
Abstract
Environmental DNA (eDNA) detection of non-native species has considerable potential to inform management decisions, including identifying the need for population control and/or eradication. An invasive species of European concern is the Asian cyprinid fish, topmouth gudgeon (Pseudorasbora parva). Here, eDNA analyses were applied at a commercial angling venue in southern England to inform operations aiming to eradicate P. parva, which had only ever been observed in one of the venue’s seven unconnected angling ponds. Eradication of P. parva was initially attempted by repeated depletion of the population using fish traps (crayfish traps fitted with 5 mm mesh netting) and the introduction of native predators over a 4-year period. The very low number of P. parva captured following these eradication efforts suggested a possible population crash. Conventional PCR analysis of water samples using species-specific primers was applied to all seven ponds to confirm that P. parva was present in only one pond, that the eradication attempt had indeed failed and that the species’ distribution in the pond appeared to be restricted to three bankside locations. The continued presence of P. parva at these locations was confirmed by subsequent trapping. Water samples from an adjacent, unconnected stream were also analysed using the eDNA methodology, but no DNA of P. parva was detected. The results suggest that further management action to eradicate P. parva be focused on the pond shown to contain the isolated P. parva population and thereby eliminate the risk of further dispersal. This study is the first to apply eDNA analysis to assess the efficacy of an eradication attempt and to provide evidence that the species was unlikely to be present in the other ponds, thus reducing the resources needed to control the species.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Surveys based on the detection of environmental DNA (eDNA) are increasingly used to detect the presence of a broad range of taxonomic groups in aquatic environments, with particular applications to species of conservation concern and non-native species (Jerde et al. 2011; Rees et al. 2014; Thomsen and Willerslev 2015). This is because eDNA-based surveys, which collect DNA shed by an organism via urine, faeces, mucus and epidermal cells into the water, tend to have greater power to detect elusive and/or rare organisms than conventional sampling approaches, e.g. bluegill sunfish (Lepomis macrochirus) (Takahara et al. 2013). This increased effectiveness, combined with relatively low financial costs and reduced impact on the environment, demonstrates that eDNA methodologies have high potential for enhancing the management of invasive fish species (Rees et al. 2014; Bylemans et al. 2016). Applications so far have included distribution assessments (Takahara et al. 2013; Keskin 2014), monitoring surveys on invasion fronts (Jerde et al. 2013; Adrian-Kalchhauser and Burkhardt-Holm 2016) and the evaluation of population eradication attempts (Dunker et al. 2016).
Eradication of potentially harmful non-native species is considered a key component of invasive species management, particularly in rapid response scenarios (UK Defra 2008; Britton et al. 2011a; Genovesi et al. 2015). Attempts to eradicate non-native fish species often involve application of a piscicide, such as rotenone (Allen et al. 2006; Britton et al. 2008), even though this practice can have substantial impacts on non-target fauna (e.g. Finlayson et al. 2010; Billman et al. 2011). In some circumstances, such as isolated water bodies, it may be possible to eradicate a fish species through a drain-down and liming of the water body (Britton et al. 2008). Other options for controlling invasive fish populations include repeated cropping by netting, trapping or electric fishing, and biological control by stocking predators (Britton et al. 2008).
Topmouth gudgeon (Pseudorasbora parva), a native species in eastern Asia, is one of the most invasive freshwater fish species in Europe, having spread across most of the continent within decades of its accidental introduction to Romania in the 1960s as a contaminant of Asian carp consignments (Gozlan et al. 2010). It arrived in England by this introduction vector in the mid-1980s (Gozlan et al. 2002). Such is the threat posed by P. parva, in particular its role as a healthy host of the rosette agent Spherotecum destruens (Gozlan et al. 2005), that it is the target of a national eradication campaign, which aims to remove all 23 known UK populations by the end of 2017 (UK EA 2014; GBNNNS 2015). P. parva is one of just two fish species currently listed as being of European Union concern under Regulation (EU) no. 1143/2014, requiring EU member states to implement management and control measures (European Union 2014). Methods which have been successfully used to eradicate local topmouth gudgeon populations include rotenone treatments (Britton et al. 2008) and repeated removals (Copp et al. 2007). Also, there are instances elsewhere in Europe where P. parva have established a population in a water body, persisted for a short period (<10 years) and then disappeared entirely (Copp et al. 2007). This suggests that the species may be susceptible to recruitment failure and local extirpation where their population numbers are dramatically reduced by either natural or human-assisted means.
