
Abstract
Topmouth gudgeon is a freshwater fish species native to East Asia. Nowadays, P. parva is spread throughout Europe which is of concern because besides being considered one of the worst aquatic Invasive Alien Species (IAS) in Europe it is also a known vector of Spherotecum destruens, the rosette-like parasite lethal to other fish species. The present study describes the development and validation of a new species-specific assay based on hydrolysis probe chemistry to detect P. parva environmental DNA (eDNA) in water samples collected in a northern region of Italy (Friuli Venezia Giulia). Water samples were collected from 55 sites in an area where partial information on the occurrence of the species is available. eDNA was isolated from all samples and the presence of P. parva eDNA was tested by means of qPCR (quantitative PCR) and microfluidic qdPCR (quantitative digital PCR) techniques. Field results for both qPCR and qdPCR were largely in agreement in terms of detection (presence/absence). Thus, we judged the presence/absence by combining the results from the two methods and found that nine sites showed “strong positive” signal of P. parva eDNA (at least 2 positive replicates), 3 showed “suspected” (only 1 positive replicate), and 42 showed “absent”. The current study shows the strong potential of the newly developed eDNA approach to be a valuable addition to the monitoring of the highly invasive topmouth gudgeon in freshwater ecosystems.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The Aichi Biodiversity Target 9 foresees that by 2020 invasive alien species and pathways are identified and prioritized, priority species are controlled or eradicated, and measures are in place to manage pathways to prevent their introduction and establishment (Leadley et al. 2014). One of the most recent techniques to monitor species of interest is through the detection of environmental DNA (eDNA), based on the analysis of shed genetic material in environmental samples (Thomsen and Willerslev 2015; Boothroyd et al. 2016; Deiner et al. 2017; Cowart et al. 2018; Manfrin et al. 2019). This technique allows the monitoring of presence/absence of one or multiple target species without the need of capturing the target organisms, but by analysing the DNA present in environmental (e.g. water, sediments, or air) samples (Ruppert et al. 2019). Being the eDNA present in trace, detection methods must be highly sensitive and at the same time protocols must be put in place to reduce the risk of false-negative and false-positive results, including the establishment of Limits Of Detection (LODs) (Klymus et al. 2020) and enzymatic inhibition control (van Doorn et al. 2009; Goldberg et al. 2016; Harper et al. 2019).
Pseudorasbora parva is a freshwater fish species native to East Asia and introduced outside of its native distribution as a contaminant of Asian carp consignments since ‘60 s (Gozlan et al. 2005). It colonizes slow-flowing waters, such as irrigation canals, lakes, ponds and rivers and tolerates low oxygen levels. These habitats are constituted by running waters, and the huge variety of suitable habitats makes topmouth gudgeon amongst the worst 100 invasive species in Europe (DAISIE 2009). It was initially introduced in Romania and Albania in 1961, the first Italian record of this species dated back to 1981 near Brunico (Bozen) (Witkowski 2011). Nowadays, P. parva is spread throughout Europe, but it has also been introduced in northern Africa and Oceania. Other than its high invasiveness, topmouth gudgeon is a vector of Spherotecum destruens, the rosette-like parasite lethal to other fish species (Gozlan et al. 2005; Spikmans et al. 2020) and this additional aspect makes mandatory the need of fast and reliable detection methods for this species along with the constitution of a shared map of distribution from diverse areas.
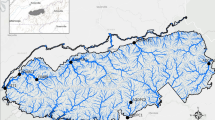
The target area of this study includes the southern portion of the FVG that identifies the resurgence belt, which is an area 2 to 8 km wide and 80 km long. In this area, the water table overlaps the topographic surface and forms numerous shallow springs and streams (Cucchi et al. 2008). The resurgence belt forms a geohydrological boundary between the upper and lower Friulian Plain. Fine deposits with a marked prevalence of silty-clayey material, are continuously present throughout the area (Cucchi et al. 1998). The resurgence waters have a temperature of 9–13 °C and a constant flow rate, during all seasons, of about 65 m3 per second; they are clear and potable waters and often rich in minerals. The characteristics of the vegetation in these areas are very varied: from dry meadows, to wet meadows to swamp like vegetation, to submerged or floating plants.
Selected sites of the Friuli Venezia Giulia (FVG) region (Italy) were screened for topmouth gudgeon’s presence as requested by Regulation (EU) No 1143/2014 of the European Parliament on the prevention and management of the introduction and spread of Invasive Alien Species (IAS), where P. parva is included in the list of invasive alien species of Union concern (Regulation implemented by EU n. 2016/1141 and n. 2017/1263) and by the Italian law No 230/2017. Upon the aforementioned legislation, each region was asked to provide a distribution map of invasive exotic species of Union concern referred to the art. 24 of EU Regulation 1143/2014.
The first record of the topmouth gudgeon in FVG dated back to 1996 when a single specimen was found in the Vipacco river, nearby station P02 of the present study (Specchi and Pizzul 1996). A number of unverified records have been reported in other areas, thus indicating the need for additional field data and targeted surveys to reveal the current spread of P. parva in the FVG Region.
