
Abstract
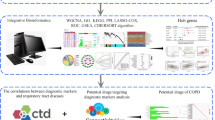
To integrate gene expression and DNA methylation data and find the potential role of DNA methylation in the invasion and replication of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). We first conducted differential expression and methylation analysis between the coronavirus disease of 2019 (COVID-19) and healthy controls. FEM was employed to identify functional epigenetic modules, from which a diagnostic model for COVID-19 was built. SKA1 and WSB1 modules were identified, with SKA1 module enriched in COVID-19 replication and transcription, and WSB1 module related to ubiquitin-protein activity. The differentially expressed or differentially methylated genes in these two modules could be used to distinguish COVID-19 from healthy controls, with AUC reaching 1 and 0.98 for SKA1 and WSB1 modules, respectively. Two epigenetically activated genes (CENPM and KNL1) from the SKA1 module were upregulated in HPV- or HBV-positive tumor samples and were found to be significantly associated with the survival of tumor patients. In conclusion, the identified FEM modules and potential signatures play an essential role in the replication and transcription of coronavirus.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has caused a widespread global pandemic resulting in over 569 million confirmed cases and more than 6 million deaths, as reported by the Johns Hopkins University Coronavirus Resource Center on July 23rd, 2022 (https://coronavirus.jhu.edu/map.html). With the increasing research on SARS-CoV-2 (Roshandel et al. 2020; Balaky et al. 2020; Zhu et al. 2020), more and more high-throughput multi-omics sequencing data are being made available on databases such as Gene Expression Omnibus (GEO), enabling integrative analyses of DNA methylation and gene expression data to identify epigenetically regulated modules or potential biomarkers. Thair et al. (2021) performed RNA-Seq on the samples infected with six viruses, including SARS-CoV-2, and identified a series of differentially expressed genes. Manuel Castro de Moura et al. (2021) analyzed the DNA methylation status of peripheral blood samples from 407 confirmed COVID-19 patients and identified 44 CpG sites associated with the severity of COVID-19. Finally, Balnis et al. (2021) compared the differentially methylated regions (DMR) between COVID-19 patients and healthy individuals, finding that the DMRs were enriched in gene promoter regions and hypomethylated in COVID-19 samples.
It's worth noting that there has not been any study that focuses on integrating DNA methylation and gene expression datasets to identify the functional epigenetic module and potential biomarkers for COVID-19. However, a supervised algorithm called FEM (Jiao et al. 2014) can be used to identify gene modules where a significant number of genes are differentially methylated and expressed simultaneously. FEM has already been applied to module discovery in many studies (Teschendorff et al. 2016; Cancer Genome Atlas Research Network et al. 2017; Ding et al. 2020a; Wang et al. 2020) and is commonly used to integrate DNA methylation and gene expression datasets (Ding et al. 2020b).
In this study, we first conducted differential expression and methylation analysis, identified two functional epigenetic modules using the FEM algorithm, and performed gene set enrichment analysis for the genes from the identified modules. Interestingly, we found that the SKA1 module is associated with virus replication and transcription, while the WSB1 module is related to the activity of ubiquitin-protein and ubiquitin-protein ligase. We also observed that two genes, CENPM and KNL1, in the SKA1 module were significantly hypomethylated and upregulated in COVID-19 samples compared with healthy individuals. To validate the associations between these two epigenetically activated genes and virus infections, we performed differential expression and survival analysis in cervical squamous cell carcinoma (CESC), liver hepatocellular carcinoma (LIHC), and oropharyngeal squamous cell carcinoma (OPSCC) tumor samples with human papillomavirus (HPV) and hepatitis B virus (HBV) positive or negative information. As expected, those two genes are upregulated in HPV- or HBV- positive group compared with the negative group, and the expression or methylation of those two genes was significantly associated with the survival of the corresponding tumor samples. Finally, we built the diagnostic modules based on the expression and methylation values of the genes from those two modules, the area under the ROC Curve (AUC) was greater than 0.98. Our results suggest that the FEM modules and the identified epigenetically activated genes may play an important role in the replication and transcription of SARS-CoV-2 and could serve as potential biomarkers and therapeutic targets for COVID-19.
Material and Method
Datasets and Preprocessing
The datasets used in this study were downloaded from GEO (https://www.ncbi.nlm.nih.gov/geo/); for RNA-Seq data, the read count data, including 62 COVID-19 patients and 24 healthy controls, were obtained under accession ID GSE152641(Thair et al. 2021), followed by the normalization with edgeR (Robinson et al. 2010). The processed Infinium Methylation EPIC DNA methylation dataset of 102 COVID-19 patients and 26 non-COVID-19 patients’ whole blood tissue samples was obtained with accession GSE174818 (Balnis et al. 2021).
Differential Analysis and Identification of Functional Epigenetic Modules
The PPI network was obtained from InBio (Li et al. 2017) and BioPlex (Huttlin et al. 2015) databases. Given the PPI network, using gene expression and DNA methylation matrix as input, the FEM algorithm was implemented to perform differential expression and methylation analysis and identify function epigenetic modules. Genes with |stat(mRNA)|≥ 1.5 and P(mRNA) ≤ 0.05 were regarded as significantly differentially expressed genes, and genes with |stat(DNAm)|≥ 1.5 and P(DNAm) ≤ 0.05 were defined as differentially methylated genes.
Genes Set Enrichment Analysis
The hypomethylated genes and the genes in the identified FEM modules were submitted to the online website DAVID (Huang et al. 2009a, b) to perform gene ontology (GO) and KEGG enrichment analysis. Terms with FDR ≤ 0.05 were considered as significantly enriched terms; for better visualization, − log10(FDR) was calculated to plot the dot plot (Fig. 3).
Diagnostic Model
The logistic regression module was built using python scikit-learn (https://scikit-learn.org/) based on the expression or the methylation beta values of the genes from SKA1 and WSB1 FEM modules. All samples were randomly split into training and test sets with a 4:1 ratio, and the test dataset was used to evaluate our model.
Analysis of the Epigenetically Activated Genes in Tumors with HPV and HBV
To validate the association of two epigenetically activated genes, CENPM and KNL1 (significantly hypomethylated and upregulated in COVID-19 samples), the cancer genome atlas (TCGA) cancer type and virus types were queried on the OncoDB database (http://oncodb.org) (Tang et al. 2022) using default parameters. We only included the figures with at least three samples and a p-value ≤ 0.05 in Fig. 5.
Result
Differential Expression and Methylation Analysis
We first performed differential expression and methylation analysis, as shown in Fig. 1A. Our analysis revealed that there were 2168 upregulated, 2110 downregulated, 105 promoter hypermethylated, and 531 promoter hypomethylated genes. We also identified 143 epigenetically activated genes and 17 epigenetically silenced genes.