To facilitate this management programme, an attempt to eradicate a P. parva population from a pond on a commercial recreational angling venue in southern England was undertaken between 2011 and 2016 using depletion and biocontrol methods. Given the requirement of such eradication attempts to undergo thorough post-operation evaluations to measure their efficacy (Britton et al. 2011a), the aim of this study was to demonstrate the potential use of eDNA analysis as a complement to conventional sampling methodologies for assessing the efficacy of fish eradication attempts. Our specific objectives were to: (1) develop a statistically-robust eDNA sampling protocol for evaluating the P. parva eradication attempt; (2) assess the efficacy of the eradication attempt using conventional and eDNA methods; and (3) determine whether or not P. parva was likely, based on eDNA analysis results, to be present in any other water bodies at the site.
Materials and methods
Primer design and testing
Species-specific primers for P. parva were designed to amplify a 350-base-pair region of the mitochondrial gene encoding cytochrome c oxidase subunit 1 (COI): forward primer (5′-3) CCTCTTCCGGAGTAGAGGCT and reverse primer (5′-3) TAGGATTGGGTCTCCTCCCC (Davison et al. 2016). Primer specificity was tested in silico against sequences of all UK freshwater fishes, using NCBI Primer-BLAST (http://www.ncbi.nlm.nih.gov/tools/primer-blast/). The primers were also tested experimentally in conventional PCRs against DNA extracts (DNeasy Blood and Tissue Kit, Qiagen, Hilden, Germany) from fish species from the same family (Cyprinidae) that are likely to occur at the study site: common carp (Cyprinus carpio), common bream (Abramis brama), roach (Rutilus rutilus) and rudd (Scardinius erythrophthalmus). Conventional PCRs were conducted using 0.1 ng of genomic DNA and none of the triplicate PCRs showed amplification for any of these species.
Testing of primer efficiency and optimisation of the PCR protocol was undertaken using DNA extracted from dorsal muscle tissue samples of P. parva. These tests showed that the primers reliably amplified P. parva DNA at a quantity of 1.5 × 10−2 ng. The ability of the primers to detect P. parva DNA reliably from water samples was confirmed in aquarium trials (1 fish in 44-L tanks) and in a field survey conducted in ponds where the species was known to occur (Davison et al. 2016).
Study site and field sampling protocol
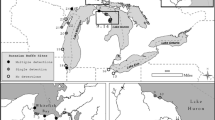
The recreational angling venue, which was located in Kent, South-east England (latitude 51° N, longitude 0° E), has no direct hydrological connections with an adjacent stream nor are any of the seven angling ponds connected (Fig. 1). A single specimen of P. parva was first captured in one of the angling ponds (area = 1.4 ha) in April 2004 but reported in the angling press to be a young grass carp Ctenopharyngodon idella (fishery owners, personal communication). An attempt to eradicate P. parva from this pond began in 2011 under the guidance of an independent fisheries consultant (commissioned by the fishery owners). From 2011 to July 2016, this consisted of intensive depletion using cylindrical fish traps (i.e. 60 by 30 cm crayfish traps with conical funnel entrance and fitted with 5 mm mesh netting). The depletion trapping was complemented by repeated, high density (116 kg ha−1) stocking of a native predatory fish, Eurasian perch (Perca fluviatilis)—a biocontrol method that has been demonstrated to exert a top-down effect on P. parva abundance (Davies and Britton 2015; Verhelst et al. 2016). Initial reports received by the authors indicated that by 2014, P. parva were no longer being captured; however, trapping data recently acquired from the venue’s owners revealed persistence of a very small number of P. parva, with the lowest capture densities occurring after predator releases (Fig. 2).

