
Abstract
Soils play an essential role in the global cycling of carbon and understanding the stabilisation mechanisms behind the preservation of soil organic carbon (SOC) pools is of globally recognised significance. Until recently, research into SOC stabilisation has predominantly focused on acidic soil environments and the interactions between SOC and aluminium (Al) or iron (Fe). The interactions between SOC and calcium (Ca) have typically received less attention, with fewer studies conducted in alkaline soils. Although it has widely been established that exchangeable Ca (CaExch) positively correlates with SOC concentration and its resistance to oxidation, the exact mechanisms behind this relationship remain largely unidentified. This synthesis paper critically assesses available evidence on the potential role of Ca in the stabilisation of SOC and identifies research topics that warrant further investigation. Contrary to the common view of the chemistry of base cations in soils, chemical modelling indicates that Ca2+ can readily exchange its hydration shell and create inner sphere complexes with organic functional groups. This review therefore argues that both inner- and outer-sphere bridging by Ca2+ can play an active role in the stabilisation of SOC. Calcium carbonate (CaCO3) can influence occluded SOC stability through its role in the stabilisation of aggregates; however, it could also play an unaccounted role in the direct sorption and inclusion of SOC. Finally, this review highlights the importance of pH as a potential predictor of SOC stabilisation mechanisms mediated by Al- or Fe- to Ca, and their respective effects on SOC dynamics.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Soil organic carbon stabilisation
Soils are the largest actively cycling terrestrial C reservoir and play an essential role in the global cycling of C. Improving our understanding of this C reservoir and modelling its dynamics are fundamental to predicting its sensitivity to future change (Brovkin and Goll 2015). However, current models suffer from large uncertainties caused by the complexities of SOC and its physicochemical interactions with the soil matrix (Friedlingstein et al. 2006). Models require further mechanistic research on the variables that drive SOC dynamics to improve their accuracy (Campbell and Paustian 2015). Therefore, understanding the mechanisms behind the accumulation and persistence of SOC is of globally recognised importance.
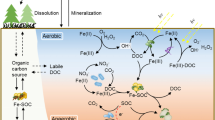
Soil organic C stabilisation broadly refers to mechanisms believed to impede the decomposition of organic matter, promoting its accumulation and persistence in soils. Conversely, decomposition refers to the progressive oxidative transformation of organic inputs, during which a fraction of the organic matter is volatilised as carbon dioxide, while residues become increasingly laden with functional groups, such as carboxyl, phenol, or hydroxyl groups (Guggenberger and Zech 1993; Oste et al. 2002; Peinemann et al. 2005). Sollins et al. (1996) originally proposed three theoretical mechanisms that confer stability to SOC: (i) an inherent recalcitrance or thermodynamic stability of soil organic matter (SOM) and its subsequent selective preservation by decomposers, (ii) the physical occlusion of SOC from decomposers, and (iii) sorption of SOC to inorganic soil components resulting in organo-mineral or organo-cation complexes.
It was previously thought that the primary mechanism behind the persistence of SOC was the selective preservation of thermodynamically stable or recalcitrant substrates by decomposers (Aber et al. 1990; Sollins et al. 1996), causing their accrual within the soil matrix. However, as hypothesised by Oades (1988), there is little evidence for the preservation of complex cell-wall materials like lignin and suberin in stable SOC pools (Gleixner et al. 1999, 2002; Rumpel and Kögel-Knabner 2011; Schmidt et al. 2011). Contrarily, recent evidence suggests that selective preservation is only relevant at the beginning of the SOC decomposition continuum (Dignac et al. 2005; Gleixner et al. 1999, 2002; Lehmann and Kleber 2015; Schmidt et al. 2011) or within organic horizons (Lemma et al. 2007; Preston et al. 2009). The stabilisation and maintenance of SOC in mineral soil horizons, over medium- to long-time periods, is now predominantly thought to be driven by specific ecosystem properties rather than the inherent recalcitrance of SOC (Schmidt et al. 2011).
The established paradigm: ecosystem properties limiting SOC decomposition
Important ecosystem properties that contribute to SOC stabilisation include:
-
(i)
the physical separation of substrates from decomposers over plurimetric to micrometric scales;
-
(ii)
interactions between SOC and cations or minerals;
-
(iii)
the occurrence of temperature or moisture conditions that are incompatible with enzymatic reactions;
-
(iv)
toxicity effects of metal ions like Al3+.
The first and second processes are the most widespread as they occur, theoretically at least, in all soils. The state of knowledge on these processes is briefly synthesised below.
Physical separation
Soil organic carbon (SOC) can be stabilised by its physical separation from decomposers, their enzymes, and the necessary components of aerobic decomposition, such as oxygen or moisture. This physical constrainment can occur over large spatial scales in biologically limiting environments, like those present in Histosols or Cryosols, where waterlogged or frozen conditions severely limit the oxidative degradation of organic substrates (Dörfer et al. 2013), but can also occur at smaller spatial scales. Mechanisms for small scale physical separation include hydrophobic interactions arranging substrates into a micellar structure (Chassin 1979), SOC inclusion within a mineral or co-genetic mineral assemblage (Bindschedler et al. 2016), SOC intercalation within phyllosilicates (Theng et al. 1986), and occlusion of SOC within pedogenic aggregates (Adu and Oades 1978). Thus, the physical constrainment of aerobic decomposition can occur over plurimetric to micrometric scales and stabilises SOC in nearly all soil environments.
Formation of soil aggregates is the most widespread microscale process that leads to the physical separation of SOC, typically labelled as occluded SOC. The relation between aggregation and the stability or accumulation of SOC has been repeatedly demonstrated (Denef et al. 2004; Moni et al. 2010; Monreal et al. 1997; Plante et al. 2002; Skjemstad et al. 1993; Virto et al. 2008, 2010). Formation of aggregates has conventionally been thought to involve the electrostatic flocculation of soil separates into stable domains 2–20 µm in size (Ghezzehei 2011), which are then bound by organic or inorganic cementing agents (Jastrow 1996; Six et al. 2004). Much emphasis has been placed on biological mechanisms that can control aggregation, such as the physical meshing of soil particles by roots and fungi or the excretion of extracellular polysaccharides/polymeric substances by microorganisms and roots (Balesdent et al. 2000; Chenu and Cosentino 2011; Six et al. 2002, 2004). In the theory of biological-controlled aggregate formation (Chenu 1989; Oades and Waters 1991; Oades 1993; Tisdall 1996), fresh SOC acts as an aggregate formation nucleus, stimulating localised activity of microorganism communities. These microorganisms excrete extracellular polysaccharide/polymeric substances that adhere to soil particles, which binds them together, creating a shell around the decomposing SOC nucleus and eventually occluding the SOC residue within (Chenu and Cosentino 2011). When driven by biology, soil structure is typically arranged into a spatial hierarchy, with distinct physical classes of aggregates that are often classified as macroaggregates (> 250 µm) or microaggregates (< 250 µm; Asano and Wagai 2014; Elliott 1986; Oades 1984; Six et al. 2000, 2004; Tisdall 1996; Tisdall and Oades 1982). These aggregate classes have different properties (size, structural stability, porosity, hydrophilicity), which confer different stabilities to the SOC occluded within (Chenu and Cosentino 2011; Dexter 1988; Kleber et al. 2007; Sutton et al. 2005; von Lützow et al. 2006; Zheng et al. 2016). It is largely accepted that in this hierarchy, microaggregates are formed within macroaggregates, which then break apart because of their weaker binding agents and larger planes of weakness, distributing microaggregates into the soil matrix (Oades 1984; Six et al. 2004; Tisdall 1996). These microaggregates are typically considered more stable because of their stronger binding agents and reduced macroporosity, increasing the stability of SOC occluded within (Denef et al. 2004; Tisdall and Oades 1982). Yet despite the recent emphasis on biological controls on soil aggregation, it should be noted that soil aggregation, its hierarchy, and the occlusion of SOC is also influenced by inorganic components of the soil matrix.
Abiotic agents, such as the composition of the mineral soil matrix, can indeed play a dominant role in aggregate formation and stability and therefore, influence occluded SOC. Polyvalent cations are known to increase aggregation in soils by flocculating negatively charged soil separates (Bronick and Lal 2005; Érika et al. 2016; Grant et al. 1992). Inorganic components can also increase the stability of aggregates through cementation, with examples including poorly crystalline minerals (Rasmussen et al. 2005), well crystallised Fe oxides (Oades and Waters 1991; Zhao et al. 2017), or carbonates (Falsone et al. 2010; Fernández-Ugalde et al. 2011, 2014; Virto et al. 2011). Inorganic components have been documented to reinforce both macroaggregates (Fernández-Ugalde et al. 2011; Virto et al. 2013) and microaggregates (Falsone et al. 2010). Some authors have pointed out that when predominantly controlled by inorganic agents, like Fe oxides in Ferralsols (Oxisols; Oades and Waters 1991), soil structure may not display the hierarchical organisation commonly associated with biology. However, when compared to biotic processes, inorganic controls on SOC occlusion have received relatively little attention recently.
Sorption
SOC can also be stabilised through sorptive interactions. These interactions include adsorption to minerals, like phyllosilicate clays, Al-, Fe-, Mn-oxides, poorly crystalline minerals, or polyvalent cations forming bridges to mineral or other organic soil constituents. A positive relationship between the resistance of SOC to chemical oxidation and the presence of specific reactive mineral species was first described by Hosking (1932). Since then, the presence of reactive minerals or metals has been repeatedly shown to correlate with increased SOC stocks (Baldock and Skjemstad 2000; Grand and Lavkulich 2011; Sokoloff 1938; Torn et al. 1997) and the resistance of SOC to microbial degradation in incubation experiments (Minick et al. 2017; Rasmussen et al. 2006; Whittinghill and Hobbie 2012). Soil organic C complexed by minerals generally exhibits older 14C-derived ages than other SOC pools (Kleber et al. 2011; Rasmussen et al. 2005; Schrumpf et al. 2013; Spielvogel et al. 2008; Trumbore 1993); thus adsorption plays a clear role in the stabilisation of SOC over long time periods. However, there is still some level of confusion within the field regarding the chemistry involved. Therefore, the main bonding mechanisms between organic and inorganic soil components are discussed briefly below.
Bonding mechanisms
There is a range of bonding mechanisms that can link organic molecules to minerals or metal cations (Table 1). The prevalence of each bonding mechanism will vary with soil texture, mineralogy, and concentration of cations. SOC can be stabilised through either inner- or outer-sphere interactions with minerals or metal cations (Sposito 2008; Sutton et al. 2005). Inner sphere complexes occur when a substance can closely approach a mineral’s surface or metal ion, usually resulting in direct chemisorption; see Eq. 1 for an example. In outer sphere interactions, water molecules prevent the direct approach or sorption of a substance to a mineral’s surface or metal ion; instead, the charges are countered through a diffuse charged zone (Oldham 2008). In soil, inner- and outer-sphere interactions act in combination to stabilise SOC over medium- to long-time periods, so that it becomes difficult, if not impossible, to ascribe SOC stabilisation in a given horizon to specific modes of interaction. However, a basic understanding of the fundamental chemical mechanisms at play is useful to inform our interpretation of operationally-defined SOC pools (see section below on sorption processes involving Ca).
Equation 1: Ligand exchange between a mineral (M) and a hydroxyl functional group on an organic substrate (R) that results in the direct and strong adsorption of SOC. Equation adapted from Huang and Schnitzer (1986).
Stabilisation by sorption
SOC can be stabilised by organo-cation or organo-mineral interactions through several mechanisms. The primary SOC stabilisation mechanism of adsorption consists of the removal of SOC from solution and transfer to a solid phase. This transfer increases the stability of SOC by reducing the chance of diffusive encounter with degrading enzymes. It can occur whenever organic compounds become adsorbed to mineral surfaces (Kaiser and Guggenberger 2000; Kalbitz et al. 2005), but also when the concentration of cations becomes sufficient for soluble organic polymers to flocculate and precipitate (Baldock and Skjemstad 2000). Much of the research on flocculation thresholds has focused on acid soils dominated by Al chemistry (Boudot 1992; Matus et al. 2006; Rasmussen et al. 2006). In these soils, it has been shown that extensive flocculation and precipitation can be expected at a C:Al ratio in the order of 10–30 or lower (Jansen et al. 2003; Scheel et al. 2007; Skjemstad et al. 1992). The flocculation of dissolved organics by other cations in natural soils has not been as extensively studied.