The monitoring of the P. parva using conventional methods such as electro-fishing can be challenging due to the environment’s characteristics where the species is potentially present, which consist of large waterbodies (Allard et al. 2014), the water temperature (Vibert 1967), deep and turbulent waters (Lyon et al. 2014). Other methods, such as using funnel traps, can be more effective in such environments, but they are also time-consuming, especially when deployed over large geographic areas. Additionally, methods that require the least interference with the environment to be surveyed are preferred when dealing with invasive species as they limit the chances of unintentional spread and therefore prove more robust from a biosecurity aspect.
To have a first reference framework of the distribution of topmouth gudgeon, we thus applied the eDNA technique intending to develop a new tool for the acquisition of information on the target species distribution in a short time span to be used to inform further targeted surveys using conventional methods. In this study a new species-specific molecular assay for P. parva was designed matching a short region of the mitochondrial Cytochrome c Oxidase I (COI). We tested it for eDNA detection in water samples using both quantitative Real-Time PCR and quantitative digital PCR (qdPCR). With this study, we aimed to compare two quantitative methods, quantitative Real Time-PCR (qPCR) and quantitative digital PCR (qdPCR), to determine whether either approach is more powerful in detecting eDNA. When the present study was first conceived, no similar assays had been published; here we provide a critical review of the systems that have since been published for the detection of P. parva eDNA.
Materials and methods
Sample sites
Water samples (1 L) were collected from 55 selected locations using sterile 50 mL disposable syringes (BDPlastipak™) and filtered in situ through Sterivex durapore PVDF 0.45 µm filters (Merck-Millipore). Filters were kept refrigerated at 4 °C in situ, and in the laboratory at − 20 °C until eDNA extraction. The eDNA extraction from Sterivex filters was performed using DNeasy PowerWater Sterivex Kit (Qiagen), according the manufacturer’s instructions. Sampling took place at 59 stations in November and December 2018 and January 2019 (Supplementary file Table S1). In this study the sampling strategy favours an increased geographical coverage (more sites in proximity to each other) than replication within sites. No field negative control was included in this study, however the possibility of false positive results due to cross-sample contamination was deemed negligible due to the use of strict sampling protocols and practice (Tables 1, 2).
Primers design and species-specificity tests
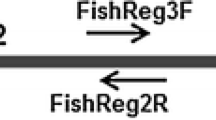
Primers and probe were designed on the mitochondrial Cytochrome c Oxidase I (COI) portion obtained from P. parva sequences stored at GenBank (Table 1, alignment details in Figure S1, SUpplementary Material) using Primer3Plus (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi) and OligoCalc (http://biotools.nubic.northwestern.edu/OligoCalc.html) to exclude possible dimers and hairpin formations between the primers Table 2.
Possible cross-species amplification was checked through BLAST and locally on a list of 22 teleost species (data not shown), including Alosa fallax, Padogobius bonelli, Salmo marmoratus, Thymallus aeliani, Rhodeus amarus, Barbus balcanicus, B. plebejus and B. caninus, Alburnus alborella, Cyprinus carpio, Phoxinus lumaireul, Squalius squalus, Telestes muticellus, Protochondrostoma genei, Leucos aula, Scardinius erythrophthalmus, Tinca tinca, Anguilla anguilla, Lepomis gibbosus, Sander lucioperca, Silurus glanis, and Gobio benacensis. These species might be present in the regional catchments and share ecological niches with P. parva. From sequence similarity searches of the target genomic region, P. parva resulted the species with highest hits, followed by Tachysurus ussuriensis a non-migratory freshwater bony-fish from Asia and Russia. Minor matches have been reported for marine species as lanternfish (Diaphus spp), coral hawkfish (Cirrhitichthys oxycephalus) and Macrognathus siamensis a tropical freshwater.
Two topmouth gudgeon specimens were caught in the external tanks outside the Aquarium of Ariis di Rivignano by using funnel traps and their genomic DNA was extracted from fins with E.Z.N.A.® Mollusc DNA Kit (Omega Bio-tek). The concentration and purity of each sample were estimated by UV light absorbance using a Nanodrop 2000 (ThermoFisher). These two samples were amplified with primers targeting a COI fragment (Leray et al. 2013) in 15 µL reactions containing 1X Primer HotMasterMix (5Prime), 0.3 µM both primers and 50 ng of gDNA in thermal reaction of: initial denaturation of 1 min at 95 °C, 5 cycles: 95 °C for 10″, 46 °C for 20″ and 72 °C for 20″, 30 cycles: 95 °C 10″, 54 °C 20″ and 72 °C 20″ followed by a final extension of 1’ at 72 °C. Purified PCRs were Sanger sequenced at an external service (Eurofins, Hamburg, Germany) for species confirmation and served as positive controls.
qPCRqPCR.