Schematic map (scale bars = 100 m) of the study site in the English county of Kent, showing location of the seven ponds and adjacent stream. In the infested lake (inset maps), pelagic sampling locations are indicated with small open circles, whereas littoral sampling locations (squares) are numbered (see Table 1), the filled squares indicating locations where positive detections of P. parva DNA occurred in the initial sampling survey (Sept 2014, inset A). Locations 1 and 10 also came up positive in Nov 2014 (inset B). See also Table 1

Numbers of topmouth gudgeon Pseudorasbora parva, calculated on a per trap per month basis, captured by fishery staff between 2011 and 2016 using fish traps (see ‘Materials and methods’) placed around the water body’s banks each sampling excursion. The arrows indicate dates of predator biocontrol release, i.e. 400, 200, 400 and 246 Eurasian perch Perca fluviatilis (left to right, respectively) of 6–9 cm total length
To ensure a statistically robust eDNA sampling protocol, an a priori power analysis was performed (http://homepage.stat.uiowa.edu/∼rlenth/Power/). This suggested that water should be collected from 24 sampling locations (12 littoral and 12 pelagic). At each sampling location, four sub-samples should be taken for analysis, and at least two PCR amplifications should be performed for each sub-sample. According to this protocol (corresponding to a doubly nested design), at a level of significance α = 0.05, statistical power (β) would equal 0.806 for the sampling zones, 0.978 for the water samples and 0.999 for the sub-samples.
Accordingly, post-eradication assessment using eDNA analysis consisted of three sampling steps (Sept 2014, Nov 2014, Feb–Mar 2015). Firstly, 24 1-L water samples were collected on 16 Sept 2014 in the infested pond, namely from 12 littoral zone locations spread equidistantly (40 m apart) around the pond shore and 12 from pelagic zone locations spaced around the water body (Fig. 1). Secondly, water sampling was undertaken during a return visit on 12 Nov 2014 at the six littoral sampling points in the infested water body closest to those where eDNA of P. parva was detected during step one. No P. parva DNA was detected in any of the 12 pelagic (mid-water) samples, so these pelagic sample locations were not considered further in the eDNA analysis. The water sampling on 12 Nov 2014 was complemented by intensive sampling, using the fish traps described above. Traps were deployed for 5 days in late Dec 2014, 10 days in early Feb and 6 days in early April 2015 (Fig. 2). Thirdly, water samples (1 L) were collected in 2015 on 17 Feb. 19 Feb and 5 March from 12 littoral zone locations in each of the other six ponds (areas of 0.5 to 2.4 ha), as well as at eight locations along the longitudinal course (1.5 km) of the small stream that runs adjacent to the ponds. Pelagic samples were not collected from the other six ponds, as this would have required movement of the boat between the water bodies, thus increasing the risk of cross contamination.
In all cases, water samples were collected using a 183-cm sampling pole with a 500-mL polypropylene sampling cup attached (Camlab Ltd., Cambridge, UK). The sampling cup was moved in a standardised manner from the bank (littoral samples) or boat (pelagic samples) to the greatest extent reached by the pole, ensuring no contact with the bottom sediment. At each sampling location, 1 L of water obtained using the sampling cup was poured into a sterilised plastic bottle. Samples were then placed in individual plastic bags and immediately refrigerated (4 °C) for transportation back to the laboratory. On each sampling day, two identical ‘blank samples’ (new sterilised bottles of de-ionised water from the laboratory), opened briefly in the field, were handled and transported in the same manner as the pond samples. Between samples, the sampling pole and cup were disinfected using Microsol 3+ (Anachem Ltd., Luton, UK) and washed with de-ionised water.