Beyond the effect of sorption on the partition of SOC between the liquid and solid phase, substrates can also be stabilised by other mechanisms, such as the toxicity effects of certain metals, the inactivation of enzymes during sorption, or steric hindrance. It has been proposed that environmental cytotoxicity could result in the stabilisation of organics complexed by some metals such as Al (Tate and Theng 1980). Al3+ is toxic and is thought to limit decomposer activity in acidic soil environments (Tonneijck et al. 2010), although evidence for this can be contradictory (Marschner and Kalbitz 2003). The extra-cellular enzymes responsible for much of SOM decomposition can also be rendered inactive by adsorption onto mineral surfaces, due to structural modifications in their conformation at the adsorption interface (Quiquampoix and Burns 2007). Steric hindrance is a general mechanism involving the lack-of-fit between a substrate and a catalyst (an enzyme) caused by changes in tertiary structure, which is a common consequence of sorptive interactions in soil (Quiquampoix and Burns 2007; Zimmerman and Ahn 2010). Therefore, the spatial arrangement of adsorbed elements along the molecular interface plays an important role in the stabilisation of SOC during adsorption.
Although it was previously proposed that adsorption occurred uniformly over mineral surfaces, resulting in a monolayer coverage (Keil et al. 1994), this has now been challenged by empirical evidence suggesting that organic loading instead occurs in distinct reactive ‘hotspots’ (Hedges and Keil 1995; Kaiser and Guggenberger 2003; Vogel et al. 2014). Vogel et al. (2014) recently utilised scanning electron microscopy and NanoSIMS to observe the direct adsorption of isotopically labelled SOM on a clay fraction during incubation. The authors found that SOM was preferentially associated with rough areas of nano-mineral clusters, including micropores, etch pits, and cracks. However, the preservation of organic C at these stabilisation ‘hotspots’ is difficult to ascribe to a single mechanism. As hypothesised by Kögel-Knabner et al. (2008), adsorption of SOC within these rough areas provides a twofold stabilisation of SOC, where the accessibility of the substrate to decomposers is reduced and the substrate is concomitantly stabilised by the aforementioned mechanisms of sorption. Therefore, at the molecular-scale, it seems that stabilisation by both physical separation and adsorption simultaneously co-occur (Fig. 1) and become indistinguishable (Chenu and Plante 2006; Kögel-Knabner et al. 2008; Vogel et al. 2014), thus questioning the conceptual segregation of the mechanisms enumerated by Sollins et al. (1996).

Occlusion and sorption co-stabilise soil organic carbon at all spatial scales, but this co-occurrence becomes more apparent at the nano-scale where they become operationally indistinguishable
Ca-mediated SOC stabilisation
Ca–SOC interactions
Research into SOC stabilisation has typically focused on acidic soil environments and the effects of Al3+ or Fe3+ or their poorly crystalline forms on SOC (Grünewald et al. 2006; Kögel-Knabner et al. 2008). Basic soil environments, and potential interactions between the Ca and C cycles have received comparatively less attention (Grünewald et al. 2006). Yet, Ca is the most abundant alkaline earth metal in the Earth’s crust, making up 2.94% of the upper continental crust (Wedepohl 1995). Furthermore, calcareous or Ca-rich soils cover more than 30% of the Earth’s surface (Bertrand et al. 2007; Chen and Barak 1982) and basic soils account for at least 12% of the world’s soil resources (Grünewald et al. 2006). Ca2+ within a soil matrix typically originates from the weathering of lithosphere or surficial formations (Dijkstra et al. 2003; Likens et al. 1998), decomposition of Ca2+-rich organic materials (Ranjbar and Jalali 2012), the lateral movement of Ca2+-rich water (Clarholm and Skyllberg 2013), atmospheric dust deposition (Dijkstra et al. 2003; Pulido-Villena et al. 2006) or anthropogenic inputs. Ca2+ is weathered with relative ease from both primary and secondary minerals (Likens et al. 1998) and has therefore typically been thought to persist or accumulate chiefly in semi-arid to arid environments. However, Ca-rich environments also exist within temperate regions on soils developed from calcareous parent material, out-of-equilibrium with climate (Slessarev et al. 2016). High Ca concentrations are also commonly found in the topsoil of acid soils derived from crystalline lithologies due to biological cycling (Cailleau et al. 2004; Federer and Hornbeck 1985; Grand & Lavkulich 2013; Likens et al. 1998; Ross et al. 1991). Therefore, Ca2+ is an environmentally ubiquitous cation that could potentially play an unaccounted role in the stabilisation of SOC.
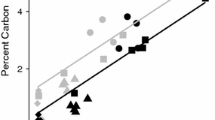
The first investigation into the interactions between Ca and SOC was published almost 80 years ago by Sokoloff (1938), who provided experimental evidence that organic matter solubility decreased upon addition of Ca when compared to Na addition. Since then, research in Ca-rich field environments has highlighted a positive correlation between exchangeable Ca2+ (CaExch) and SOC concentration (see Fig. 2; Bertrand et al. 2007; Bruckert et al. 1986; Clough and Skjemstad 2000; Duchaufour 1982; Gaiffe et al. 1984; Oades 1988; Paul et al. 2003; Shang and Tiessen 2003). As an example, Yang et al. (2016) recently established that alpine grassland soils in the Neotropics contained nearly double the concentration of SOC (405.3 ± 41.7 t ha−1) when developed on Ca-rich, calcareous lithology than their acidic counterparts (226.0 ± 5.6 t ha−1). Similarly, O’Brien et al. (2015) and Li et al. (2017) demonstrated that CaExch was the strongest explanatory variable for SOC concentration of grasslands. However, identification of the exact mechanisms responsible for this widespread correlation remain elusive.

adapted from Gaiffe and Schmitt (1980)
Positive relationship between exchangeable calcium (centimoles of charge per kg) and soil organic carbon concentration (%) in the Jura Mountain range,
A simple case of co-occurrence?
The positive correlation between CaExch and SOC could be explained by their simple co-occurrence, as an increase in SOC concentration generally increases the cation exchange capacity (CEC) of a soil (Yuan et al. 1967). Calcium is a plant macronutrient and there is evidence that Ca also has a localised positive effect on net primary productivity (NPP) and SOM inputs through above-ground and below-ground biomass (Briedis et al. 2012b; Carmeis Filho et al. 2017; Paradelo et al. 2015). This localised effect on NPP has been shown to positively influence the accumulation of SOC in limed Ferralsols (Oxisols; Briedis et al. 2012b; Carmeis Filho et al. 2017) and could explain a portion of the differences in SOC concentration observed between acidic and calcareous soils. However, these explanations fail to account for the decrease in respiration rate (per unit C) associated with Ca prevalence and observed in multiple field studies (Groffman et al. 2006; Hobbie et al. 2002) or incubation experiments (Minick et al. 2017; Whittinghill and Hobbie 2012). These results are at first glance counter-intuitive since the addition of Ca2+ to an edaphic ecosystem is also linked to a change in decomposer communities composition from fungi- to bacterial dominance (Blagodatskaya and Anderson 1999; Rousk et al. 2009, 2010) and an improvement in conditions for bacterial decomposition (Illmer and Schinner 1991; Ivarson 1977; Zelles et al. 1987), at least partially due to the buffering of soil pH to circumneutral levels (Narendrula-Kotha and Nkongolo 2017). It would thus be expected that the rate of enzymatic decomposition of SOC would increase when Ca2+ saturates the exchange complex (Andersson et al. 1999; Chan and Heenan 1999; Thirukkumaran and Morrison 1996). This could be the case in the organic (litter) layer (Minick et al. 2017), but is generally not observed in the mineral soil. Contrary to hypotheses formulated in both studies, Hobbie et al. (2002) and Groffman et al. (2006) found that microbial respiration was actually lower in Ca-rich environments, even though physicochemical conditions for microbial activity were improved. Furthermore, lab experiments have shown that CaExch concentration is correlated with a reduction in SOC leaching losses (Minick et al. 2017), photo-oxidation (7%; Clough and Skjemstad 2000) and respiration as CO2 during incubation (Minick et al. 2017; Whittinghill and Hobbie 2012). Therefore, CaExch seems to be linked to a reduction in the propensity of C substrates for decomposition that is not solely linked to its effects on NPP or microbial ecology.
Consequently, this review will investigate the potential mechanisms behind the stabilisation and accumulation of SOC mediated by Ca and its mineral forms, namely their influence on:
-
(i)
aggregation and the occlusion of SOC;
-
(ii)
inclusion of SOC within pedogenic- or biogenic-CaCO3;
-
(iii)
organo-mineral and organo-cation interactions.
Mechanisms for Ca-mediated SOC stabilisation
Occlusion: Ca and aggregation
The Ca ion and aggregates
It is widely accepted that Ca2+ has a significant positive effect on aggregation and soil structural stability and therefore, indirectly influences the accumulation and occlusion of SOC. Early authors demonstrated an influence of Ca2+ on soil aggregation (Martin et al. 1955; Peterson 1947). This dependence was further investigated by Gaiffe et al. (1984) who demonstrated that the removal of CaExch and its replacement by KExch led to a disruption of aggregates. As theorised by Edwards and Bremner (1967), one of the main mechanisms thought to be behind this stabilisation is the flocculation of negatively charged separates by outer sphere interactions involving Ca2+, which is explored further in the following section on sorption processes. This process operates in the bulk soil and it has also been hypothesised that Ca2+ could play a role in flocculating particles in the gut of certain earthworm species, leading to the formation of ‘Ca humates’ (Satchell 1967). This was supported by the results of Shipitalo and Protz (1989) who utilised micromorphology and chemical pre-treatments to infer that Ca probably played a role in flocculating particles within earthworm casts of certain species, stabilising the microaggregates within them. Another mechanism for the stabilisation of aggregates in Ca-rich environments involves the formation of complexes between Ca and high-molecular weight organic compounds such as root mucilages or microbial polysaccharides/polymeric substances. It has been shown that these substances readily complex Ca2+ and create gel-like structures that bind aggregates (Czarnes et al. 2000; de Kerchove and Elimelech 2007; Erktan et al. 2017; Gessa and Deiana 1992). In particular, galacturonic acids, a common root mucilage, display a high affinity towards Ca, which links polymer chains to form an adhesive matrix (de Kerchove and Elimelech 2007). Czarnes et al. (2000) also showed that these polygalacturonic acid gels increase the hydrophobicity of aggregates, thereby increasing their stability during wetting and drying cycles. Further investigation is needed to analyse the role that adhesive Ca-mucilage matrices play in aggregate stabilisation and the occlusion of SOC.
The effects of carbonate on aggregates
Interactions between Ca-bearing secondary minerals and soil structure have been extensively covered in the literature because of the use of liming (CaCO3 addition) in agriculture. There have been many experiments that have documented the positive effects of the addition of calcite/aragonite (CaCO3) or gypsum (CaSO4.2H2O) on the structure of non-calcareous soils (Armstrong and Tanton 1992; Baldock et al. 1994; Briedis et al. 2012a; Grant et al. 1992; Grünewald et al. 2006; Inagaki et al. 2017; Kaiser et al. 2014; Melvin et al. 2013; Muneer and Oades 1989a; Paradelo et al. 2016). Some authors have also assessed the effects of CaCO3 removal from calcareous soils on aggregate stability, finding that the treatment reduced soil structural stability and increased porosity (Falsone et al. 2010; Muneer and Oades 1989c; Toutain 1974; Virto et al. 2011). Furthermore and reminiscent of the work of Oades and Waters (1991) on Fe oxides in Ferralsols (Oxisols), Fernández-Ugalde et al. (2011) demonstrated that the hierarchical model of aggregation was partially disrupted by carbonate. In the semi-arid Mediterranean soils of their study, the authors showed that the abundance of CaCO3 controlled macroaggregate turnover and increased their stability, to the extent that the usual disruption of macroaggregates, leading to the release of constituent microaggregates, was prevented (Fernández-Ugalde et al. 2011; Oades 1984).
