vs 2,94 ± 0.9 at T0 and T2 respectively) likewise for the ratio 22:6/20:5 (6163 ± 3251 vs 7009 ± 3521 at T0 and T2 respectively), it may suggest a dose response with increasing CLA levels in CSF by increasing its levels in plasma. In addition, we found both in experimental animals (Banni et al. 1999) and more recently in humans (manuscript in preparation) that CLA plasma levels were linearly correlated to its dietary intake.
The additional therapeutic effect of LO + CLA is that LO inhibits VLCFA accumulation, mainly in peripheral tissues due to the slow passage through the blood–brain barrier of erucic acid. In addition, CLA may induce the remaining activity of ALDP, and thereby peroxisomal beta oxidation, in peripheral tissues and brain, as confirmed by the increased 22:6/20:5 ratio both in plasma and in CSF. Indirect evidence of the increased peroxisomal beta oxidation, is the significant increase of oleic acid. A contribution of the increased plasma oleic acid level, after LO + CLA intake, might derive, other than to dietary trioleate in LO, from peroxisomal beta oxidation of erucic acid, as previously demonstrated (Golovko and Murphy 2006). As a matter of fact, we should not expect a large increase after treatment, because of the already high intake of olive oil in the Italian diet (Leclercq et al. 2009). In the synergistic action of LO and CLA, the former may prevent the formation of 26:0, by inhibiting elongases. On the other hand CLA, by enhancing peroxisomal beta oxidation both in peripheral tissues and in the brain, may contribute to shortening 24:0, therefore re-establishing the physiological role for catabolism of eicosanoids and lipid peroxidation products. Inflammation is known to be a crucial factor for ALD pathogenesis (Ito et al. 2001). This concerted action results in an improvement of the SEPs. A better SEP is a sign of neurological improvement which implies an ongoing physiological recovery. The evident decrease of IL-6 concentrations in CSF, in three out of five patients after LO + CLA administration, could represent the biochemical support of this finding. In the present study, four out five patients showed a prolonged central somatosensory conduction time to lower limb stimulation before treatment, confirming a previous report in ALD carriers (Restuccia et al. 1997). In our study, tibial nerve SEPs fully recovered after treatment in three out four patients who showed abnormal tibial nerve SEPs at baseline. This result is particularly important, when compared with the findings of a previous study performed in adult ALD patients, in which no SEP improvement was obtained by dietary LO supplementation (Restuccia et al. 1999).
A major limitation of this study is the number of patients who, as women, are only partially affected by ALD. The results need to be confirmed in a larger number of subjects and to be related to a clinical improvement, not evident in our patients. Nonetheless, the ethical committee would not allow a larger trial with ALD patients without this preliminary data.
In conclusion, we hypothesize that the synergistic action of LO + CLA can improve the progressive somatosensory conduction impairment in ALD carriers. The results of this pilot study are encouraging for planning a larger multi-centre trial, that could help open the field for new therapeutic strategies, not only for ALD, but also for other neurodegenerative diseases where inflammation plays a central role.
References
Alasnier C, Berdeaux O, Chardigny JM, Sebedio JL (2002) Fatty acid composition and conjugated linoleic acid content of different tissues in rats fed individual conjugated linoleic acid isomers given as triacylglycerols small star, filled. J Nutr Biochem 13:337–345
Angioni E, Lercker G, Frega NG et al. (2002) UV spectral properties of lipids as a tool for their identification. Eur J Lipid Sci Technol 104:59–64
Asheuer M, Bieche I, Laurendeau I et al. (2005) Decreased expression of ABCD4 and BG1 genes early in the pathogenesis of X-linked adrenoleukodystrophy. Hum Mol Genet 14:1293–1303
Baes M, Aubourg P (2009) Peroxisomes, myelination, and axonal integrity in the CNS. Neuroscientist 15:367–379
Banks WA, Kastin AJ, Gutierrez EG (1994) Penetration of interleukin-6 across the murine blood–brain barrier. Neurosci Lett 179:53–56
Banni S, Carta G, Contini MS et al. (1996) Characterization of conjugated diene fatty acids in milk, dairy products, and lamb tissues. J Nutr Biochem 7:150–155