Laboratory protocol
Within 24 h of collection, the water samples were filtered through a 0.4-μm pore size polycarbonate filter of diameter 47 mm (Isopore, EMD Millipore, Darmstadt, Germany) using a vacuum pump (EMD Millipore). From each sampling location, four sub-samples of 100 mL were filtered. Between filtration of samples from each location, the filtering equipment was sterilised using Microsol 3+ and washed with de-ionised water, and at regular intervals during filtration, de-ionised water was run through the filtration system, with these samples analysed to detect any potential cross contamination. The filters were immediately frozen at −80 °C. DNA extraction from the filters took place within 3 months from initial sampling using a PowerWater DNA Isolation Kit (MO BIO, Carlsbad, CA, USA).
Conventional PCR amplifications were performed in 20 μL reaction mixtures, containing 6 μL of DNA template, 0.5 μM of each primer, 10 μL (=50 units) HotStar Taq Plus DNA polymerase (Qiagen Fast Cycling PCR Kit) and 2 μL CoralLoad Fast Cycling Dye (Qiagen). The cycling conditions employed were an initial denaturation step at 95 °C for 5 min, followed by 32 cycles of denaturation (96 °C; 5 s), annealing (61 °C; 5 s) and extension (68 °C; 12 s), with a final extension at 72 °C for 1 min. Amplified PCR products were visualised using electrophoresis on 2% agarose gel, stained with SYBR Gold Nucleic Acid Gel Stain (Invitrogen, Paisley, UK). Three replicate PCRs were conducted for each 100 mL sub-sample, with each one including a negative control (de-ionised water) and a positive control (tissue-extracted P. parva DNA). To confirm the identity of sequences amplified from the pond samples, PCR products from the positive sampling points were purified (Nucleospin Gel and PCR Cleanup) and sequenced by a commercial service (Eurofins Genomic Services Ltd., Wolverhampton, UK).
To confirm that negative results were not detection errors (‘false negatives’) caused by PCR inhibition, additional PCRs were conducted using the PCR protocol described previously (Jane et al. 2015; Adrian-Kalchhauser and Burkhardt-Holm 2016). PCRs were performed using an eDNA sample (6 μL) from a single location within each pond that was spiked with 2 μL of genomic P. parva DNA (0.01 ng/μL). The strength of the resultant electrophoresis gel band was compared visually with that from the same quantity of P. parva DNA amplified in de-ionised water alone (i.e. without sample). As these PCRs indicated the presence of inhibition, a further set of PCRs were undertaken in which the extracted samples (one sub-sample from each sampling location) were re-analysed following a 1:5 dilution in de-ionised water, a technique used to combat inhibition by diluting the inhibitory compounds (McKee et al. 2015). Three replicate PCRs were conducted on these diluted samples. To assess whether inhibition was still occurring following the 1:5 dilution, three replicate PCRs per pond were conducted in which a spike of 0.02 ng of tissue-extracted P. parva DNA was added.
Filtration, extraction, PCR preparation and post-PCR analysis were undertaken in separate rooms of a laboratory dedicated to molecular biology, observing strict anti-contamination procedures (no transfer of equipment between rooms; changing of labcoats when moving between rooms; thorough cleaning of all equipment and surfaces before and after use).
Results
In the initial sampling step, of the infested water body only, P. parva DNA was detected at 3 of the 12 littoral zone locations (Table 1). These sampling locations came from adjacent locations at one end of the pond (Fig. 1). DNA of P. parva was not detected in any of the 100-mL sample replicates collected from the pelagic zone. Spiking tests indicated a small level of inhibition occurring in pelagic and littoral samples. Two samples contained the minimum quantity of DNA required for sequencing, which confirmed the identity of the eDNA as that of P. parva. Both sequences showed a 100% match with 34 sequences of P. parva registered in the Genbank database (e.g. accession number HQ960448).