There are several mechanisms by which CaCO3 could positively affect aggregate stability and the occlusion of SOC. CaCO3 is easily weathered and acts as an abundant source of Ca2+, thus encouraging the flocculation of soil separates and aggregation through the mechanisms listed above (Baldock and Skjemstad 2000; Clough and Skjemstad 2000; Wuddivira and Camps-Roach 2007). Carbonate ions are also capable of reprecipitation with Ca2+ under the right environmental conditions, forming secondary CaCO3 crystals (from micrite to sparite) that cement aggregates (Fernández-Ugalde et al. 2011, 2014; Shang and Tiessen 2003; Virto et al. 2013). This mechanism was analysed in detail by Falsone et al. (2010), who utilised N2 adsorption and Hg porosimetry to demonstrate that this formation of secondary CaCO3 crystals decreased aggregate porosity in the 2–50 nm range and thus, decreased the accessibility of intra-microaggregate SOC to decomposers. Certain earthworm species have also been shown to cement particles that pass through their gut with a mixture of poorly crystalline biogenic carbonates (calcite, vaterite, aragonite) excreted from their calciferous glands (Brinza et al. 2014; Edwards and Bohlen 1995). The cementing effect of carbonates on aggregates is well-documented in arid soils in which large concentrations of pedogenic carbonates are found (Fernández-Ugalde et al. 2011, 2014; Virto et al. 2013). However, it may also play a role in humid or sub-humid environments where carbonate-rich parent materials are continually getting dissolved and locally reprecipitated, but this still needs to be investigated further.
Although it is widely accepted that the occurrence of CaCO3 positively affects soil structure and offers favourable conditions for the stabilisation of SOC by occlusion, its actual consequence on occluded SOC stocks is less clear. In a recent review, Paradelo et al. (2015) concluded that while CaCO3 addition had a clear positive effect on soil structure, its effect on occluded SOC stocks was uncertain. Positive (Egan et al. 2018; Muneer and Oades 1989a, b), non-existent (Paradelo et al. 2016) or negative effects (Chan and Heenan 1999) of CaCO3-amendment on occluded SOC have indeed been reported. In some instances, it may be difficult to disentangle the integrative effects of agricultural management from the simple effects of CaCO3 additions. In natural, unamended soils, Fernández-Ugalde et al. (2014) showed that carbonates had a positive effect on occluded SOC stocks. This finding needs to be replicated in a range of natural soils, as differences in initial conditions (e.g. texture, mineralogy, organic inputs and their distribution) could reasonably result in different outcomes.
Inclusion
Inclusion is defined as the envelopment of SOC within a mineral or cogenetic mineral assemblage that leads to its physical protection (Babel 1975). Stabilisation of SOC by inclusion works through a similar mechanism to intercalation or occlusion, by physically separating a substrate from decomposers. SOC may be trapped within any form of pedogenic carbonates, but its inclusion may not be quantitatively important when carbonate formation chiefly occurs through abiotic processes. Diaz et al. (2016) recently dated small concentrations of SOC (0.1–0.5%) included within pedogenic carbonate nodules in Cameroon with C14 measurement, recording ages ranging between 8000 and 13,000 years. This highlights the potential of this mechanism to stabilise SOC over long time periods, but probably only in small concentrations.
Calcium carbonate is one of the most abundant biominerals on Earth and can be synthesised by a wide range of terrestrial organisms (Skinner and Jahren 2007). Biomineralisation of CaCO3 can either be induced within cells, mediated by biological activity that stimulates physicochemical precipitation, or initiated by the presence of an existing biological matrix that initiates crystal nucleation and growth in the extracellular environment (Bindschedler et al. 2016). During each of these forms of biomineralisation, SOC can become included and encapsulated within the crystal structure (Verrecchia et al. 1995). A few specific examples of biogenic carbonate forms include calcified root cells, fungal filaments and rhizoliths (calcified roots; e.g. Becze-Deàk et al. 1997; Jaillard et al. 1991; Monger et al. 1991), calcified earthworm biospheriods (Barta 2011), and the mineralisation of bacterial or fungal organic templates (Bindschedler et al. 2014; Cailleau et al. 2009). Another mechanism for the inclusion of SOC could be biomineralisation pathways such as the oxalate-carbonate pathway (Verrecchia 1990). The oxalate-carbonate pathway involves biomineralisation of CaCO3 during the bacterial catabolism of calcium oxalate-rich SOC produced by plants or fungi. It thus intimately links SOC to the nucleation site of CaCO3 biomineralisation and could allow its inclusion within the crystal matrix in both acidic (Cailleau et al. 2004, 2005; Verrecchia et al. 2006) and calcareous soils (Rowley et al. 2016). However, there has been very little direct quantification of the concentrations or 14C ages of SOC included within biogenic carbonate forms, which could potentially contain much higher SOC concentrations than abiotically-formed pedogenic carbonates. Therefore, further investigation is now needed to quantify the inclusion of SOC within biogenic carbonate and its role in SOC dynamics.
Sorption: Ca, minerals and organics
Organo-mineral interactions with calcite
Lithogenic and pedogenic CaCO3 could also play a key role in the stabilisation of SOC via adsorption. Most of the research into direct organo-calcite interactions has focused on the interactions between DOC and calcite in sorption experiments. Earlier work by Suess (1970) and Carter (1978) showed that DOC could be directly adsorbed onto CaCO3, while Suzuki (2002) more recently showed that CaCO3 was an effective adsorbant of DOC from black tea solutions, possibly due to its high point of zero charge (9.5; Grünewald et al. 2006). Thomas et al. (1993b) more specifically studied the affinity of different synthetic carbonates for common organic functional groups and demonstrated that calcite, dolomite, and magnesite all sorbed a wide range of organic compounds, which included carboxylic acids, alcohols, sulphates, sulfonates, amines, amino acids and carboxylated polymers. Interestingly, interaction with DOC has been shown to modify carbonate precipitation equilibria, by inhibiting either further crystal precipitation (Inskeep and Bloom 1986; Reddy et al. 1990; Reynolds 1978) or the dissolution of sorbent minerals (Thomas et al. 1993a). Jin and Zimmerman (2010) established that CaCO3 obtained from aquifers preferentially adsorbed dissolved organic matter with a high molecular weight, which the authors attributed to a form of outer sphere interaction. It has been theorised that the kinetics of DOC adsorption by carbonates may be biphasic, occurring through an initial rapid phase of outer sphere interactions, which is then followed by a slower phase of inner sphere and hydrophobic interactions that in turn protect the carbonate surface from dissolution (Jin and Zimmerman 2010; Lee et al. 2005; Thomas et al. 1993b). While these DOC adsorption experiments have provided interesting insight into potential CaCO3–SOC interactions, there has been relatively little direct research on the adsorption of SOC by different forms of calcite/aragonite in soils. Measurements of soil carbonate content commonly differentiate between a reactive and a total pool (Pansu and Gautheyrou 2006), but there is little evidence for the role of these operationally-defined pools in adsorption of SOC. Further research should focus on the effects of the supposedly reactive, poorly crystalline or amorphous CaCO3 pool on the adsorption of SOC in natural environments.
Outer sphere processes
Irrespective of their carbonate content, many soils have significant concentrations of free Ca2+ which may also contribute to the stabilisation of SOC. The widely observed correlation between CaExch and SOC has led to the implicit assumption that Ca2+ predominantly affects SOC through weak outer sphere interactions (von Lützow et al. 2006), such as those contributing to the retention of exchangeable cations (Table 1). This form of cation bridging by Ca2+ has been highlighted as an important component of SOC stabilisation by many authors and is well-documented (Clough and Skjemstad 2000; Edwards and Bremner 1967; Oades 1988). As illustrated in the lyotropic series, cations’ outer sphere (exchangeable) behaviour is related to the size of their hydration shell and valence. This is confirmed by chemical modelling, which indicates that exchangeable bridges by Ca2+ typically have a larger residence time than those of monovalent cations, like Na+, because the charge-to-hydration radius ratio of Na+ prevents it from efficiently countering the repulsion between negatively-charged surfaces (Iskrenova-Tchoukova et al. 2010; Sutton et al. 2005). Thus, Ca2+ is a fundamental flocculating agent of natural systems because of its ability to form efficient outer sphere bridge units.
However, it is interesting to note that Al3+ and H+ rate higher than Ca2+ on the lyotropic series and should thus cause similar or higher levels of apparent flocculation in soils in which they are abundant, such as most acid soils. Yet, it is widely observed that colloidal mobility is enhanced in acidic environments where Al3+ and H+ dominate and there is little or no Ca2+ present, such as those associated with the formation of Luvisols (Lavkulich and Arocena 2011). It is also worth considering that the innate reversibility of outer sphere interactions should mean that exchangeable Ca bridges would not be inherently persistent in natural soils. These considerations lead us to explore the possibility that interactions between Ca and SOC are not solely attributable to outer sphere (exchangeable) processes and that, despite its correlative association with SOC, CaExch may not be solely responsible for the bulk of SOC stabilisation in Ca-rich soils.
Inner sphere processes
It is generally observed that each cation has a different range of interactions in soils. For instance, trivalent Fe is seldom found in large amounts as a free ion in soil, as it very readily hydrolyses to form insoluble precipitates under most environmentally-relevant conditions. Al3+ also hydrolyses into insoluble hydroxides at slightly acidic to basic pH, while in acidic soils, it is found to participate both in outer sphere, cation exchange and inner sphere, ligand exchange reactions. Ca2+ is thought to retain its hydration shell and behave strictly like an exchangeable cation, as are other ‘base’ cations such as Mg2+, K+ and Na+ (Essington 2004). However in soil science, the fundamental controls on the propensity of each cation to form inner sphere complexes with SOM are not as well understood as the affinity of cations for non-specific exchange sites.
One of the reasons for this is that there are many factors that can influence inner sphere complexation of SOM by ions in the soil matrix, including characteristics of cations (ionic potential, electronegativity, polarisability of their electron cloud, hydrated radii, propensity to retain their hydration shell), of ligands (amount and type of organic functional groups) and of the environment (pH, ionic strength, solution composition, metal-to-ligand ratio, pressure and temperature conditions; Essington 2004). Cations can be broadly split into three classes (Class A, B, and intermediate/C) based upon the polarisability of their electron cloud, which in turn, indicates how likely they are to form inner sphere complexes with specific ligands (Ahrland et al. 1958; Pearson 1963; Schwarzenbach 1961). Class A cations are weakly polarisable and tend to form complexes with O-containing ligands, such as carboxylate functional groups through ionic bonding. On the other hand, Class B cations have a labile electron cloud and tend to form complexes with N- or S-bearing ligands through more covalent bonding (Langmuir 1997; Sposito 2008). Al3+ and the base cations, including Ca2+, are considered group A cations, indicating that they may theoretically form inner sphere complexes with widely-occurring O-bearing ligands such as carboxylate groups (Sposito 2008). However, each cation’s actual behaviour in soil cannot be predicted from one or a couple of first-principles only, as it results from the interaction of several factors. For instance, Na is not generally seen to engage in inner sphere complexation in soils, while K only does so in the interlayer of specific phyllosilicates. Advanced chemical modelling can offer insight into these issues.
Authors have modelled the interactions between dissolved organic C (DOC) and Ca2+ in an attempt to investigate their molecular-scale interactions (Aristilde and Sposito 2008; Benedetti et al. 1995). These models suggest that Ca2+ can bind to SOC through both inner sphere and outer sphere processes (Bogatko et al. 2013; Iskrenova-Tchoukova et al. 2010; Kalinichev and Kirkpatrick 2007; Sutton et al. 2005). Sutton et al. (2005) modelled the complexation of deprotonated carboxyl groups by Ca2+ and found that their interactions were predominantly inner sphere (75%). The model of Kalinichev and Kirkpatrick (2007) also confirmed that Ca2+ could form direct cation bridges with carboxylate and to a lesser extent, phenolic and other –OH functional groups, unlike Mg2+, whose hydration water is more tightly held (Dontsova and Norton 2002; Kalinichev and Kirkpatrick 2007; Tipping 2005). Chemical modelling thus indicates that Ca2+ can interact with SOC through inner- and outer-sphere processes, thereby potentially increasing SOC stability against decomposition or leaching (Minick et al. 2017).