Quantitative real time PCR (qPCR)
A C1000 thermocycler equipped with a CFX96 head (BioRad) was used to perform qPCRs. Amplification mix contained the following reagents at final concentrations: 1X KAPA Probe Force master mix (Kapa Biosystems), 0.4 µM each primer and 0.2 µM probe, 2 µL of eDNA (or 1 µL of gDNA) and water at a final volume of 15 µL. Each field sample was run at a minimum of 3 technical replicates. All extracts were tested in triplicates, with the exception of some samples which were tested up to 6 times for the purpose of further validation of positive signals The thermal profile was as follows: 98 °C 3′, 50 cycles at 95 °C 10″ and 64 °C 30″. Quantitative analysis has been run with the CFX Maestro software 1.1. v 4.1 (Bio-Rad). No amplification was observed on a total of 19 negative technical controls (No Template Controls, NTCs).
Microfluidic-based quantitative digital PCR (qdPCR)
Quantitative digital PCR (qdPCR) was carried out using qdPCR 37 K™ Integrated Fluidic Circuits (IFCs) on a Biomark™ HD system (Fluidigm). Each reaction mixture (total volume 6 μl) comprised 1.8 μL of eDNA sample (or 1 μL of gDNA), 0.6 μL 20 × GE sample loading reagent, 3 μL 2 × KAPA Probe Force master mix and 0.3 uL of pre-mixed assay oligonucleotides, whereby the forward and reverse primers had a final concentration of 0.5 µM and the probe a final concentration of 0.25 µM. IFCs were primed and loaded using an MX IFC Controller (Fluidigm), whereby each sample/reaction mixture was partitioned in 770 parallel nano chambers before executing qdPCR on a Biomark HD platform following the same thermal conditions reported above. NTCs were included in all runs and showed no evidence of contamination. Image and end-point data analysis was executed with Fluidigm Digital PCR Analysis v. 4.1.2 software. Number of positive partitions per sample was confirmed by inspecting amplification curves by eye and concentration of targets in template DNA was estimated as follows:
whereby the “number of estimated count of copies per panel” is corrected according to a Poisson distribution, the “volume of reaction mix per panel” is 0.6545 μL and the “reaction mix dilution factor” is the total volume of the reaction mix (6 μL) divided by the volume of template DNA (1.8 μL).
Detection criteria
To estimate the Limit of Detection (LOD) for both techniques, genomic DNA from two P. parva specimens was quantified and used to create serial dilutions from 1/10th up to 1/106 of the initial concentration (actual range 160–45 ng µL−1). Each dilution was amplified at a minimum of 5 replicates for qPCR and 3 replicates for qdPCR. LOD was identified as the lowest concentration of the target analyte that can be detected with a 95% detection rate as the standard confidence level by using the R script for LOD calculation (Klymus et al. 2020) in R Studio, v. 1.0.143.
Additionally, a sample consisting of 1 L filtered water filtered from a 700 L tank containing 10 P. parva juveniles (density 20 g/m3) at the Aquarium of Ariis di Rivignano (hereinafter named P00), was tested up to two dilutions of its initial concentration, namely 1/100
and 1/1000th and used to further validate the ability of the new assay to detect P. parva eDNA (Fig. 1 and Table S2, Supplementary Material). The results from both methods were used to judge the presence/absence of the target species. The numbers of replicates and sites tested for the two methods were different due to practical limitations.

Standard curve plot on qPCR positive control samples PCRs. On y-axis the quantitative cycles (Cq), on x-axis the Log of the starting quantity of each dilution. gDNA_1 is reported in black circles, gDNA_2 in green ones and eDNA from the tank containing P. parva is reported in blue. This latest sample analytical quantification has not been acquired, but based on the amplification cycles, a starting quantity has been estimated and the series dilution plotted on this graph. E = reaction efficiency, R^2 = coefficient of determination and y-int = y intercept. Figure edited with Maestro software (Bio Rad)
For the samples collected in the field, detection of P. parva eDNA was considered as “strong evidence” when at least two technical replicate were positive and showed a sigmoidal amplification curve (up to 45 Ct, Troth et al. 2020); whereas it was considered “suspected eDNA presence” when only one of the techniques showed at least one positive technical replicate, and “absent eDNA” when the sample was negative to both methods.