In the second sampling step, repeat sampling and eDNA analysis of water from the locations where P. parva eDNA had been detected in step one provided further confirmation of the species’ presence. This corroborated the trapping data recently acquired from the venue’s owners (Fig. 2).
In the third sampling step, all sample replicates from the other six angling ponds and from the adjacent small stream proved negative for P. parva eDNA. Spiking tests indicated a small level of inhibition occurring in all six ponds. Following the 1:5 dilution of extracted samples to combat the detected inhibition, no further inhibition was detected. All samples that had previously shown negative for P. parva DNA (i.e. previously negative littoral locations and pelagic locations from the infested pond, and all samples from the a priori non-infested ponds) also proved to be negative following the 1:5 dilution. These results suggest that the level of inhibition occurring in the samples was not sufficient to mask the presence of DNA during the first analysis.
Discussion
The current study demonstrates that eDNA surveys are a valuable method for post-evaluation of eradication attempts, with equal, if not greater, power to detect remnant populations of target species than conventional survey methods. Water samples subjected to eDNA analysis confirmed the persistence of a small population of P. parva in the infested pond, such as reported in detection studies elsewhere (Britton et al. 2011b). In the other water bodies, eDNA analysis corroborated trapping results for the other six angling ponds and electrofishing results for the adjacent stream, which indicate it is unlikely the species was present at the time of sampling.
Small-bodied fishes at low population densities can often be difficult to detect, and imperfect detection using conventional methods (i.e. electric fishing and trapping) has previously been demonstrated for P. parva in 100 m2 ponds (Britton et al. 2011b). At low population abundances, eDNA surveys may represent the most effective method of confirming the presence of a fish species. For example, eDNA sampling detected the presence of European weather loach (Misgurnus fossilis) in a location where it had not been recorded for 13 years using traditional methods, including fish traps, electrofishing and seine nets (Sigsgaard et al. 2015). In the present study, the spatial heterogeneity of the positive eDNA detections is likely to reflect the heterogeneous distribution of the target species, which has been recorded previously (Li et al. 2010; Davison et al. 2016). The lack of detections from the open water sampling locations is indicative of a distribution favouring shallow vegetated areas in the littoral zone (as previously shown for P. parva: Li et al. 2010) or an alternative favoured habitat type that is present in only a few isolated locations around the pond. The trapping of 78 specimens in the vicinity of these sampling points (7 months after the initial water sample collection), suggests that a small, localised population in this area was the most likely source of the detected eDNA.
Spatial heterogeneity of eDNA is common in lentic water bodies (e.g. Eichmiller et al. 2014), emphasising the need for sufficient water samples to be collected (with adequate spatial coverage) to increase the likelihood of detection of localised species in low abundance. In the present study, only five positive detections resulted from 96 sub-samples of water from 24 locations in the infested lake. Detection power could potentially have been improved by modifying the PCR protocol, such as increasing the number of cycles (Rameckers et al. 1997). The sensitivity of detection could arguably be increased by using quantitative real-time PCR (qPCR) protocols, for which higher levels of sensitivity have been reported (Tréguier et al. 2014; Biggs et al. 2015). However, in mesocosm trials, no difference between qPCR and conventional PCR was found in the detection of DNA of target species present at low density (Nathan et al. 2014). A practical consideration is that conventional PCR is financially less costly than qPCR, and therefore more likely to be available to those tasked with the management of invasive species (Davison et al. 2016).