Building empirical evidence for Ca–SOC interactions
While models predict that Ca2+ can form both inner- and outer-sphere bridges with SOC, empirical evidence of these associations in natural environments remains scarce. Density fractionation, which separates free, occluded and mineral-associated SOC (Golchin et al. 1994) has the potential to offer insight. When performed sequentially (Sollins et al. 2009), density fractionation can separate SOC fractions associated with different minerals; furthermore, because the method uses extremely concentrated salt solutions (usually Na polytungstate), outer sphere associations are not expected to survive the treatment, meaning that only strong (inner sphere) association with minerals are considered. Wen et al. (2017) recently showed that there was more SOC associated with calcite-rich than with dolomite-rich heavy fractions, possibly corroborating modelling predictions of stronger SOC association with Ca than Mg (Kalinichev and Kirkpatrick 2007). The occluded fraction was however not separated from the mineral-associated fraction, so that the results remain somewhat equivocal. Further density fractionation studies analysing the relative role of occlusion and sorption for SOC accumulation in Ca-rich soils would undoubtedly prove informative. Density fractionation is however a costly and time-consuming technique (Poeplau et al. In review) and may be difficult to use in calcareous soils, since polytungstate left in contact with free Ca for extended periods can precipitate as insoluble Ca-metatungstate. Methods applicable to the bulk soil would constitute useful complements to fractionation approaches.
Selective extractions on bulk soil have typically been used to analyse the effects of cation pools on SOC stocks. As indicated in Fig. 2, the operationally defined CaExch pool, extracted by salt solutions, represents a reactive and abundant pool of Ca2+ that is regularly correlated with SOC concentration (Bruckert et al. 1986; Gaiffe and Schmitt 1980; Li et al. 2017; O’Brien et al. 2015), thus highlighting its potential as an indicator variable for the measurement of Ca2+ interacting with SOC. However, by definition, CaExch only represents Ca2+ engaged in outer sphere interactions. The selective chemical extraction of the inner sphere Ca pool, corresponding to pyrophosphate extractions for Al and Fe in acidic soil environments (Bascomb 1968; Parfitt and Childs 1988; Rasmussen et al. 2006), is challenging due to the insolubility of most chelating agents once complexed by Ca (e.g., Ca-pyrophosphate or Ca-oxalate). In a recent study, van der Heijden et al. (2017) isolated a “non-crystalline pool of Ca” in acidic, base-poor soils, which may have included a significant contribution of Ca complexed by SOM, but the extract (dilute oxalic + nitric acid) was not specific to organic complexes. Extraction with other chelating agents that remain soluble in their Ca form (e.g. ethylenediaminetetraacetic acid, EDTA; Bélanger et al. 2008) or with salts of strong cation complexants (e.g. copper chloride; Barra et al. 2001; Juo and Kamprath 1979) may be informative; however, these extractants could also attack the mineral CaCO3 pool, making their use difficult in calcareous soils.
Alternatively, X-ray absorption spectroscopy (XAS) could eventually be used to investigate the coordination environment of Ca2+–SOC complexes under different environmental conditions. As an example, Martin-Diaconescu et al. (2015) have recently successfully probed the coordination environment of synthetic Ca complexes. While powerful, these direct spectroscopic techniques require the use of synchrotron light source and can only be applied to small amounts of samples with limited compositional complexity. We are still lacking a method that allows for the routine assessment of inner sphere Ca–SOM complexes and their relative importance in different soils, which constitutes a significant research gap given the potential for inner sphere interactions to stabilise SOM with increased efficiency (Mikutta et al. 2007), and perhaps through ecosystem disturbance events (Basile-Doelsch et al. 2009; Grand and Lavkulich 2012).
Implications for conceptual models of SOC cycling
Despite the growing body of evidence supporting a major role for specific soil minerals and cations in SOC stabilisation (Doetterl et al. 2015), soil mineralogy and geochemistry are largely absent from leading models of SOC cycling. The following section will discuss a few processes that have the potential to improve representations of SOC stabilisation, with particular emphasis on Ca-rich soils.
Digressing from the expected profile-scale depth distributions
In addition to their influence on the total amount of SOC retained in soil, polyvalent cations are suspected to influence its vertical distribution in soil profiles. Current pedometric approaches to mapping soil C acknowledge the importance of accounting for soil type when estimating the vertical distribution of SOC (Kempen et al. 2011; Wiese et al. 2016). Polyvalent cations can indeed cause departures from the generally assumed exponential decline of SOC concentration with depth (Hilinski 2001). A classical example involves Podzols characterised by the effective translocation of Al–SOC complexes to deep soil horizons (Ferro-Vázquez et al. 2014; Grand and Lavkulich 2011). Contrastingly, Calcisols have an accumulation of Ca-saturated SOC in surface horizons (Yang et al. 2016). This accumulation of Ca-saturated SOC is likely caused by the complexation, flocculation, and precipitation of organic acids and clays in the presence of Ca, precluding their translocation to subsoil horizons. Two of the most common low molecular weight organic acids (oxalic and citric acids) in soil notably form sparingly soluble salts in the presence of Ca (calcium oxalate Ksp ≈ 10−8.5; Certini et al. 2000), preventing their translocation, whereas their Al and Fe counterparts are highly soluble (Gadd 1999). The fundamental differences in how polyvalent cations modulate SOC inputs, stability and depth distributions highlight the critical importance of accounting for geochemical factors when modelling SOC dynamics.
Preferential sorption
Integrating a geochemical dimension into conceptual models of SOM dynamics is also important because the formation of organo-mineral complexes appears to be a preferential process, with selectivity exhibited by both the organic and mineral component (Spielvogel et al. 2008). Very little is actually known about the preferential nature of organo-mineral interactions in soils. To date, there is some evidence within the literature that specific organic compounds such as N-rich microbial metabolites, microbial cell-wall fragments, and possibly pyrogenic C may be selectively sorbed by soil minerals (Brodowski et al. 2007; Jin and Zimmerman 2010; Miltner et al. 2012; Scheel et al. 2008; Schurig et al. 2013; Spielvogel et al. 2008). Furthermore, it has been suggested that some reactive mineral surfaces, such as those of Al and Fe phases, may be disproportionally involved in the sorption of specific classes of organics, such as proteins, lignin, and phenolic compounds (Heckman et al. 2013; Kögel-Knabner et al. 2008; Xiao et al. 2015). However, there have been very few studies looking at the potential preferential sorption of organic compounds in Ca-rich edaphic environments.
If molecular scale stabilisation of SOC by Ca2+ predominantly occurs through inner- or outer-sphere cation bridging, then it should preferentially target functional groups such as carboxyls and phenols. Römkens and Dolfing (1998) and Kaiser (1998) accordingly demonstrated that Ca2+ preferentially flocculated and precipitated high molecular weight DOC compounds, which contained higher proportions of carboxylic and phenolic functional groups. There is also evidence for the preferential adsorption of negatively charged products of lignin degradation (syringyl units; Grünewald et al. 2006) and DOC (Jin and Zimmerman 2010; Jin et al. 2014) by calcite. The implications of this hypothesis for our understanding of SOC dynamics could be profound. It could potentially provide a mechanism to bridge the two competing hypotheses of SOC persistence, i.e. biochemical recalcitrance vs. mineral association, since organic compounds with different compositions could have different probabilities for sorptive preservation. This perspective is highly relevant to SOC modelling. As an example, the CENTURY model assumes universal preservation of lignin in stable SOC pools (Parton 1996; Parton et al. 2015), which has been questioned by experimental evidence (Gleixner et al. 1999, 2002). Accruing evidence on selective sorption of specific compounds to minerals or cations, including lignin derivatives, could speak in favour of considering SOC biochemical composition as a predictor of residence time, but the parametrisation would have to be adjusted for different geochemical environments.
Conversely, Minick et al. (2017) demonstrated that high additions of Ca at the Hubbard Brook experimental forest specifically reduced the mineralisation of 13C-depleted SOC, which should represent a relatively fresh pool, little affected by oxidative transformation (Rumpel and Kögel-Knabner 2011), thus contradicting the hypothesis that Ca2+ preferentially stabilises oxidised SOC. Yet stabilisation of 13C-depleted SOC could simply imply that occlusion was the predominant mechanism of SOC stabilisation at the Hubbard Brook forest. SOC occlusion could limit the mineralisation of 13C-depleted sources because aggregates occlude a heterogeneous mixture of both 13C-enriched, decomposed and relatively fresh, 13C-depleted particulate-organic matter (Poeplau et al. In review). However, this still requires further evidence. Future investigation should specifically focus on the relative importance of occlusion and adsorption for SOC stabilisation, as influenced by the geochemical environment (dominant free cation) and the composition of organic components (esp. prevalence of functional groups).
pH: the master variable for SOC stabilisation mechanisms?
As pH shifts from acidic to basic conditions, so does the importance of SOC stabilisation by different polyvalent cations and their mineral forms, moving from Al3+ or Fe3+ to Ca2+, respectively (Tipping 2005). The prevalence of each cation is indeed largely linked to soil pH due to the buffering capacity of primary and secondary minerals, notably calcite and Al oxides (Oste et al. 2002; Slessarev et al. 2016). As each cation is associated with different SOC stabilisation mechanisms (Fig. 3), this shift in pH could arguably be used to predict the concentration and types of SOC being stabilised in each environment. Therefore, we propose the following conceptual model: in acidic environments, complexation of organic ligands by free Al3+ and Fe3+ as well as their mineral forms (Kalbitz and Kaiser 2008; Scheel et al. 2008) and the cementation of aggregates by Fe oxides (Oades and Waters 1991; Zhao et al. 2017) are likely to control SOC stabilisation. There could also be a localised effect of Ca in the topsoil of these acidic environments caused by biological cycling and resulting accumulation of Ca (Clarholm and Skyllberg 2013). As soil pH increases above 6, Ca2+ becomes more prevalent and consequently, there should be increased evidence of SOC stabilisation by inner- and outer-sphere Ca2+ bridging or Ca-mediated aggregation (Kayler et al. 2011). As soil pH increases further to pH > 8.3, excess Ca2+ will begin to precipitate as CaCO3, reducing the stabilisation by free Ca2+ at the expense of CaCO3-mediated mechanisms (Lindsay 1979). When soil pH starts to increase beyond pH 9.5, soils will become increasingly sodic and dominated by Na+, which tends to disperse soil separates, reducing occlusion (Wong et al. 2010) and sorption of SOC (Iskrenova-Tchoukova et al. 2010; Sutton et al. 2005), and consequently weaken SOC stabilisation.

The shifting role of polyvalent cations in the stabilisation of SOC with increasing pH. A ‘window of opportunity’ for microbial decomposition is highlighted in green according to the proposition of (Clarholm and Skyllberg 2013). Thresholds are based on values available in the literature and it is expected that adjustments will occur as more results become available. Al–Fe oxides refer to true oxides as well as oxyhydroxides and their poorly crystalline forms. OCP oxalate-carbonate pathway
As indicated by Fig. 3, stabilisation of SOC by polyvalent cations is expected to be weakest at near-neutral levels of pH, which also coincides with optimum conditions for bacterial mineralisation (Groffman et al. 2006; Illmer and Schinner 1991; Ivarson 1977; Whittinghill and Hobbie 2012; Zelles et al. 1987). This was suggested previously by Clarholm and Skyllberg (2013) as a “window of opportunity” (highlighted in green; Fig. 3) for C mineralisation. Taking this reasoning a step further, we hypothesize that low and high pH environments will afford different capacities for SOC stabilisation. Given the documented efficiency of sorption by Al and Fe forms and of aggregation by Ca, we postulate that adsorption by Al–Fe oxides generally dominates SOC stabilisation at low pH, stabilising SOC for long time periods; but as the pH increases beyond the “window of opportunity”, it could be expected that the dominant stabilisation mechanism would be occlusion within aggregates, potentially involving larger amounts of SOC but for shorter durations. However, due to the relatively limited body of work on Ca-mediated SOC stabilisation mechanisms, these hypotheses currently remain speculative; the dominant SOC stabilisation mechanisms associated with each cation, the amount of SOC they can affect and the relative strength of the conferred protection still require confirmation. Nonetheless, pH has the potential to act as a fundamental indicator of the mechanisms controlling SOC stabilisation. Therefore, this review suggests that SOC models should consider incorporating pH as a master variable to represent the effects of different stabilisation mechanisms by polyvalent cations and their mineral forms on the accumulation and persistence of SOC.