Banni S, Angioni E, Casu V et al. (1999) Decrease in linoleic acid metabolites as a potential mechanism in cancer risk reduction by conjugated linoleic acid. Carcinogenesis 20:1019–1024
Banni S, Carta G, Angioni E et al. (2001) Distribution of conjugated linoleic acid and metabolites in different lipid fractions in the rat liver. J Lipid Res 42:1056–1061
Banni S, Petroni A, Blasevich M et al. (2004a) Detection of conjugated C16 PUFAs in rat tissues as possible partial beta-oxidation products of naturally occurring conjugated linoleic acid and its metabolites. Biochim Biophys Acta 1682:120–127
Banni S, Petroni A, Blasevich M et al. (2004b) Conjugated linoleic acids (CLA) as precursors of a distinct family of PUFA. Lipids 39:1143–1146
Banni S, Montisci R, Sanfilippo R et al. (2010) Physiological response to lipid peroxidation in ischemia and reperfusion during carotid endarterectomy. Lipids Health Dis 9:41
de Waart DR, Koomen GC, Wanders RJ (1994) Studies on the urinary excretion of thromboxane B2 in Zellweger patients and control subjects: evidence for a major role for peroxisomes in the beta-oxidative chain-shortening of thromboxane B2. Biochim Biophys Acta 1226:44–48
Desmedt JE, Cheron G (1980) Central somatosensory conduction in man: neural generators and interpeak latencies of the far-field components recorded from neck and right or left scalp and earlobes. Electroencephalogr Clin Neurophysiol 50:382–403
Di Biase A, Salvati S, Avellino C et al. (1998) X-linked adrenoleukodystrophy: first report of the Italian Study Group. Ital J Neurol Sci 19:315–319
Diczfalusy U, Alexson SE (1990) Identification of metabolites from peroxisomal beta-oxidation of prostaglandins. J Lipid Res 31:307–314
Douar AM, Mosser J, Sarde CO, Lopez J, Mandel JL, Aubourg P (1994) X-linked adrenoleukodystrophy gene: identification of a candidate gene by positional cloning. Biomed Pharmacother 48:215–218
Fa M, Diana A, Carta G et al. (2005) Incorporation and metabolism of c9, t11 and t10, c12 conjugated linoleic acid (CLA) isomers in rat brain. Biochim Biophys Acta 1736:61–66
Ferdinandusse S, Denis S, Mooijer PAW et al. (2001) Identification of the peroxisomal beta-oxidation enzymes involved in the biosynthesis of docosahexaenoic acid. J Lipid Res 42:1987–1995
Ferdinandusse S, Meissner T, Wanders RJ, Mayatepek E (2002) Identification of the peroxisomal beta-oxidation enzymes involved in the degradation of leukotrienes. Biochem Biophys Res Commun 293:269–273
Folch J, Lees M, Sloane-Stanley GH (1957) A simple method for the isolation and purification of total lipid from animal tissues. J Biol Chem 226:497–509
Golovko MY, Murphy EJ (2006) Uptake and metabolism of plasma-derived erucic acid by rat brain. J Lipid Res 47:1289–1297
Hamilton JA, Hillard CJ, Spector AA, Watkins PA (2007) Brain uptake and utilization of fatty acids, lipids and lipoproteins: application to neurological disorders. J Mol Neurosci 33:2–11
Heim P, Claussen M, Hoffmann B et al. (1997) Leukodystrophy incidence in Germany. Am J Med Genet 71:475–478
Ho JK, Moser H, Kishimoto Y, Hamilton JA (1995) Interactions of a very long chain fatty acid with model membranes and serum albumin. Implications for the pathogenesis of adrenoleukodystrophy. J Clin Invest 96:1455–1463
Hu FB, Stampfer MJ, Manson JE et al. (1997) Dietary fat intake and the risk of coronary heart disease in women. N Engl J Med 337:1491–1499
Hunt WT, Kamboj A, Anderson HD, Anderson CM (2010) Protection of cortical neurons from excitotoxicity by conjugated linoleic acid. J Neurochem 115:123–130
Iannone A, Petroni A, Murru E et al. (2009) Impairment of 8-iso-PGF(2ALPHA) isoprostane metabolism by dietary conjugated linoleic acid (CLA). Prostaglandins Leukot Essent Fatty Acids 80:279–287
Ito M, Blumberg BM, Mock DJ et al. (2001) Potential environmental and host participants in the early white matter lesion of adreno-leukodystrophy: morphologic evidence for CD8 cytotoxic T cells, cytolysis of oligodendrocytes, and CD1-mediated lipid antigen presentation. J Neuropathol Exp Neurol 60:1004–1019
Jedlitschky G, Mayatepek E, Keppler D (1993) Peroxisomal leukotriene degradation: biochemical and clinical implications. Adv Enzyme Regul 33:181–194
Kirk EP, Fletcher JM, Sharp P, Carey B, Poulos A (1998) X-linked adrenoleukodystrophy: the Australasian experience. Am J Med Genet 76:420–423