The lack of detection of P. parva DNA in the six other lakes on site serves to corroborate the species’ absence in angler’s catches and conventional surveys undertaken before and after the eDNA survey (fishery owners, personal communication). Indeed, no P. parva were observed or captured in the adjacent stream during an electrofishing survey carried out a few months after the water samples for eDNA analysis were collected (Environment Agency, personal communication). Whilst caution is always needed when declaring a species to be absent on the basis of absence of detection, regardless of the survey method used (Mackenzie 2005; Kéry and Schmidt 2008), the statistically rigorous sampling protocol used here suggests that it is unlikely that P. parva is present in the other nearby, but unconnected, ponds and the stream. PCR-inhibiting compounds in the water are a potential cause of false negatives, but in this case study, the detected inhibition was not sufficient to affect the results. It does demonstrate, however, the importance of incorporating steps in laboratory protocols to assess the extent of inhibition, and if necessary to overcome inhibition by methods such as dilution of samples or addition of bovine serine albumin (Deiner et al. 2015; McKee et al. 2015).
The risk of false positives also needs to be considered when basing management decisions on the results of eDNA surveys. Positive detections should not necessarily be taken as an indication of presence of live organisms, as DNA could enter the water from other sources, e.g. decaying corpses or bird faeces (Merkes et al. 2014; Dunker et al. 2016). Before costly management action is taken, ‘ground truthing’ (i.e. capture of live individuals) is recommended to corroborate eDNA detection, such as was the case in the present study.
The present study demonstrates the applicability of eDNA surveys to assess the efficacy of eradication attempts in aquatic environments, providing additional support for studies elsewhere in which eDNA analysis was reported to be more sensitive than conventional methods for detecting species present in low abundance. Accurate assessments of the success of eradications is important; the continuation of a monitoring programme after the final individuals have been removed can be costly, whilst conversely the premature declaration of success and resultant cessation of monitoring can be even more costly and potentially nullify previous efforts (Rout et al. 2009, 2014). Surveys based on eDNA analysis are therefore an important tool to assist the decision-making process as regards the management of non-native species, both for early detection and rapid response, as well as for the assessment of eradication success. To this end, a nested quantitative PCR protocol is currently being tested in still and running waters for such applications to enhance the sensitivity of the analysis.
References
Adrian-Kalchhauser I, Burkhardt-Holm P (2016) An eDNA assay to monitor a globally invasive fish species from flowing freshwater. PLoS One 11:e0147558
Allen Y, Kirby S, Copp GH, Brazier M (2006) Toxicity of rotenone to topmouth gudgeon Pseudorasbora parva for eradication of this non-native species from a tarn in Cumbria, England. Fish Manag Ecol 13:337–340
Biggs J, Ewald N, Valentini A, Gabouriaud C, Griffiths RA, Foster J, Wilkinson JW, Arnell A, Brotherton P, Williams P (2015) Using eDNA to develop a national citizen science-based monitoring programme for the great crested newt (Triturus cristatus). Biol Conserv 183:19–28
Billman HG, St-Hilaire S, Kruse CG, Peterson TS, Peterson CR (2011) Toxicity of the piscicide rotenone to Columbia spotted frog and boreal toad tadpoles. Trans Amer Fish Soc 140:919–927
Britton JR, Brazier M, Davies GD, Chare SI (2008) Case studies on eradicating the Asiatic cyprinid Pseudorasbora parva from fishing lakes in England to prevent their riverine dispersal. Aquat Conserv Mar Freshw Ecosys 18:867–876