Conclusions
Although an addition of Ca2+ generally improves microbial conditions for decomposition by increasing pH and reducing stress from H+, it can counter-intuitively reduce respiration rates through the stabilisation of SOC. The main mechanisms behind Ca-mediated SOC stabilisation are likely linked to the ability of Ca2+ to bridge negatively charged surfaces. Chemical modelling indicates that Ca2+ can bridge SOC and minerals through both inner- and outer-sphere interactions, but this still requires empirical confirmation. When scaled up, Ca bridging also positively affects soil structure; yet surprisingly little is known about the implication for the amount and stability of occluded SOC. The relative prevalence of occlusion and adsorption for SOC stabilisation in Ca-influenced soils needs to be determined, as it could have important consequences for the stabilisation of SOC in terms of its maximum amount, mean residence time but also composition. Indeed, there is some evidence that adsorption could preferentially involve some classes of organic compounds whereas occlusion may be relatively indiscriminate, at least at the macroscopic level. Soil pH could also play a fundamental role in predicting the occurrence of these stabilisation mechanisms and should be considered for inclusion in current SOC models. In order to better represent interactions between the C and Ca cycle in conceptual and numerical models of SOC cycling, we suggest that further mechanistic investigation should focus on the quantification of the relative prevalence and strength of each stabilisation mechanism and their variation across pH thresholds.
References
Aber JD, Melillo JM, McClaugherty CA (1990) Predicting long-term patterns of mass loss, nitrogen dynamics, and soil organic matter formation from initial fine litter chemistry in temperate forest ecosystems. Can J Bot 68(10):2201–2208
Adu JK, Oades JM (1978) Physical factors influencing decomposition of organic materials in soil aggregates. Soil Biol Biochem 10(2):109–115
Ahmed E, Holmström SJM (2014) Siderophores in environmental research: roles and applications. Microb Biotechnol 7(3):196–208
Ahrland S, Chatt J, Davies NR (1958) The relative affinities of ligand atoms for acceptor molecules and ions. Q Rev 12(3):265–276
Andersson S, Nilsson I, Valeur I (1999) Influence of dolomitic lime on DOC and DON leaching in a forest soil. Biogeochemistry 47(3):295–315
Aristilde L, Sposito G (2008) Molecular modeling of metal complexation by a fluoroquinolone antibiotic. Environ Toxicol Chem 27(11):2304–2310
Armstrong ASB, Tanton TW (1992) Gypsum applications to aggregated saline—sodic clay topsoils. J Soil Sci 43(2):249–260
Asano M, Wagai R (2014) Evidence of aggregate hierarchy at micro- to submicron scales in an allophanic Andisol. Geoderma 216:62–74
Babel U (1975) Micromorphology of soil organic matter. In: Gieseking JE (ed) Soil components: organic components, vol 1. Springer, Berlin, pp 369–473
Baldock JA, Skjemstad JO (2000) Role of the soil matrix and minerals in protecting natural organic materials against biological attack. Org Geochem 31(7–8):697–710
Baldock J, Aoyama M, Oades J, Grant C (1994) Structural amelioration of a South Australian red-brown earth using calcium and organic amendments. Soil Res 32(3):571–594
Balesdent J, Chenu C, Balabane M (2000) Relationship of soil organic matter dynamics to physical protection and tillage. Soil Tillage Res 53(3–4):215–230
Barra CM, Curtius AJ, de Campos RC, Perez DV (2001) Evaluation of four aluminum extraction methods using selected Brazilian soils. Commun Soil Sci Plant Anal 32(11–12):1969–1980
Barta G (2011) Secondary carbonates in loess-paleosoil sequences: a general review. Central Eur J Geosci 3(2):129–146
Bascomb CL (1968) Distribution of pyrophosphate extractable iron and organic carbon in soils of various groups. J Soil Sci 19(2):251–268
Basile-Doelsch I, Brun T, Borschneck D, Masion A, Marol C, Balesdent J (2009) Effect of landuse on organic matter stabilized in organomineral complexes: a study combining density fractionation, mineralogy and δ13C. Geoderma 151(3):77–86
Becze-Deàk J, Langohr R, Verrecchia EP (1997) Small scale secondary CaCO3 accumulations in selected sections of the European loess belt. Morphological forms and potential for paleoenvironmental reconstruction. Geoderma 76(3):221–252
Bélanger N, Paré D, Hendershot WH (2008) Chapter 27—determining nutrient availability in forest soils. In: Carter MR, Gregorich EG (eds) Soil sampling and methods of analysis. Canadian Society of Soil Science, Pinawa, p 1224
Benedetti MF, Milne CJ, Kinniburgh DG, Van Riemsdijk WH, Koopal LK (1995) Metal ion binding to humic substances: application of the non-ideal competitive adsorption model. Environ Sci Technol 29(2):446–457
Bertrand I, Delfosse O, Mary B (2007) Carbon and nitrogen mineralization in acidic, limed and calcareous agricultural soils: apparent and actual effects. Soil Biol Biochem 39(1):276–288
Bindschedler S, Cailleau G, Braissant O, Milliere L, Job D, Verrecchia EP (2014) Unravelling the enigmatic origin of calcitic nanofibres in soils and caves: purely physicochemical or biogenic processes? Biogeosciences 11(10):2809–2825
Bindschedler S, Cailleau G, Verrecchia E (2016) Role of fungi in the biomineralization of calcite. Minerals 6(2):41
Blagodatskaya EV, Anderson T-H (1999) Adaptive responses of soil microbial communities under experimental acid stress in controlled laboratory studies. Appl Soil Ecol 11(2–3):207–216
Bogatko S, Cauët E, Bylaska E, Schenter G, Fulton J, Weare J (2013) The aqueous Ca2+ system, in comparison with Zn2+, Fe3+, and Al3+: an ab initio molecular dynamics study. Chem Eur J 19(9):3047–3060
Boudot J-P (1992) Relative efficiency of complexed aluminum noncrystalline Al hydroxide, allophane and imogolite in retarding the biodegradation of citric acid. Geoderma 52(1):29–39
Briedis C, de Moraes Sá JC, Caires EF, de Fátima Navarro J, Inagaki TM, Boer A, de Oliveira Ferreira A, Neto CQ, Canalli LB, Bürkner dos Santos J (2012a) Changes in organic matter pools and increases in carbon sequestration in response to surface liming in an oxisol under long-term no-till. Soil Sci Soc Am J 76(1):151–160
Briedis C, Sá JCdM, Caires EF, Navarro JdF, Inagaki TM, Boer A, Neto CQ, Ferreira AdO, Canalli LB, Santos JBd (2012b) Soil organic matter pools and carbon-protection mechanisms in aggregate classes influenced by surface liming in a no-till system. Geoderma 170:80–88
Brinza L, Schofield PF, Hodson ME, Weller S, Ignatyev K, Geraki K, Quinn PD, Mosselmans JFW (2014) Combining µXANES and µXRD mapping to analyse the heterogeneity in calcium carbonate granules excreted by the earthworm Lumbricus terrestris. J Synchrotron Radiat 21(Pt 1):235–241
Brodowski S, Amelung W, Haumaier L, Zech W (2007) Black carbon contribution to stable humus in German arable soils. Geoderma 139(1–2):220–228
Bronick CJ, Lal R (2005) Soil structure and management: a review. Geoderma 124(1–2):3–22
Brovkin V, Goll D (2015) Land unlikely to become large carbon source. Nat Geosci 8(12):893
Bruckert S, Gaiffe M, Duquet B, Tavant Y, Tavant H (1986) Rôle du flux de calcium sur la stabilisation de la matière organique des sols. Ann Sci Univ France Comté Besançon 4(6):25–29
Cailleau G, Braissant O, Verrecchia EP (2004) Biomineralization in plants as a long-term carbon sink. Naturwissenschaften 91(4):191–194
Cailleau G, Braissant O, Dupraz C, Aragno M, Verrecchia EP (2005) Biologically induced accumulations of CaCO3 in orthox soils of Biga, Ivory Coast. Catena 59(1):1–17
Cailleau G, Verrecchia EP, Braissant O, Emmanuel L (2009) The biogenic origin of needle fibre calcite. Sedimentology 56(6):1858–1875
Campbell EE, Paustian K (2015) Current developments in soil organic matter modeling and the expansion of model applications: a review. Environ Res Lett 10(12):123004
Carmeis Filho ACA, Penn CJ, Crusciol CAC, Calonego JC (2017) Lime and phosphogypsum impacts on soil organic matter pools in a tropical oxisol under long-term no-till conditions. Agric Ecosyst Environ 241:11–23
Carter P (1978) Adsorption of amino acid-containing organic matter by calcite and quartz. Geochim Cosmochim Acta 42(8):1239–1242
Certini G, Corti G, Ugolini FC (2000) Vertical trends of oxalate concentration in two soils under Abies alba from Tuscany (Italy). J Plant Nutr Soil Sci 163(2):173–177
Chan KY, Heenan DP (1999) Lime-induced loss of soil organic carbon and effect on aggregate stability. Soil Sci Soc Am J 63(6):1841–1844
Chassin P (1979) Determination of the angle of contact of humic acids diols solutions—consequences on the mechanisms of aggregate destruction. Ann Agron 30(6):481–491
Chen Y, Barak P (1982) Iron nutrition of plants in calcareous soils. In: Brady NC (ed) Advances in agronomy. Academic Press, Waltham, pp 217–240
Chenu C (1989) Influence of a fungal polysaccharide, scleroglucan, on clay microstructures. Soil Biol Biochem 21(2):299–305
Chenu C, Cosentino D (2011) Microbial regulation of soil structural dynamics. In: Ritz K, Young IM (eds) The architecture and biology of soils: life in inner space. CABI, Cambridge, pp 37–70
Chenu C, Plante AF (2006) Clay-sized organo-mineral complexes in a cultivation chronosequence: revisiting the concept of the ‘primary organo-mineral complex’. Eur J Soil Sci 57(4):596–607
Clarholm M, Skyllberg U (2013) Translocation of metals by trees and fungi regulates pH, soil organic matter turnover and nitrogen availability in acidic forest soils. Soil Biol Biochem 63:142–153
Clough A, Skjemstad JO (2000) Physical and chemical protection of soil organic carbon in three agricultural soils with different contents of calcium carbonate. Aust J Soil Res 38(5):1005–1016
Czarnes S, Hallett PD, Bengough AG, Young IM (2000) Root- and microbial-derived mucilages affect soil structure and water transport. Eur J Soil Sci 51(3):435–443
de Kerchove AJ, Elimelech M (2007) Formation of polysaccharide gel layers in the presence of Ca2+ and K+ ions: measurements and mechanisms. Biomacromol 8(1):113–121
Denef K, Six J, Merckx R, Paustian K (2004) Carbon sequestration in microaggregates of no-tillage soils with different clay mineralogy. Soil Sci Soc Am J 68(6):1935–1944
Dexter AR (1988) Advances in characterization of soil structure. Soil and Tillage Research 11(3):199–238
Diaz N, Dietrich F, King GE, Valla PG, Sebag D, Herman F, Verrecchia EP (2016) A 20-ka reconstruction of a Sahelo-Sudanian paleoenvironment using multi-method dating on pedogenic carbonate. In: EGU general assembly conference abstracts, vol 18, p 4243
Dignac MF, Bahri H, Rumpel C, Rasse DP, Bardoux G, Balesdent J, Girardin C, Chenu C, Mariotti A (2005) Carbon-13 natural abundance as a tool to study the dynamics of lignin monomers in soil: an appraisal at the Closeaux experimental field (France). Geoderma 128(1–2):3–17