Differential analysis. A Distribution of the differentially expressed and methylated genes: the epigenetically activated genes (hypomethylated and upregulated, stat(DNAm) < − 1.5 and stat(mRNA) > 1.5 and p-value < = 0.05), and epigenetically silenced genes (hypermethylated and downregulated, stat(DNAm) > 1.5 and stat(mRNA) < − 1.5 and p-value < = 0.05) are shown as purple dots. Size of points represents the min(− log10(PDNAm), − log10(PmRNA)). B GO enrichment analysis result for the hypomethylated genes
Moreover, we found that the hypermethylated genes were enriched in response to lipopolysaccharide. In contrast, the hypomethylated genes were enriched in functions such as protein binding, protein homodimerization activity, focal adhesion, and so on, as depicted in Fig. 1B.
Our findings were consistent with a previous study (Thair et al. 2021), which showed that upregulated genes were enriched in G-protein coupled receptor signaling pathway, neuroactive ligand-receptor interaction, inflammatory response, and DNA replication-dependent nucleosome assembly. In contrast, downregulated genes were enriched in rRNA processing, viral transcription, viral process, RNA transport, et al. (Supplementary Table S1).
Functional Epigenetic Modules
To further investigate the relationship between gene expression and DNA methylation, we used the FEM algorithm (Jiao et al. 2014) to integrate these two omic data. Our analysis identified two significant functional epigenetic modules SKA1 (p-value = 0.003) and WSB1 (p-value = 0.043), comprising 79 and 60 genes, respectively (Fig. 2 and Supplementary Table S2).

Functional epigenetic modules. Module SKA1 (A) and WSB1 (B), The node color illustrates the DNA methylation difference (blue means high methylation, and yellow indicates low methylation), while the edge color shows differentially expressed genes (red represents genes with elevated expression levels in COVID-19 and green represents genes with low expression). The size of the nodes is correlated with the degrees of the nodes in the network
After performing enrichment analysis for the genes in these two modules, we were surprised to find that the genes in the SKA1 module were enriched in the biological process of virus DNA replicating, including sister chromatid cohesion, cell division, mitotic nuclear division, chromosome segregation, cell cycle, and viral transcription (Fig. 3A). These results suggest that the SKA1 module may play a potential role in the replication and transcription of SARS-CoV-2.