Inhibition test
To evaluate possible inhibitor effects on the amplification of eDNA samples, an “internal amplification control” (IAC) was produced (namely IAC 1 in van Doorn et al. 2009). First, double-strand IAC (dsIAC) was generated in a 25 µL PCR mix containing 1X AccuStart II PCR ToughMix (Quantabio), 0.4 µM each primer, and 50 ng of single-strand (ssIAC), by using the following thermal profile: initial denaturation 95 °C for 2′, 35 cycles: 95 °C for 30″, 61 °C for 30″, 72 °C for 1′ and a final extension of 72 °C for 10′. The product was checked in a 3% TAE MetaPhor agarose gel and purified with 1.7X Mag-Bind® TotalPure NGS (Omegabiotek). The purified product was quantified in triplicate with Qubit® dsDNA BR Assay Kit (Thermofisher). dsIAC was run in duplicate alongside a standard dilution series from 3 × 105 to 3 × 102 DNA copy number. Each reaction was performed in a final volume of 15 µL containing 1X KAPA Probe Force master mix (Kapa Biosystems), 0.3 µM of the reverse primer and 0.15 µM of the forward primer, 1X EvaGreen® Dye (Biotium) and 1 µL selected IAC dilution, by using the following thermal profile: initial denaturation 95 °C for 2′, 35 cycles: 95 °C for 30″, 68 °C for 30″, followed by a melting curve from 65 °C to 95° with a temperature increment of 0.5 °C every 5″. Triplicates PCR reactions containing 3 × 104 IAC copy numbers and 1µL of eDNA samples randomly taken from the sampled stations were run for inhibition tests.
Statistical analysis
To measure inter-rater reliability between the two approaches, Coehn’s kappa coefficient (k) was calculated using Cts from positive amplification and setting the tolerance = 0. Pearson’s correlation coefficient (r) was calculated to measure linear correlation between the two sets of eDNA data obtained with qPCR and qdPCR. Both statistics were run in RStudio v 1.0.143. The final statistical outputs are provided in the Supplementary file S5.
Results
The detection limit of the qPCR was comparable between the two positive controls and resulted in a dilution of 1/105 for both DNA_1 and _2. Even though the second positive control was onefold less concentrated in respect to the DNA_1, the detection limit resulted from the qPCR is the same for both the positive genomic controls (Fig. 1). The newly developed assay also successfully detected the presence of target eDNA in the water sample collected from a tank containing P. parva (Table S2, Supplementary Material), further validating the effectiveness of this approach.
Comparable results were obtained in qdPCR (Table 3 and for raw data see Table S2, Supplementary Material). Positive controls, along with their 1/10th dilution reached the saturation of the dPCR samples with 100% of positive partitions (> 11,839 in Table S2, Supplementary Material). Higher variability among replicates was observed at the strongest dilutions with no amplification observed at dilutions 1/106.
When applying a curve‐fitting modelling approach for determining LOD (Klymus et al. 2020), the LOD of our target resulted in being of 4.50E-04 ng µL−1/reaction.
Inhibition test was run using a target Internal Amplification Control (IAC 1) (van Doorn et al. 2009) with an increased Tannealing of 68 °C verified upon gradient PCR from 60° to 68 °C. The IAC at 3*104 copy number was amplified at an average Ct 23.77 (± 0.32), and the same IAC dilution within random eDNA samples was amplified at an average Ct 25.45 (± 0.36), resulting in an amplification delay of 1.7 cycles. Fifty-five sites were analysed in this study to detect P. parva’s presence and to outline a distribution map of this species (Fig. 2), both qPCR and qdPCR results were used to judge the presence/absence of the target species. Nine sites showed strong evidence of P. parva eDNA with at least two positive amplifications among replicates, forty-three negatives, and three were classified with “suspected eDNA presence”, since only 1 positive technical replicate was amplified and results obtained were not confirmed by both the methods applied.

Map of the Friuli Venezia Region (white region in the Italy image) alongside the stations monitored through eDNA. In red the sites resulted positive to the presence of P. parva, in green those where the eDNA target was absent and in yellow stations that are considered as “suspected eDNA presence”
In particular, P48 and P50 have been amplified in a single replicate by mean of qPCR, but not with qdPCR, while P28 resulted positive in 1 out of 3 replicates in qdPCR, but not in qPCR (Table 4, for threshold cycle details refer to Table S3, Supplementary Material).
The site SA (Fig. 2, nearby P00 that represents the aquarium Insitute where P. parva juveniles were kept) stands for Stella in Ariis, which is a river surrounding the aquarium Institute. We checked P. parva in its waters to control possible presence of the species due to the proximity to the aquarium. The site P36 resulted negative in both qPCR and dPCR, even though two specimens of P. parva were captured by electrofishing technique approximately one and half year after the eDNA survey was conducted (i.e. on July 23rd 2020 by ETPI; Zanetti personal communication). The different number of replicates within the same PCR method are due to an initial amplification of 3 replicates for each sample, then the number of replicates was increased for the samples worth additional investigation as the cases: 1 positive replicate out of 3; negative amplification in one of the two PCR methods or, as in P36, negative amplification despite the capture of two specimens in the same river waters but at different times.
The two approaches showed moderate agreement in terms of presence/absence (86% agreement and Cohen's Kappa = 0.6), whereas in terms of quantification a moderate correlation was also found (Pearson's R2 = 0.7123).