Dijkstra FA, Van Breemen N, Jongmans AG, Davies GR, Likens GE (2003) Calcium weathering in forested soils and the effect of different tree species. Biogeochemistry 62(3):253–275
Doetterl S, Stevens A, Six J, Merckx R, Van Oost K, Casanova Pinto M, Casanova-Katny A, Munoz C, Boudin M, Zagal Venegas E, Boeckx P (2015) Soil carbon storage controlled by interactions between geochemistry and climate. Nature Geosci 8(10):780–783
Dontsova KM, Norton LD (2002) Clay dispersion, infiltration, and erosion as influenced by exchangeable Ca and Mg. Soil Sci 167(3):184–193
Dörfer C, Kühn P, Baumann F, He J-S, Scholten T (2013) Soil organic carbon pools and stocks in permafrost-affected soils on the Tibetan Plateau. PLoS ONE 8(2):e57024
Duchaufour R (1982) Pedology: pedogenesis and classification. Springer, Dordrecht
Edwards CA, Bohlen PJ (1995) Biology and ecology of earthworms. Springer, Dordrecht
Edwards AP, Bremner JM (1967) Microaggregates in soil. J Soil Sci 18(1):64
Egan G, Crawley MJ, Fornara DA (2018) Effects of long-term grassland management on the carbon and nitrogen pools of different soil aggregate fractions. Sci Total Environ 613–614(Suppl C):810–819
Elliott ET (1986) Aggregate structure and carbon, nitrogen, and phosphorus in native and cultivated soils. Soil Sci Soc Am J 50(3):627–633
Érika AdS, Geraldo CdO, Carla EC, José MdL, Laura BBdM, Pedro ANB (2016) Stability of soil aggregates in Latosols and Cambisols via standard method and sonification. Afr J Agric Res 11(39):3894–3903
Erktan A, Balmot J, Merino-Martín L, Monnier Y, Pailler F, Coq S, Abiven S, Stokes A, Le Bissonnais Y (2017) Immediate and long-term effect of tannins on the stabilization of soil aggregates. Soil Biol Biochem 105:197–205
Essington ME (2004) Soil and water chemistry. An intergrative approach. CRC Press, Boca Raton
Falsone G, Catoni M, Bonifacio E (2010) Effects of calcite on the soil porous structure: natural and experimental conditions. Agrochimica 54(1):1–12
Federer CA, Hornbeck JW (1985) The buffer capacity of forest soils in new England. Water Air Soil Pollut 26(2):163–173
Fernández-Ugalde O, Virto I, Barré P, Gartzia-Bengoetxea N, Enrique A, Imaz MJ, Bescansa P (2011) Effect of carbonates on the hierarchical model of aggregation in calcareous semi-arid Mediterranean soils. Geoderma 164(3–4):203–214
Fernández-Ugalde O, Virto I, Barré P, Apesteguía M, Enrique A, Imaz MJ, Bescansa P (2014) Mechanisms of macroaggregate stabilisation by carbonates: implications for organic matter protection in semi-arid calcareous soils. Soil Res 52(2):180–192
Ferro-Vázquez C, Nóvoa-Muñoz JC, Costa-Casais M, Klaminder J, Martínez-Cortizas A (2014) Metal and organic matter immobilization in temperate Podzols: a high resolution study. Geoderma 217–218:225–234
Friedlingstein P, Cox P, Betts R, Bopp L, Von Bloh W, Brovkin V, Cadule P, Doney S, Eby M, Fung I, Bala G, John J, Jones C, Joos F, Kato T, Kawamiya M, Knorr W, Lindsay K, Matthews HD, Raddatz T, Rayner P, Reick C, Roeckner E, Schnitzler KG, Schnur R, Strassmann K, Weaver AJ, Yoshikawa C, Zeng N (2006) Climate-carbon cycle feedback analysis: results from the (CMIP)-M-4 model intercomparison. J Clim 19(14):3337–3353
Gadd GM (1999) Fungal production of citric and oxalic acid: importance in metal speciation, physiology and biogeochemical processes. In: Poole RK (ed) Advances in microbial physiology. Academic Press, New York, pp 47–92
Gaiffe M, Schmitt A (1980) Sols et végétation à l’étage montagnard dans les forêts du Jura Central. Science du sol: Bulletin l’Association Francaise pour l’Étude du sol 4:265–296
Gaiffe M, Duquet B, Tavant H, Tavant Y, Bruckert S (1984) Biological stability and physical stability of a clay-humus complex placed under different conditions of calcium or potassium saturation. Plant Soil 77(2–3):271–284
Gessa C, Deiana S (1992) Ca-polygalacturonate as a model for a soil-root interface: II. Fibrillar structure and comparison with natural root mucilage. Plant Soil 140(1):1–13
Ghezzehei TA (2011) Soil structure. In: Huang PA, Li Y, Sumner ME (eds) Handbook of soil sciences. CRC Press, Boca Raton, pp 1–18
Gleixner G, Bol R, Balesdent J (1999) Molecular insight into soil carbon turnover. Rapid Commun Mass Spectrom 13(13):1278–1283
Gleixner G, Poirier N, Bol R, Balesdent J (2002) Molecular dynamics of organic matter in a cultivated soil. Org Geochem 33(3):357–366
Golchin A, Oades JM, Skjemstad JO, Clarke P (1994) Study of free and occluded particulate organic-matter in soils by solid-state C-13 CP/MAS NMR-spectroscopy and scanning electron microscopy. Aust J Soil Res 32(2):285–309
Grand S, Lavkulich LM (2011) Depth distribution and predictors of soil organic carbon in Podzols of a forested watershed in southwestern Canada. Soil Sci 176(4):164–174
Grand S, Lavkulich LM (2012) Effects of forest harvest on soil carbon and related variables in Canadian spodosols. Soil Sci Soc Am J 76(5):1816–1827
Grand S, Lavkulich LM (2013) Potential influence of poorly crystalline minerals on soil chemistry in Podzols of southwestern Canada. Eur J Soil Sci 64(5):651–660
Grant CD, Dexter AR, Oades JM (1992) Residual effects of additions of calcium compounds on soil structure and strength. Soil Tillage Res 22(3):283–297
Groffman PM, Fisk MC, Driscoll CT, Likens GE, Fahey TJ, Eagar C, Pardo LH (2006) Calcium additions and microbial nitrogen cycle processes in a Northern Hardwood Forest. Ecosystems 9(8):1289–1305
Grünewald G, Kaiser K, Jahn R, Guggenberger G (2006) Organic matter stabilization in young calcareous soils as revealed by density fractionation and analysis of lignin-derived constituents. Org Geochem 37(11):1573–1589
Guggenberger G, Zech W (1993) Dissolved organic carbon control in acid forest soils of the Fichtelgebirge (Germany) as revealed by distribution patterns and structural composition analyses. Geoderma 59(1):109–129
Heckman K, Grandy AS, Gao X, Keiluweit M, Wickings K, Carpenter K, Chorover J, Rasmussen C (2013) Sorptive fractionation of organic matter and formation of organo-hydroxy-aluminum complexes during litter biodegradation in the presence of gibbsite. Geochim Cosmochim Acta 121:667–683
Hedges JI, Keil RG (1995) Sedimentary organic-matter preservation—an assessment and speculative synthesis. Mar Chem 49(2–3):81–115
Hilinski T (2001) Implementation of exponential depth distribution of organic carbon in the CENTURY model. Colorado State University, Department of Soil and Crop Science, Fort Collins
Hobbie SE, Miley TA, Weiss MS (2002) Carbon and nitrogen cycling in soils from acidic and nonacidic tundra with different glacial histories in Northern Alaska. Ecosystems 5(8):0761–0774
Hosking JS (1932) The influence of hydrogen-ion concentration on the decomposition of soil organic matter by hydrogen peroxide. J Agric Sci 22:92–100
Huang PM, Schnitzer M (1986) Interactions of soil minerals with natural organics and microbes. The Soil Science Society of America, Madison
Illmer P, Schinner F (1991) Effects of lime and nutrient salts on the microbiological activities of forest soils. Biol Fertil Soils 11(4):261–266
Inagaki TM, de Moraes Sá JC, Caires EF, Gonçalves DRP (2017) Why does carbon increase in highly weathered soil under no-till upon lime and gypsum use? Sci Total Environ 599–600:523–532
Inskeep WP, Bloom PR (1986) Kinetics of calcite precipitation in the presence of water-soluble organic ligands1. Soil Sci Soc Am J 50(5):1167–1172
Iskrenova-Tchoukova E, Kalinichev AG, Kirkpatrick RJ (2010) Metal cation complexation with natural organic matter in aqueous solutions: molecular dynamics simulations and potentials of mean force. Langmuir 26(20):15909–15919
Israelachvili JN (2011) 6—Van der Waals forces. Intermolecular and Surface Forces, 3rd edn. Academic Press, San Diego, pp 107–132
Ivarson KC (1977) Changes in decompostion rate, microbial population and carbohydrate content of an acid peat bog after liming and reclamation. Can J Soil Sci 57(2):129–137
Jaillard B, Guyon A, Maurin AF (1991) Structure and composition of calcified roots, and their identification in calcareous soils. Geoderma 50(3):197–210
Jansen B, Nierop KGJ, Verstraten JM (2003) Mobility of Fe(II), Fe(III) and Al in acidic forest soils mediated by dissolved organic matter: influence of solution pH and metal/organic carbon ratios. Geoderma 113(3):323–340
Jastrow JD (1996) Soil aggregate formation and the accrual of particulate and mineral-associated organic matter. Soil Biol Biochem 28(4–5):665–676
Jin J, Zimmerman AR (2010) Abiotic interactions of natural dissolved organic matter and carbonate aquifer rock. Appl Geochem 25(3):472–484
Jin J, Zimmerman AR, Moore PJ, Martin JB (2014) Organic and inorganic carbon dynamics in a karst aquifer: Santa Fe River Sink-Rise system, north Florida, USA. J Geophys Res Biogeosci 119(3):340–357
Juo ASR, Kamprath EJ (1979) Copper chloride as an extractant for estimating the potentially reactive aluminum pool in acid soils1. Soil Sci Soc Am J 43(1):35–38
Kaiser K (1998) Fractionation of dissolved organic matter affected by polyvalent metal cations. Org Geochem 28(12):849–854
Kaiser K, Guggenberger G (2000) The role of DOM sorption to mineral surfaces in the preservation of organic matter in soils. Org Geochem 31(7–8):711–725
Kaiser K, Guggenberger G (2003) Mineral surfaces and soil organic matter. Eur J Soil Sci 54(2):219–236
Kaiser M, Ghezzehei TA, Kleber M, Myrold DD, Berhe AA (2014) Influence of calcium carbonate and charcoal applications on organic matter storage in silt-sized aggregates formed during a microcosm experiment. Soil Sci Soc Am J 78(5):1624–1631
Kalbitz K, Kaiser K (2008) Contribution of dissolved organic matter to carbon storage in forest mineral soils. J Plant Nutr Soil Sci 171(1):52–60
Kalbitz K, Schwesig D, Rethemeyer J, Matzner E (2005) Stabilization of dissolved organic matter by sorption to the mineral soil. Soil Biol Biochem 37(7):1319–1331
Kalinichev AG, Kirkpatrick RJ (2007) Molecular dynamics simulation of cationic complexation with natural organic matter. Eur J Soil Sci 58(4):909–917
Kayler ZE, Kaiser M, Gessler A, Ellerbrock RH, Sommer M (2011) Application of delta C-13 and delta N-15 isotopic signatures of organic matter fractions sequentially separated from adjacent arable and forest soils to identify carbon stabilization mechanisms. Biogeosciences 8(10):2895–2906
Keil RG, Tsamakis E, Fuh CB, Giddings JC, Hedges JI (1994) Mineralogical and textural controls on the organic composition of coastal marine-sediments—hydrodynamic separation using splitt-fractionation. Geochim Cosmochim Acta 58(2):879–893
Kempen B, Brus DJ, Stoorvogel JJ (2011) Three-dimensional mapping of soil organic matter content using soil type-specific depth functions. Geoderma 162(1–2):107–123
Kleber M, Sollins P, Sutton R (2007) A conceptual model of organo-mineral interactions in soils: self-assembly of organic molecular fragments into zonal structures on mineral surfaces. Biogeochemistry 85(1):9–24