Latruffe N, Vamecq J (1997) Peroxisome proliferators and peroxisome proliferator activated receptors (PPARs) as regulators of lipid metabolism. Biochimie 79:81–94
Leclercq C, Arcella D, Piccinelli R, Sette S, Le Donne C, Turrini A (2009) The Italian National Food Consumption Survey INRAN-SCAI 2005–06: main results in terms of food consumption. Public Health Nutr 12:2504–2532
Leung LK, Glauert HP (1996) Reduction of the concentrations of prostaglandins E2 and F2alpha, and thromboxane B2 in cultured rat hepatocytes treated with the peroxisome proliferator ciprofibrate. Toxicol Lett 85:143–149
MacLullich AM, Edelshain BT, Hall RJ et al. (2011) Cerebrospinal fluid interleukin-8 levels are higher in people with hip fracture with perioperative delirium than in controls. J Am Geriatr Soc 59:1151–1153
Mayatepek E, Lehmann WD (1996) 12- and 15-hydroxyeicosatetraenoic acid are excreted in the urine of peroxisome-deficient patients: evidence for peroxisomal metabolism in vivo. Pediatr Res 39:146–149
Mayatepek E, Tiepelmann B (1996) Defective degradation of leukotrienes in peroxisomal-deficient human hepatocytes. Biochem Biophys Res Commun 227:131–134
Moser HW, Moser AB, Naidu S, Bergin A (1991) Clinical aspects of adrenoleukodystrophy and adrenomyeloneuropathy. Dev Neurosci 13:254–261
Mosser J, Douar AM, Sarde CO et al. (1993) Putative X-linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters. Nature 361:726–730
Moya-Camarena SY, Vanden Heuvel JP, Blanchard SG, Leesnitzer LA, Belury MA (1999) Conjugated linoleic acid is a potent naturally occurring ligand and activator of PPARalpha. J Lipid Res 40:1426–1433
Poulos A, Gibson R, Sharp P, Beckman K, Grattan-Smith P (1994) Very long chain fatty acids in X-linked adrenoleukodystrophy brain after treatment with Lorenzo's oil. Ann Neurol 36:741–746
Reddy JK, Hashimoto T (2001) Peroxisomal beta-oxidation and peroxisome proliferator-activated receptor alpha: an adaptive metabolic system. Ann Rev Nutr 21:193–230
Restuccia D, Di Lazzaro V, Valeriani M, Colosimo C, Tonali P (1993) N24 spinal response to tibial nerve stimulation and magnetic resonance imaging in lesions of the lumbosacral spinal cord. Neurology 43:2269–2275
Restuccia D, Valeriani M, Di Lazzaro V, Tonali P, Mauguiere F (1994) Somatosensory evoked potentials after multisegmental upper limb stimulation in diagnosis of cervical spondylotic myelopathy. J Neurol Neurosurg Psychiatr 57:301–308
Restuccia D, Di Lazzaro V, Valeriani M et al. (1997) Neurophysiological abnormalities in adrenoleukodystrophy carriers. Evidence of different degrees of central nervous system involvement. Brain 120(Pt 7):1139–1148
Restuccia D, Di Lazzaro V, Valeriani M et al. (1999) Neurophysiologic follow-up of long-term dietary treatment in adult-onset adrenoleukodystrophy. Neurology 52:810–816
Schaumburg HH, Powers JM, Raine CS, Suzuki K, Richardson EP Jr (1975) Adrenoleukodystrophy. A clinical and pathological study of 17 cases. Arch Neurol 32:577–591
Sikorski AM, Hebert N, Swain RA (2008) Conjugated linoleic acid (CLA) inhibits new vessel growth in the mammalian brain. Brain Res 1213:35–40
Valeriani M, Restuccia D, Le Pera D, Barba C, Tonali P (2000) Scalp distribution of the earliest cortical somatosensory evoked potential to tibial nerve stimulation: proposal of a new recording montage. Clin Neurophysiol 111:1469–1477
Vrethem M, Kvarnstrom M, Stenstam J et al. (2011) Cytokine mapping in cerebrospinal fluid and blood in multiple sclerosis patients without oligoclonal bands. Mult Scler doi:10.1177/1352458511424903
Yamada T, Ishida T, Kudo Y, Rodnitzky RL, Kimura J (1986) Clinical correlates of abnormal P14 in median SEPs. Neurology 36:765–771
Zinkham WH, Kickler T, Borel J, Moser HW (1993) Lorenzo's oil and thrombocytopenia in patients with adrenoleukodystrophy. New Eng J Med 328:1126–1127
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Jean-Marie Saudubray
Competing interest: None declared.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Cappa, M., Bizzarri, C., Petroni, A. et al. A mixture of oleic, erucic and conjugated linoleic acids modulates cerebrospinal fluid inflammatory markers and improve somatosensorial evoked potential in X-linked adrenoleukodystrophy female carriers. J Inherit Metab Dis 35, 899–907 (2012). https://doi.org/10.1007/s10545-011-9432-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-011-9432-3