Discussion
The eDNA protocol described in this study proved to be effective at providing baseline data on the potential occurrence and distribution of topmouth gudgeon in a relatively large geographical area, the FVG region in the North East of Italy. This indirect (eDNA) monitoring approach was applied in the laboratory as well as in the field, and provided a number of significant advantages over conducting an initial survey of the same spatial scale using direct conventional methods (e.g. trapping), including: (i) a shorter time of execution (estimated to be at least half of direct methods, based on personal experience of the personnel involved); (ii) relatively lower costs (e.g. when factoring in personnel costs of a conventional survey); (iii) negligible levels of invasiveness on non-target organisms (e.g. compared to electrofishing and trapping); and (iv) reduced biosecurity risks (less likely to spread unwanted IAS or water borne pathogens).
In the present study two different PCR techniques were compared to determine whether one was more sensitive than the other. Comparable results in terms of sensitivity (LOD) and detection in field samples were observed when employing the new molecular assay in qPCR and qdPCR systems, which highlights the transferability of this new maker across different screening platforms. Nonetheless, some differences were observed, 1 out 55 sampled sites (P28) gave a positive hit for qdPCR, but resulted negative to qPCR, whereas 2 out 55, namely P48 and P50 were positive in a single replicate in qPCR, but negative with dPCR. Among positive samples, three sites, namely P22, P27 and P29 were amplified with qPCR, but not in qdPCR (Table 4). This discrepancy is probably due to manual error since the comparative analysis showed similar results for both the PCR methods and amplifications in qPCR were perfectly sigmoidal and ranged cycle threshold from 35.46 to 38.15 (Table S3, Supplementary Material).
Although, we did not accurately quantify each eDNA by serial dilution for each sample because the intent was to obtain a qualitative result (absence/presence) for each sampled site. However, serial dilutions were performed on two genomic DNAs and on 1 eDNA from a tank containing P. parva juveniles and the quantification aspect was used to provide reference for estimating LOD and identifying signal below or above LOD.
The fact that the two approaches were moderately in agreement indicates that when possible additional field sample replicates and increased technical replicates should be performed. In addition, also the use of both techniques would likely increase power of detection especially in conditions close to LOD, even if this latter solution is not feasible for many laboratories. In the cases of “suspected eDNA presence” detection (5.5% of tested sites), discordance between qPCR and qdPCR methodologies reported in this study is likely to do with the fact that the number of target eDNA copy numbers is very close to the LOD. However, given the high sensitivity of the methods used and adopting a conservative approach in the case of IAS surveillance, the cases of “suspected eDNA presence” are to be considered as true positives, which would prompt a follwo up targeted survey to confirm the presence of the target organism. In future surveys, to address this uncertainty and to increase power of detection, it is recommended to use at least one of the methods described here and increase the number of field replicate samples (as opposed to qdPCR/qPCR technical replicates). Within the context of a surveillance and monitoring programme for aquatic IAS, the eDNA approach provides an effective first step to direct subsequent efforts using direct methods, hence enabling more effective use of financial resources. From an IAS surveillance and management point of view, despite some unresolved sites (5.5% of the total sites), this study provided conclusive results from 9 sites (16% of the total sites analysed). At site P36, the eDNA analysis was negative, although two specimens of P. parva were caught by electrofishing in 2020. This discrepancy cannot be attributed to the eDNA analysis, for which additional replicates were run on this specific sample, but may be due to late arrival of this species following eDNA monitoring. eDNA detection is therefore recommended for targeted direct surveys to confirm the presence and determine relative abundance of P. parva. Since eDNA can travel downstream from its source, the direct surveys should start at the point of eDNA detection and when possible continue upstream or at relevant hydrographical and physical points.
The presence of inhibitors led to inaccurate target quantification and some false-negative results (Jane et al. 2015; McKee et al. 2015). A delay of 1.7 Cq cycles in qPCR tests was the result of an inhibitory effect, most probably due to substances such as humic acid co-extracted with the eDNA samples (Jane et al. 2015). Thanks to the dilution effect of using small (nano) reaction volumes, it is expected that qdPCR will provide an advantage towards samples that are potentially affected by enzymatic inhibitors (Hoshino and Inagaki 2012). However, even though the effect of potential inhibition was not directly tested when using qdPCR, the similarity between qPCR and qdPCR suggest that inhibitory effects may be relatively small in this study. In future studies, it is nonetheless recommended to improve the current eDNA protocol with the inclusion of a dedicated environmental PCR mastermix (Uchii et al. 2019).
The topmouth gudgeon is a species already monitored through eDNA with the scope of providing information for invasive fish eradication plans (Davison et al. 2017, 2019; Robinson et al. 2019). Davison and colleagues used a COI fragment of 350 bp designed on P. parva (2017) and a second target of 101 bp (2019), while Robinson et al. (2019) used a 16S mtDNA fragment of 147 bp. The present assay was designed before the P. parva eDNA publications, and the new marker was designed to serve the purpose of monitoring the target areas, and the shorter length is expected to make it more effective, even though a comparison was not done. Furthermore, since detecting such organisms is very important as they can alter an entire ecosystem, protocols that are more sensitive and approaches that are more conservative should be prioritized.