Kleber M, Nico PS, Plante A, Filley T, Kramer M, Swanston C, Sollins P (2011) Old and stable soil organic matter is not necessarily chemically recalcitrant: implications for modeling concepts and temperature sensitivity. Glob Change Biol 17(2):1097–1107
Kögel-Knabner I, Guggenberger G, Kleber M, Kandeler E, Kalbitz K, Scheu S, Eusterhues K, Leinweber P (2008) Organo-mineral associations in temperate soils: integrating biology, mineralogy, and organic matter chemistry. J Plant Nutr Soil 171(1):61–82
Langmuir D (1997) Aqueous environmental geochemistry. Prentice Hall, Upper Saddle River
Lavkulich LM, Arocena JM (2011) Luvisolic soils of Canada: genesis, distribution, and classification. Can J Soil Sci 91(5):781–806
Lee YJ, Elzinga EJ, Reeder RJ (2005) Cu(II) adsorption at the calcite–water interface in the presence of natural organic matter: kinetic studies and molecular-scale characterization. Geochim Cosmochim Acta 69(1):49–61
Lehmann J, Kleber M (2015) The contentious nature of soil organic matter. Nature 528(7580):60–68
Lemma B, Nilsson I, Kleja DB, Olsson M, Knicker H (2007) Decomposition and substrate quality of leaf litters and fine roots from three exotic plantations and a native forest in the southwestern highlands of Ethiopia. Soil Biol Biochem 39(9):2317–2328
Li D, Wen L, Yang L, Luo P, Xiao K, Chen H, Zhang W, He X, Chen H, Wang K (2017) Dynamics of soil organic carbon and nitrogen following agricultural abandonment in a karst region. J Geophys Res Biogeosci 122:230–242
Likens GE, Driscoll CT, Buso DC, Siccama TG, Johnson CE, Lovett GM, Fahey TJ, Reiners WA, Ryan DF, Martin CW, Bailey SW (1998) The biogeochemistry of calcium at Hubbard Brook. Biogeochemistry 41(2):89–173
Lindsay W (1979) Chemical equilibria in soils. Wiley, New York
Marschner B, Kalbitz K (2003) Controls of bioavailability and biodegradability of dissolved organic matter in soils. Geoderma 113(3–4):211–235
Martin JP, Martin WP, Page JB, Raney WA, de Ment JD (1955) Soil aggregation. In: Norman AG (ed) Advances in agronomy. Academic Press, Waltham, pp 1–37
Martin-Diaconescu V, Gennari M, Gerey B, Tsui E, Kanady J, Tran R, Pécaut J, Maganas D, Krewald V, Gouré E, Duboc C, Yano J, Agapie T, Collomb M-N, DeBeer S (2015) Ca K-edge XAS as a probe of calcium centers in complex systems. Inorg Chem 54(4):1283–1292
Matus F, Amigo X, Kristiansen SM (2006) Aluminium stabilization controls organic carbon levels in Chilean volcanic soils. Geoderma 132(1):158–168
Melvin AM, Lichstein JW, Goodale CL (2013) Forest liming increases forest floor carbon and nitrogen stocks in a mixed hardwood forest. Ecol Appl 23(8):1962–1975
Mikutta R, Mikutta C, Kalbitz K, Scheel T, Kaiser K, Jahn R (2007) Biodegradation of forest floor organic matter bound to minerals via different binding mechanisms. Geochim Cosmochim Acta 71(10):2569–2590
Mikutta R, Lorenz D, Guggenberger G, Haumaier L, Freund A (2014) Properties and reactivity of Fe-organic matter associations formed by coprecipitation versus adsorption: clues from arsenate batch adsorption. Geochim Cosmochim Acta 144:258–276
Miltner A, Bombach P, Schmidt-Brucken B, Kastner M (2012) SOM genesis: microbial biomass as a significant source. Biogeochemistry 111(1–3):41–55
Minick KJ, Fisk MC, Groffman PM (2017) Soil Ca alters processes contributing to C and N retention in the Oa/A horizon of a northern hardwood forest. Biogeochemistry 132:343–357
Monger HC, Daugherty LA, Lindemann WC, Liddell CM (1991) Microbial precipitation of pedogenic calcite. Geology 19(10):997–1000
Moni C, Rumpel C, Virto I, Chabbi A, Chenu C (2010) Relative importance of sorption versus aggregation for organic matter storage in subsoil horizons of two contrasting soils. Eur J Soil Sci 61(6):958–969
Monreal CM, Schulten HR, Kodama H (1997) Age, turnover and molecular diversity of soil organic matter in aggregates of a Gleysol. Can J Soil Sci 77(3):379–388
Muneer M, Oades JM (1989a) The role of Ca-organic interactions in soil aggregate stability.1. Laboratory studies with glucose-C-14, CaCO3 and CaSO4.2H2O. Aust J Soil Res 27(2):389–399
Muneer M, Oades JM (1989b) The role of Ca-organic interactions in soil aggregate stability.2. Field studies with C-14-labelled straw, CaCO3 and CaSO4.2H2O. Aust J Soil Res 27(2):401–409
Muneer M, Oades JM (1989c) The role of Ca-organic interactions in soil aggregate stability. 3. Mechanisms and models. Aust J Soil Res 27(2):411–423
Narendrula-Kotha R, Nkongolo KK (2017) Microbial response to soil liming of damaged ecosystems revealed by pyrosequencing and phospholipid fatty acid analyses. PLoS ONE 12(1):e0168497
Oades JM (1984) Soil organic matter and structural stability: mechanisms and implications for management. Plant Soil 76(1/3):319–337
Oades JM (1988) The retention of organic matter in soils. Biogeochemistry 5(1):35–70
Oades JM (1993) The role of biology in the formation, stabilization and degradation of soil structure. Geoderma 56(1):377–400
Oades J, Waters A (1991) Aggregate hierarchy in soils. Soil Res 29(6):815–828
O’Brien SL, Jastrow JD, Grimley DA, Gonzalez-Meler MA (2015) Edaphic controls on soil organic carbon stocks in restored grasslands. Geoderma 251–252:117–123
Oldham KB (2008) A Gouy–Chapman–Stern model of the double layer at a (metal)/(ionic liquid) interface. J Electroanal Chem 613(2):131–138
Oste LA, Temminghoff EJM, Riemsdijk WHV (2002) Solid-solution partitioning of organic matter in soils as influenced by an increase in pH or Ca concentration. Environ Sci Technol 36(2):208–214
Pansu M, Gautheyrou J (2006) Handbook of soil analysis: mineralogical, organic and inorganic methods. Springer, Berlin
Paradelo R, Virto I, Chenu C (2015) Net effect of liming on soil organic carbon stocks: a review. Agric Ecosyst Environ 202:98–107
Paradelo R, van Oort F, Barre P, Billiou D, Chenu C (2016) Soil organic matter stabilization at the pluri-decadal scale: insight from bare fallow soils with contrasting physicochemical properties and macrostructures. Geoderma 275:48–54
Parfitt RL, Childs CW (1988) Estimation of forms of Fe and Al—a review, and analysis of contasting soils by dissolution and mossbauer methods. Aust J Soil Res 26(1):121–144
Parton WJ (1996) The CENTURY model. In: Powlson DS, Smith P, Smith JU (eds) Evaluation of soil organic matter models: using existing long-term datasets. Springer, Berlin, pp 283–291
Parton WJ, Del Grosso SJ, Plante AF, Adair EC, Lutz SM (2015) Modeling the dynamics of soil organic matter and nutrient cycling. In: Paul EA (ed) Soil microbiology, ecology and biochemistry, 4th edn. Academic Press, Boston, pp 505–537
Paul EA, Morris SJ, Six J, Paustian K, Gregorich EG (2003) Interpretation of soil carbon and nitrogen dynamics in agricultural and afforested soils. Soil Sci Soc Am J 67(5):1620–1628
Pearson RG (1963) Hard and soft acids and bases. J Am Chem Soc 85(22):3533–3539
Peinemann N, Guggenberger G, Zech W (2005) Soil organic matter and its lignin component in surface horizons of salt-affected soils of the Argentinian Pampa. Catena 60(2):113–128
Peterson JB (1947) Calcium linkage, a mechanism in soil granulation. Soil Sci Soc Am J 12:29–34
Plante AF, Feng Y, McGill WB (2002) A modeling approach to quantifying soil macroaggregate dynamics. Can J Soil Sci 82(2):181–190
Preston CM, Nault JR, Trofymow JA (2009) Chemical changes during 6 years of decomposition of 11 litters in some canadian forest sites. Part 2. 13C abundance, solid-state 13C NMR spectroscopy and the meaning of “lignin”. Ecosystems 12(7):1078–1102
Pulido-Villena E, Reche I, Morales-Baquero R (2006) Significance of atmospheric inputs of calcium over the southwestern Mediterranean region: high mountain lakes as tools for detection. Glob Biogeochem Cycles 20(2):1–8
Quiquampoix H, Burns RG (2007) Interactions between proteins and soil mineral surfaces: environmental and health consequences. Elements 3(6):401–406
Ranjbar F, Jalali M (2012) Calcium, magnesium, sodium, and potassium release during decomposition of some organic residues. Commun Soil Sci Plant Anal 43(4):645–659
Rasmussen C, Torn MS, Southard RJ (2005) Mineral assemblage and aggregates control carbon dynamics in a California conifer forest. Soil Sci Soc Am J 69(6):1711–1721
Rasmussen C, Southard RJ, Horwath WR (2006) Mineral control of organic carbon mineralization in a range of temperate conifer forest soils. Glob Change Biol 12(5):834–847
Reddy KJ, Lindsay WL, Workman SM, Drever JI (1990) Measurement of calcite ion activity products in soils. Soil Sci Soc Am J 54(1):67–71
Reynolds RC (1978) Polyphenol inhibition of calcite precipitation in Lake Powell 1. Limnol Oceanogr 23(4):585–597
Römkens PFAM, Dolfing J (1998) Effect of Ca on the solubility and molecular size distribution of DOC and Cu binding in soil solution samples. Environ Sci Technol 32(3):363–369
Ross DS, Bartlett RJ, Magdoff FR (1991) Exchangeable cations and the pH-independent distribution of cation exchange capacities in Spodosols of a forested watershed. In: Wright RJ, Baligar VC, Murrmann RP (eds) Proceedings of the second international symposium on plant-soil interactions at low pH, 24–29 June 1990, Beckley, WV. Springer, Dordrecht, pp 81–92
Rousk J, Brookes PC, Baath E (2009) Contrasting soil pH effects on fungal and bacterial growth suggest functional redundancy in carbon mineralization. Appl Environ Microbiol 75(6):1589–1596
Rousk J, Baath E, Brookes PC, Lauber CL, Lozupone C, Caporaso JG, Knight R, Fierer N (2010) Soil bacterial and fungal communities across a pH gradient in an arable soil. Int Soc Microb Ecol J 4(10):1340–1351
Rowley MC, Estrada-Medina H, Tzec-Gamboa M, Rozin A, Cailleau G, Verrecchia EP, Green I (2016) Moving carbon between spheres, the potential oxalate-carbonate pathway of Brosimum alicastrum Sw Moraceae. Plant Soil 412:465–479
Rumpel C, Kögel-Knabner I (2011) Deep soil organic matter-a key but poorly understood component of terrestrial C cycle. Plant Soil 338(1–2):143–158
Satchell JE (1967) Lumbricidae. In: Burgess A, Raw F (eds) Soil biology. Academic Press, London, pp 259–322
Scheel T, Dörfler C, Kalbitz K (2007) Precipitation of dissolved organic matter by aluminum stabilizes carbon in acidic forest soils. Soil Sci Soc Am J 71(1):64–74
Scheel T, Jansen B, Van Wijk AJ, Verstraten JM, Kalbitz K (2008) Stabilization of dissolved organic matter by aluminium: a toxic effect or stabilization through precipitation? Eur J Soil Sci 59(6):1122–1132
Schmidt MWI, Torn MS, Abiven S, Dittmar T, Guggenberger G, Janssens IA, Kleber M, Kögel-Knabner I, Lehmann J, Manning DAC, Nannipieri P, Rasse DP, Weiner S, Trumbore SE (2011) Persistence of soil organic matter as an ecosystem property. Nature 478(7367):49–56
Schrumpf M, Kaiser K, Guggenberger G, Persson T, Kögel-Knabner I, Schulze ED (2013) Storage and stability of organic carbon in soils as related to depth, occlusion within aggregates, and attachment to minerals. Biogeosciences 10(3):1675–1691