Britton JR, Gozlan RE, Copp GH (2011a) Managing non-native fish in the environment. Fish Fish 12:256–274
Britton JR, Pegg J, Gozlan RE (2011b) Quantifying imperfect detection in an invasive pest fish and the implications for conservation management. Biol Conserv 144:2177–2181
Bylemans J, Furlan EM, Pearce L, Daly T, Gleeson DM (2016) Improving the containment of a freshwater invader using environmental DNA (eDNA) based monitoring. Biol Invasions 18:3081–3089
Copp GH, Wesley KJ, Verreycken H, Russell IC (2007) When an ‘invasive’ fish species fails to invade! Example of the topmouth gudgeon Pseudorasbora parva. Aquat Invasions 2:107–112
Davies GD, Britton JR (2015) Assessing the efficacy and ecology of biocontrol and biomanipulation for managing invasive pest fish. J Appl Ecol 52:1264–1273
Davison PI, Créach V, Liang W-J, Andreou D, Britton JR, Copp GH (2016) Laboratory and field validation of a simple method for detecting four species of non-native freshwater fish using environmental DNA. J Fish Biol 89:1782–1793
Deiner K, Walser J-C, Mächler E, Altermatt F (2015) Choice of capture and extraction methods affect detection of freshwater biodiversity from environmental DNA. Biol Conserv 183:53–63
Dunker KJ, Sepulveda AJ, Massengill RL, Olsen JB, Russ OL, Wenburg JK, Anotovich A (2016) Potential of environmental DNA to evaluate northern pike (Esox lucius) eradication attempts: an experimental test and case study. PLoS One 11:e0162277
Eichmiller JJ, Bajer PG, Sorenson PW (2014) The relationship between the distribution of common carp and their environmental DNA in a small lake. PLoS One 9:e112611
European Union (2014) Regulation (EU) no 1143/2014 of the European parliament and of the Council of 22 October 2014 on the prevention and management of the introduction and spread of invasive alien species. Official Journal of the European Union 4.11.2014, L317: 35–55
Finlayson B, Somer WL, Vinson MR (2010) Rotenone toxicity to rainbow trout and several mountain stream insects. N Amer J Fish Manag 30:102–111
GBNNNS (2015) The Great Britain Invasive Non-native Species Strategy August 2015. The Great Britain Non-native Species Secretariat, York. www.gov.uk/government/uploads/system/uploads/attachment_data/file/455526/gb-non-native-species-strategy-pb14324.pdf Accessed 15 August 2016
Genovesi P, Carboneras C, Vila M, Walton P (2015) EU adopts innovative legislation on invasive species: a step towards a global response to biological invasions? Biol Invasions 17:1307–1311
Gozlan RE, Andreou D, Asaeda T, Beyer K, Bouhadad R, Burnard D, Caiola N, Cakić P, Djikanović V, Esmaeili HR, Falka I, Golicher D, Harka A, Jeney G, Kováč V, Musil J, Nocita A, Povž M, Poulet N, Virbickas T, Wolter C, Tarkan AS, Tricarico E, Trichkova TA, Verreycken H, Witkowski A, Zhang C-G, Zweimueller I, Britton JR (2010) Pan-continental invasion of Pseudorasbora parva: towards a better understanding of freshwater fish invasions. Fish Fish 11:315–340
Gozlan RE, Pinder AC, Shelley J (2002) Occurrence of the Asiatic cyprinid Pseudorasbora parva in England. J Fish Biol 61:298–300
Gozlan RE, St-Hilaire S, Feist SW, Martin P, Kent ML (2005) Biodiversity: disease threat to European fish. Nature 435:1046
Jane SF, Wilcox TM, McKelvey KS, Young MK, Schwartz MK, Lowe WL, Letcher BH, Whiteley AR (2015) Distance, flow and PCR inhibition: eDNA dynamics in two headwater streams. Mol Ecol Resourc 15:216–227
Jerde CL, Chadderton WL, Mahon AR, Renshaw MA, Corush J, Budny ML, Mysorekar S, Lodge DM (2013) Detection of Asian carp DNA as part of a Great Lakes basin-wide surveillance program. Can J Fish Aquat Sci 70:522–526
Jerde CL, Mahon AR, Chadderton WL, Lodge DM (2011) “Sight-unseen” detection of rare aquatic species using environmental DNA. Conserv Let 4:150–157
Kéry M, Schmidt B (2008) Imperfect detection and its consequences for monitoring for conservation. Comm Ecol 9:207–216
Keskin E (2014) Detection of invasive freshwater fish species using environmental DNA survey. Biochem Syst Ecol 56:68–74