For selecting our marker, the GenBank database was searched for the most used DNA data for species identification of P. parva, finding 169 available sequences for COI and 29 for 16S. It was decided to design a primer system targeting a 79 bp fragment of the COI gene, which is smaller than the one proposed by Davison and colleagues (2019), based on hydrolysis probe chemistry. In silico analysis performed on all the COI sequences available in GenBank (169 sequences, at the time of writing this manuscript) highlighted that both ours and Davison primer sets are able to detect the majority of P. parva sequences, with few differences. Roughly 17 sequences did not match 100% with our forward, probe and reverse primers, while the Davison’s systems presented: for the system of 350 bp an optimal matching for the forward primer (only 2 excluded out of 169), but less matching sequences to the reverse primer (31 excluded out of 169 sequences) and for the system of 101 bp, the forward did not match with 3, the probe with 16 and the reverse did not match 100% 90 out of 169 sequences (Supplementary material, Table S4). Substantial optimization was carried out to refine qPCR conditions towards high specificity to the target region and aiming for highest stringency. For instance, the work done by Davison on P. parva used a conventional PCR with an annealing temperature (Tann) of 62 °C; in contrast, Robinson performed a quantitative PCR-High Resolution Melt (qPCR-HRM) with a Tann of 61 °C. We worked with a probe-based system with a Tann of 64 °C, since higher Tann improves oligonucleotides stringency (Lorenz, 2012; Veldhoen et al., 2016; Langlois et al., 2020). The availability of multiple markers for eDNA detection of P. parva is however an advantage, because of the possibility to incorporate them into a multi-marker simultaneous eDNA detection system and hence establishing more robust eDNA protocols for IAS surveillance.
At present, the detection of P. parva within the target area, is confined in the southern part of the region. However, eDNA shedding rates are expected to vary throughout an organism’s life cycle, with peaks generally associated to spawning times and/or higher biomass (Rees et al. 2014; Tillotson et al. 2018; Thalinger et al. 2019); thus, future eDNA efforts should be carried out during the Spring and Summer seasons, when the spawning activity of P. parva takes place (Záhorská et al. 2009).
eDNA is a technique that complements conventional doirect sampling/capture methods, for determining P. parva distribution within a given area to inform management decisions on eradication procedures. Subsequently, the FVG region is implementing environmental monitoring plans by means of eDNA with the aim of targeting key species, but also to acquire, on a regional scale, the map of distribution of target communities within freshwater ecosystems. Establishing a management strategy based on positive or suspected positive eDNA samples is key to surveillance programmes, whereby decision making depends on the level of risk that managers are willing to accept (Sepulveda et al. 2020). Thus, if a similar framework was to be adopted for the surveillance of P. parva in this region, a “suspected positive” result would warrant a revisit to the site and subsequent water sample collection.
Concerning the distribution of P. parva alongside the FVG watercourses, the current picture delineates the presence of this fish in some rivers and streams only in the south part of the region and in watercourses nearby Slovenia. Further monitoring campaigns will be part of a regional environmental monitoring plan to collect data on the presence/absence of P. parva, alongside other target species by monitoring eDNA composition.
Change history
06 August 2022
Missing Open Access funding information has been added in the Funding Note
References
Allard L, Greouillet G, Khazraie K, Tudesque K, Vigouroux R, Brosse S (2014) Electrofishing efficiency in low conductivity neotropical streams: towards a non-destructive fish sampling method. Fish Man Ecol 21:234–243
Boothroyd M, Mandrak NE, Fox M, Wilson CC (2016) Environmental DNA (eDNA) detection and habitat occupancy of threatened spotted gar (Lepisosteus oculatus). Aquat Conserv 26:1107–1119. https://doi.org/10.1002/aqc.2617
Cowart DA, Breedveld KGH, Ellis MJ, Hull JM, Larson ER (2018) Environmental DNA (eDNA) applications for the conservation of imperiled crayfish (Decapoda: Astacidea) through monitoring of invasive species barriers and relocated populations. J Crustacean Biol. https://doi.org/10.1093/jcbiol/ruy007