Schurig C, Smittenberg RH, Berger J, Kraft F, Woche SK, Goebel MO, Heipieper HJ, Miltner A, Kaestner M (2013) Microbial cell-envelope fragments and the formation of soil organic matter: a case study from a glacier forefield. Biogeochemistry 113(1–3):595–612
Schwarzenbach G (1961) The general, selective, and specific formation of complexes by metallic cations. Adv Inorg Chem Radiochem 3:257–285
Shang C, Tiessen H (2003) Soil organic C sequestration and stabilization in karstic soils of Yucatan. Biogeochemistry 62(2):177–196
Shipitalo MJ, Protz R (1989) Chemistry and micromorphology of aggregation in earthworm casts. Geoderma 45(3):357–374
Six J, Paustian K, Elliott ET, Combrink C (2000) Soil structure and organic matter I. Distribution of aggregate-size classes and aggregate-associated carbon. Soil Sci Soc Am J 64(2):681–689
Six J, Conant RT, Paul EA, Paustian K (2002) Stabilization mechanisms of soil organic matter: implications for C-saturation of soils. Plant Soil 241(2):155–176
Six J, Bossuyt H, Degryze S, Denef K (2004) A history of research on the link between (micro)aggregates, soil biota, and soil organic matter dynamics. Soil Tillage Res 79(1):7–31
Skinner HCW, Jahren AH (2007) 8.04—biomineralization A2—Holland, Heinrich D. In: Turekian KK (ed) Treatise on geochemistry. Pergamon, Oxford, pp 1–69
Skjemstad JO, Fitzpatrick RW, Zarcinas BA, Thompson CH (1992) Genesis of Podzols on coastal dunes in southern Queensland. 2. Geochemistry and forms of elements as deduced from various soil extraction procedures. Aust J Soil Res 30(5):615–644
Skjemstad JO, Janik LJ, Head MJ, McClure SG (1993) High energy ultraviolet photo-oxidation: a novel technique for studying physically protected organic matter in clay- and silt-sized aggregates. J Soil Sci 44(3):485–499
Slessarev EW, Lin Y, Bingham NL, Johnson JE, Dai Y, Schimel JP, Chadwick OA (2016) Water balance creates a threshold in soil pH at the global scale. Nature 540(7634):567–569
Sokoloff VP (1938) Effect of neutral salts of sodium and calcium on carbon and nitrogen of soils. J Agric Res 57:0201–0216
Sollins P, Homann P, Caldwell BA (1996) Stabilization and destabilization of soil organic matter: mechanisms and controls. Geoderma 74(1–2):65–105
Sollins P, Kramer MG, Swanston C, Lajtha K, Filley T, Aufdenkampe AK, Wagai R, Bowden RD (2009) Sequential density fractionation across soils of contrasting mineralogy: evidence for both microbial- and mineral-controlled soil organic matter stabilization. Biogeochemistry 96(1–3):209–231
Spaccini R, Piccolo A, Conte P, Haberhauer G, Gerzabek MH (2002) Increased soil organic carbon sequestration through hydrophobic protection by humic substances. Soil Biol Biochem 34(12):1839–1851
Spielvogel S, Prietzel J, Kögel-Knabner I (2008) Soil organic matter stabilization in acidic forest soils is preferential and soil type-specific. Eur J Soil Sci 59(4):674–692
Sposito G (2008) The chemistry of soils. Oxford University Press, Oxford
Suess E (1970) Interaction of organic compounds with calcium carbonate—I. Association phenomena and geochemical implications. Geochim Cosmochim Acta 34(2):157–168
Sutton R, Sposito G, Diallo MS, Schulten H-R (2005) Molecular simulation of a model of dissolved organic matter. Environ Toxicol Chem 24(8):1902–1911
Suzuki S (2002) Black tea adsorption on calcium carbonate: a new application to chalk powder for brown powder materials. Colloids Surf A 202(1):81–91
Tate KR, Theng BKG (1980) Organic matter and its interactions with inorganic soil constiuents. In: Theng BKG (ed) Soils with variable charge. Soil Bureau, Lower Hutt, pp 225–249
Theng BKG, Churchman GJ, Newman RH (1986) The occurrence of interlayer clay-organic complexes in two New Zealand soils. Soil Sci 142(5):262–266
Thirukkumaran CM, Morrison IK (1996) Impact of simulated acid rain on microbial respiration, biomass, and metabolic quotient in a mature sugar maple (Acersaccharum) forest floor. Can J For Res 26(8):1446–1453
Thomas MM, Clouse JA, Longo JM (1993a) Adsorption of organic compounds on carbonate minerals. 3. Influence on dissolution rates. Chem Geol 109(1–4):227–237
Thomas MM, Clouse JA, Longo JM (1993b) Adsorption of organic compounds on carbonate minerals: 1. Model compounds and their influence on mineral wettability. Chem Geol 109(1–4):201–213
Tipping E (2005) Modelling Al competition for heavy metal binding by dissolved organic matter in soil and surface waters of acid and neutral pH. Geoderma 127(3–4):293–304
Tisdall JM (1996) Formation of soil aggregates and accumulation of soil organic matter. In: Carter MRS, Stewart BA (eds) Structure and organic matter storage in agricultural soils. CRC Press, Boca Raton, pp 57–86
Tisdall JM, Oades JM (1982) Organic-matter and water-stable aggregates in soils. J Soil Sci 33(2):141–163
Tonneijck FH, Jansen B, Nierop KGJ, Verstraten JM, Sevink J, De Lange L (2010) Towards understanding of carbon stocks and stabilization in volcanic ash soils in natural Andean ecosystems of northern Ecuador. Eur J Soil Sci 61(3):392–405
Torn MS, Trumbore SE, Chadwick OA, Vitousek PM, Hendricks DM (1997) Mineral control of soil organic carbon storage and turnover. Nature 389(6647):170–173
Toutain F (1974) Etude écologique de l’humidificaton dans les hetraies acidiphiles. In. Université de Nancy, Nancy
Trumbore SE (1993) Comparison of carbon dynamics in tropical and temperate soils using radiocarbon measurements. Glob Biogeochem Cycles 7(2):275–290
van der Heijden G, Legout A, Mareschal L, Ranger J, Dambrine E (2017) Filling the gap in Ca input-output budgets in base-poor forest ecosystems: the contribution of non-crystalline phases evidenced by stable isotopic dilution. Geochim Cosmochim Acta 209:135–148
Verrecchia EP (1990) Litho-diagenetic implications of the calcium oxalate-carbonate biogeochemical cycle in semiarid Calcretes, Nazareth, Israel. Geomicrobiol J 8(2):87–99
Verrecchia EP, Freytet P, Verrecchia KE, Dumont J-L (1995) Spherulites in calcrete laminar crusts; biogenic CaCO3 precipitation as a major contributor to crust formation. J Sediment Res 65(4a):690
Verrecchia EP, Braissant O, Cailleau G (2006) The oxalate–carbonate pathway in soil carbon storage: the role of fungi and oxalotrophic bacteria. Cambridge University Press, Cambridge
Virto I, Barre P, Chenu C (2008) Microaggregation and organic matter storage at the silt-size scale. Geoderma 146(1–2):326–335
Virto I, Moni C, Swanston C, Chenu C (2010) Turnover of intra- and extra-aggregate organic matter at the silt-size scale. Geoderma 156(1–2):1–10
Virto I, Gartzia-Bengoetxea N, Fernandez-Ugalde O (2011) Role of organic matter and carbonates in soil aggregation estimated using laser diffractometry. Pedosphere 21(5):566–572
Virto I, Barré P, Enrique A, Poch RM, Fernández-Ugalde O, Imaz MJ, Bescansa P (2013) Micromorphological analysis on the influence of the soil mineral composition on short-term aggregation in semi-arid Mediterranean soils. Span J Soil Sci 3(2):116–129
Vogel C, Mueller CW, Hoeschen C, Buegger F, Heister K, Schulz S, Schloter M, Kögel-Knabner I (2014) Submicron structures provide preferential spots for carbon and nitrogen sequestration in soils. Nat Commun 5(2947):1–7
von Lützow M, Kögel-Knabner I, Ekschmitt K, Matzner E, Guggenberger G, Marschner B, Flessa H (2006) Stabilization of organic matter in temperate soils: mechanisms and their relevance under different soil conditions - a review. Eur J Soil Sci 57(4):426–445
Wedepohl HK (1995) The composition of the continental crust. Geochim Cosmochim Acta 59(7):1217–1232
Wen L, Li D, Chen H, Wang K (2017) Dynamics of soil organic carbon in density fractions during post-agricultural succession over two lithology types, southwest China. J Environ Manag 201:199–206
Whittinghill KA, Hobbie SE (2012) Effects of pH and calcium on soil organic matter dynamics in Alaskan tundra. Biogeochemistry 111(1–3):569–581
Wiese L, Ros I, Rozanov A, Boshoff A, de Clercq W, Seifert T (2016) An approach to soil carbon accounting and mapping using vertical distribution functions for known soil types. Geoderma 263:264–273
Wong VNL, Greene RSB, Dalal RC, Murphy BW (2010) Soil carbon dynamics in saline and sodic soils: a review. Soil Use Manag 26(1):2–11
Wuddivira MN, Camps-Roach G (2007) Effects of organic matter and calcium on soil structural stability. Eur J Soil Sci 58(3):722–727
Xiao J, Wen Y, Li H, Hao J, Shen Q, Ran W, Mei X, He X, Yu G (2015) In situ visualisation and characterisation of the capacity of highly reactive minerals to preserve soil organic matter (SOM) in colloids at submicron scale. Chemosphere 138:225–232
Poeplau C, Don A, Six J, Kaiser M, Benbi D, Chenu C, Cotrufo FM, Derrien D, Gioacchini P, Grand S, Gregorich E, Griepentrog M, Gunina A, Haddix M, Kuzyakov Y, Kühnel A, Macdonald LM, Soong J, Trigalet S, Vermeire M-L, Rovira P, van Wesemaell B, Wiesmeier M, Yeasmin S, Yevdokimov I, Nieder R (in review) Isolating soil organic carbon fractions with varying turnover rates – A comprehensive comparison of fractionation methods
Yang S, Cammeraat E, Jansen B, Cerli C, Kalbitz K (2016) Organic carbon stabilization of soils formed on acidic and calcareous bedrocks in Neotropical alpine grassland, Peru. EGU General Assembly 2016, held 17-22 April, 2016 in Vienna, Austria
Yuan TL, Gammon N, Leighty RG (1967) Relative contribution of organic and clay fractions to cation-exchange capacity of sandy soils from several soil groups. Soil Sci 104(2):123–128
Zelles L, Scheunert I, Kreutzer K (1987) Bioactivity in limed soil of a spruce forest. Biol Fertil Soils 3(4):211–216
Zhao J, Chen S, Hu R, Li Y (2017) Aggregate stability and size distribution of red soils under different land uses integrally regulated by soil organic matter, and iron and aluminum oxides. Soil Tillage Res 167:73–79
Zheng W, Morris EK, Lehmann A, Rillig MC (2016) Interplay of soil water repellency, soil aggregation and organic carbon. A meta-analysis. Geoderma 283:39–47
Zimmerman AR, Ahn M-Y (2010) Organo-mineral–enzyme interaction and soil enzyme activity. In: Shukla G, Varma A (eds) Soil enzymology. Soil biology, vol 22. Springer, Berlin, pp 271–292
Acknowledgements
A special acknowledgement must be paid to Dr. Michèle Gaiffe for allowing us to modify her previous diagram and include it in the paper. Thanks also to Dr. Pascal Vittoz, Dr. David Sebag, Prof. Jasquelin Peña, Magali Matteodo, Dr. Nathalie Diaz, Finaritra Randevoson, and Fabienne Dietrich for enjoyable and profitable conversations regarding the manuscript. Special thanks to the Canton de Vaud and IDYST for financially supporting this work and the “Conseil de coordination scientifique du Vallon de Nant” for research permissions.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: Marc G. Kramer.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Rowley, M.C., Grand, S. & Verrecchia, É.P. Calcium-mediated stabilisation of soil organic carbon. Biogeochemistry 137, 27–49 (2018). https://doi.org/10.1007/s10533-017-0410-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10533-017-0410-1