Li W, Zhang T, Li Z (2010) Spatial distribution and abundance of small fishes in Xiaosihai Lake, a shallow lake along the Changjiang (Yangtze) River, China. Chin J Oceanol Limnol 28:470–477
Mackenzie D (2005) Was it there? Dealing with imperfect detection for species presence/absence data. Austr NZ J Stat 47:65–74
McKee AM, Spear SF, Pierson TW (2015) The effect of dilution and the use of a post-extraction nucleic acid purification column on the accuracy, precision and inhibition of environmental DNA samples. Biol Conserv 183:70–76
Merkes CM, McCalla SG, Jensen NR, Gaikowski MP, Amberg JJ (2014) Persistence of DNA in carcasses, slime and avian feces may affect interpretation of environmental DNA data. PLoS One 9:e113346
Nathan LM, Simmons M, Wegleitner B, Jerde CL, Mahon AR (2014) Quantifying environmental DNA signals for aquatic invasive species across multiple detection platforms. Environ Sci Technol 48:12800–12806
Rameckers J, Hummel S, Herrmann B (1997) How many cycles does a PCR need? Determinations of cycle numbers depending on the number of targets and the reaction efficiency factor. Naturwissenschaften 84:259–262
Rees HC, Maddison BC, Middleditch DJ, Patmore JR, Gough KC (2014) The detection of aquatic animal species using environmental DNA– a review of eDNA as a survey tool in ecology. J Appl Ecol 51:1450–1459
Rout TM, Kirkwood R, Sutherland DR, Murphy S, McCarthy MA (2014) When to declare successful eradication of an invasive predator? Anim Conserv 17:125–132
Rout TM, Thompson CJ, McCarthy MA (2009) Robust decisions for declaring eradication of invasive species. J Appl Ecol 46:782–786
Sigsgaard EE, Carl H, Møller PR (2015) Monitoring the near-extinct European weather loach in Denmark based on environmental DNA from water samples. Biol Conserv 183:46–52
Takahara T, Minamoto T, Doi H (2013) Using environmental DNA to estimate the distribution of an invasive fish species in ponds. PLoS One 8:e56584
Thomsen PF, Willerslev E (2015) Environmental DNA—an emerging tool in conservation for monitoring past and present biodiversity. Biol Conserv 183:4–18
Tréguier A, Paillisson J-M, Dejean T, Valentini A, Schlepfer MA, Roussel JM (2014) Environmental DNA surveillance for invertebrate species: advantages and technical limitations to detect invasive crayfish Procambarus clarkii in freshwater ponds. J Appl Ecol 51:871–879
UK Defra (2008) The Invasive Non-Native Species Framework Strategy for Great Britain –Protecting our natural heritage from invasive species. Product code: PB 13075. Department for Environment, Food and Rural Affairs, London. 42 pp. www.nonnativespecies.org/downloadDocument.cfm?id=99 Accessed 15 August 2016
UK EA (2014) Topmouth gudgeon eradication programme. UK Environment Agency Progress update: July 2014. www.nonnativespecies.org/downloadDocument.cfm?id=1133 Accessed 15 August 2016
Verhelst P, Boets P, Van Thuyne G, Verreycken H, Goethals PL, Mouton AM (2016) The distribution of an invasive fish species is highly affected by the presence of native fish species: evidence based on species distribution modelling. Biol Invasions 18:427–444
Acknowledgements
This study was funded by the UK Department of Environment, Food and Rural Affairs. The authors wish to thank Bedwell Fisheries Services for providing background information on their eradication attempt, and the owners and staff of the fishery, in particular K. Pallet, for permission to collect water samples and for providing the fish trapping data. The authors also thank Dr. D. Andreou for providing tissue-extracted DNA from other fish species and Dr. W.-J. Liang for advice.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: J. Antonio Baeza
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Davison, P.I., Copp, G.H., Créach, V. et al. Application of environmental DNA analysis to inform invasive fish eradication operations. Sci Nat 104, 35 (2017). https://doi.org/10.1007/s00114-017-1453-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00114-017-1453-9