Cucchi F, Giorgetti F, Gemiti F, Massari G, Oberti S (1998) Caratterizzazione geochimica delle falde acquifere della pianura friulana (Geochemical caractherisation of the Friulian Plain aquifers). Atti Acque Sotterranee: Risorsa Invisibile 44:61–71
Cucchi F, Franceschini G, Zini L (2008) Hydrogeochemical investigations and groundwater provinces of the Friuli Venezia Giulia Plain aquifers, northeastern Italy. Environ Geol 55:985–999. https://doi.org/10.1007/s00254-007-1048-4
DAISIE (2009) Handbook of alien species in Europe. Springer, Dordrecht
Davison PI, Falcou-Préfol M, Copp GH, Davies GD, Vilizzi L, Créach V (2019) Is it absent or is it present? Detection of a non-native fish to inform management decisions using a new highly-sensitive eDNA protocol. Biol Inv 21:2549–2560. https://doi.org/10.1007/s10530-019-01993-z
Davison PI, Copp GH, Créach V, Vilizzi L, Britton JR (2017) Application of environmental DNA analysis to inform invasive fish eradication operations. Sci Nat. https://doi.org/10.1007/s00114-017-1453-9
Deiner K, Bik HM, Mächler E, Seymour M, Lacoursière-Roussel A, Altermatt F, Creer S et al (2017) Environmental DNA metabarcoding: transforming how we survey animal and plant communities. Mol Ecol 26(21):5872–5895. https://doi.org/10.1111/mec.14350
van Doorn R, Klerks MM, van Gent-Pelzer MP, Speksnijder AG, Kowalchuk GA, Schoen CD (2009) Accurate quantification of microorganisms in PCR-inhibiting environmental DNA extracts by a novel internal amplification control approach using Biotrove OpenArrays. App Env Micr. https://doi.org/10.1128/AEM.00796-09
Goldberg CS, Turner CR, Deiner K, Klymus KE, Thomsen PF, Murphy MA, Spear SF et al (2016) Critical considerations for the application of environmental DNA methods to detect aquatic species. MEE 7:1299–1307
Gozlan RE, St-Hilaire S, Feist SW, Martin P, Kent ML (2005) Biodiversity: disease threat to European fish. Nature 435:1046–1046. https://doi.org/10.1038/4351046a
Harper LR, Buxton AS, Rees HC, Rein KB, Halfmaerten D, Read DS, Watson HV et al (2019) Prospects and challenges of environmental DNA (eDNA) monitoring in freshwater ponds. Hydrobiologia 826:25–41. https://doi.org/10.1007/s10750-018-3750-5
Hoshino T, Inagaki F (2012) Molecular quantification of environmental DNA using microfluidics and digital PCR. Syst Appl Microbiol 35(6):390–395. https://doi.org/10.1016/j.syapm.2012.06.006
Jane SF, Wilcox TM, McKelvey KS, Young MK, Schwartz MK, Lowe WH, Letcher BH et al (2015) Distance, flow and PCR inhibition: eDNA dynamics in two headwater streams. Mol Ecol Res 15:216–227
Kavlick F (2018) Development of a universal internal positive control. BioTech 65:275–280. https://doi.org/10.2144/btn-2018-0034
Klymus KE, Ruiz Ramos DV, Thompson NL, Richter CA (2020) Development and testing of species-specific quantitative PCR assays for environmental DNA applications. J vis Exp 165:e61825. https://doi.org/10.3791/61825
Langlois VS, Allison MJ, Bergman LC, To TA, Helbing CC (2020) The need for robust qPCR-based eDNA detection assays in environmental monitoring and species inventories. Environ DNA. https://doi.org/10.1002/edn3.164
Leadley PW, Krug CB, Alkemade R, Pereira HM, Sumaila UR, Walpole M, Marques A et al. (2014) Progress towards the Aichi biodiversity targets: an assessment of biodiversity trends, policy scenarios and key actions. Secretariat of the Convention on Biological Diversity, Montreal, Canada. Technical Series No. 78, pp. 13
Leray M, Yang JY, Meyer CP, Mills SC, Agudelo N, Ranwez V, Boehm JT, Machida RJ. (2013) A new versatile primer set targeting a short fragment of the mitochondrial COI region for metabarcoding metazoan diversity: application for characterizing coral reef fish gut contents. Front Zool 10: 34. https://doi.org/10.1186/1742-9994-10-34
Lorenz TC (2012) Polymerase chain reaction: basic protocol plus troubleshooting and optimization strategies Jove, 63, e3998
Lyon JP, Bird T, Nicol S, Kearns J, O’Mahony J, Todd CR, Cowx IG et al (2014) Efficiency of electrofishing in turbid lowland rivers: implications for measuring temporal change in fish populations. Can J Fish Aquat 71:878–886
Manfrin C, Souty-Grosset C, Anastácio PM, Reynolds JD, Giulianini PG (2019) Detection and control of invasive freshwater crayfish: from traditional to innovative methods. Diversity. https://doi.org/10.3390/d11010005
McKee AM, Spear SF, Pierson TW (2015) The effect of dilution and the use of a post-extraction nucleic acid purification column on the accuracy, precision, and inhibition of environmental DNA samples, Biol Cons https://doi.org/10.1016/j.biocon.2014.11.031
Rees HC, Maddison BC, Middleditch DJ, Patmore JRM, Gough KC (2014) The detection of aquatic animal species using environmental DNA—a review of eDNA as a survey tool in ecology. J App Ecol. https://doi.org/10.1111/1365-2664.12306
Robinson CV, Garcia de Leaniz C, Rolla M, Consuegra S (2019) Monitoring the eradication of the highly invasive topmouth gudgeon (Pseudorasbora parva) using a novel eDNA assay. Environ DNA. https://doi.org/10.1002/edn3.12
Ruppert MK, Kline RJ, Rahman MS (2019) Past, present, and future perspectives of environmental DNA (eDNA) metabarcoding: a systematic review in methods, monitoring, and applications of global eDNA. Glob Ecol Conserv. https://doi.org/10.1016/j.gecco.2019.e00547
Sepulveda AJ, Nelson NM, Jerde CL, Luikart G (2020) Are environmental DNA methods ready for aquatic invasive species management? Trends Ecol Evol 35(8):668–678. https://doi.org/10.1016/j.tree.2020.03.011SpecchiM,PizzulE(1996)Pseudorasboraparva(Schlegel,1842)(Osteichthyes,Cyprinidae)nelleacquedelFriuli-VeneziaGiulia.Primasegnalazione.Gortania.AttiMuseoFriul.diStoriaNat.17:145-147
Specchi M, Pizzul E (1996) Pseudorasbora parva (Schlegel, 1842) (Osteichthyes, Cyprinidae) in the Friuli-Venezia Giulia freshwaters (North-Eastern Italy), first observation. Gortania, Atti Museo Friul. di Storia Nat 17:145–147
Spikmans F, Lemmers P, op den Camp HJM, van Haren E, Kappen F, Blaakmeer A, van der Velde G et al (2020) Impact of the invasive alien topmouth gudgeon (Pseudorasbora parva) and its associated parasite Sphaerothecum destruens on native fish species. Biol Inv, https://doi.org/10.1007/s10530-019-02114-6
Thalinger B, Wolf E, Traugott M, Wanzenböck J (2019) Monitoring spawning migrations of potamodromous fish species via eDNA. Sci Rep. https://doi.org/10.1038/s41598-019-51398-0
Thomsen PF, Willerslev E (2015) Environmental DNA—an emerging tool in conservation for monitoring past and present biodiversity. Biol Cons. https://doi.org/10.1016/j.biocon.2014.11.019
Tillotson MD, Kelly RP, Duda JJ, Hoy M, Kralj J, Quinn TP (2018) Concentrations of environmental DNA (eDNA) reflect spawning salmon abundance at fine spatial and temporal scales. Biol Cons. https://doi.org/10.1016/j.biocon.2018.01.030
Troth CR, Burian A, Mauvisseau Q, Bulling M, Nightingale J, Mauvisseau C, Sweet MJ (2020) Development and application of eDNA-based tools for the conservation of white clawed crayfish. Sci Tot Environ. https://doi.org/10.1016/j.scitotenv.2020.141394
Uchii K, Doi H, Okahashi T, Katano I, Yamanaka H, Sakata MK, Minamoto T (2019) Comparison of inhibition resistance among PCR reagents for detection and quantification of environmental DNA. Environ DNA. https://doi.org/10.1002/edn3.37
Veldhoen N, Hobbs J, Ikonomou G, Hii M, Lesperance M, Helbing CC (2016) Implementation of novel design features for qPCR-based eDNA assessment. PLoS ONE. https://doi.org/10.1371/journal.pone.0164907
Vibert R (1967) Fishing with electricity—its application to biology and management—FAO-EIFAC, Fishing News Ltd., London: pp. 275
Witkowski A (2011) NOBANIS—Invasive Alien Species Fact Sheet—Pseudorasbora parva. In Online Database of the European Network on Invasive Alien Species—NOBANIS. Wrocław, Poland: Museum of Natural History, Wrocław University
Záhorská E, Kováč V, Falka I, Beyer K, Katina S, Copp GH, Gozlan RE (2009) Morphological variability of the Asiatic cyprinid, topmouth gudgeon Pseudorasbora parva, in its introduced European range. J Fish Biol. https://doi.org/10.1111/j.1095-8649.2008.02121.x
Acknowledgements
This work was supported by Servizio biodiversità della Direzione centrale risorse agroalimentari, forestali ed ittiche. The datasets generated during during the current study are available from the corresponding author on reasonable request. Authors would like to thank Messrs Francesco Bregant and Maurizio Angeli for their precious support on water filtering.
Funding
Open access funding provided by Università degli Studi di Trieste within the CRUI-CARE Agreement. This research was entirely funded by the Friuli Venezia Giulia Region.
Author information
Authors and Affiliations
Contributions
MZ and EP planned the stations suitable for the environmental monitoring; CM and MZ collected and extracted the eDNA; CM, AP and PGG designed the primers, planned and performed the qPCR experiments, CM and LM planned and performed the qdPCR experiments. CM, MZ, LM, PGG and AP written and revised the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Authors declare not to have any conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Manfrin, C., Mirimin, L., Zanetti, M. et al. Highly sensitive environmental DNA detection of topmouth gudgeon, Pseudorasbora parva: a comparison of qPCR and microfluidic qdPCR. Biol Invasions 24, 2121–2133 (2022). https://doi.org/10.1007/s10530-022-02761-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10530-022-02761-2