Enrichment results for the genes in module SKA1 (A) and WSB1 (B)
In contrast, the genes in the WSB1 module were found to be enriched in protein polyubiquitination and protein ubiquitination-related biological processes and KEGG pathways (Fig. 3B). This indicates that the WSB1 module may be involved in protein regulation and signaling pathways related to ubiquitination.
Development of a Diagnostic Model for COVID-19 and Validation of Potential Markers
After identifying two modules that may be associated with the replication and transcription of SARS-CoV-2 and protein ubiquitination, we built a diagnostic model to test whether the genes in those two modules (SKA1 and WBS1) can distinguish COVID-19 samples from healthy controls. We randomly split the whole data into training and test sets, then implemented a logistic regression classifier to train the model in the training set and validate it in the test set. As expected, the AUC of this model was 1 and 0.79 for the gene expression and DNA methylation-based models, respectively (Fig. 4A and B). The heatmap (Fig. 4C and D) clearly showed a pattern for the expression and DNA methylation of SKA1 module genes between COVID-19 and healthy control samples. Similarly, the classifier for the WSB1 module had a good performance of AUC 0.83 and 0.99 (Supplementary Figure S1). The learning curves show that the training and validation accuracy become closer as the training size (number of samples) increases (Supplementary Figure S2), indicating that our model was not overfitting.

Diagnostic module for COVID-19 based on the genes in SKA1 module. The ROC curve for the diagnostic models based on gene expression (A) and DNA methylation (B) data of the genes in SKA1 module. Heatmap of the gene expression (C) and DNA methylation (D) of genes in SKA1 module between COVID-19 and healthy control samples. Gene expression and DNA methylation values were scaled from 0 to 1 by rows (genes). Scatter plot of the differentially methylated and differentially expressed genes in SKA1 (E) and WSB1 modules (F). The genes with labels (CENPM, KNL1, RBCK1, CCNF, and UNKL) are significantly epigenetically activated genes (hypomethylated and upregulated genes)
We noticed five epigenetically activated genes in the SKA1 and WSB1 modules, including CENPM, KNL1, RBCK1, CCNF, and UNKL, which were significantly hypomethylated and overexpressed in COVID-19 samples. Since the SKA1 module was enriched in the replication and transcription of SARS-CoV-2, we hypothesized that these CENPM and KNL1 might also be associated with the replication and transcription of viruses. To investigate this hypothesis, we examined the differential expression status of CENPM and KNL1 in six major oncoviruses across TCGA cancer types using OncoDB (see “Methods” section).
The OncoDB analysis showed that among all TCGA cancer types, the differential expression of CENPM was observed in HPV-positive CESC, OPSCC, and HBV-positive LIHC tumor samples. Specifically, compared with virus-negative samples, both CENPM and KNL1 are significantly overexpressed in virus-positive samples in the corresponding virus type (Fig. 5, p-value < 0.01). In addition, a study by Xiao et al. (Xiao et al. 2019) also reported overexpression of CENPM in hepatitis B virus (HBV)-related liver tissues compared with normal tissues, which is consistent with our findings.

Associations between HPV, HBV in CESC, OPSCC, LIHC, and genes CENPM and KNL1. In each TCGA cancer type, the expression of CENPM and KNL1 in virus-positive and virus-negative groups are shown as boxplots (upper panel). The lower panel shows the Kaplan–Meier survival curve of the overall survival based on the gene expression (CENPM on CESC: HPV, LIHC: HBV, and KNL1 on OPSCC: HPV) or DNA methylation (CENPM on OPSCC: HPC and KNL1 on CESC: HPV) of CENPM or KNL1
Finally, we examined the association between the gene expression or DNA methylation of CENPM and KNL1 and the overall survival probability of tumor samples stratified by virus-positive in the corresponding TCGA cancer types. As anticipated, we found that CENPM and KNL1 are significantly associated with the survival of HPV-positive CESC and OPSCC, as well as HBV-positive LIHC tumor samples (p-value ≤ 0.05). Our results suggest that genes CENPM and KNL1 are involved in the replication and transcription of SARS-CoV-2, as well as in the process of HPV and HBV in tumors.
Discussion
DNA methylation is a critical biomarker in many diseases, including cancer (Ding et al. 2019). Various studies have investigated the DNA methylation or gene expression profiles in COVID-19. However, to date, no research has focused on the combined analysis of gene expression and DNA methylation simultaneously and identifying functional epigenetic modules that are differentially methylated and expressed.
Here we identified two significantly functional epigenetic modules by integrating Methylation EPIC DNA methylation array and RNA-Seq datasets using FEM. SKA1 module was found to be closely associated with the cell cycle, DNA replication, and transcription of SARS-CoV-2, while module WSB1 is related to protein ubiquitination. Ubiquitin modifications can regulate the innate immune response by affecting the related regulatory proteins, altering their stability via the ubiquitin–proteasome pathway, or directly regulating their activity. It has been reported that viruses, including coronaviruses, often use modulation of ubiquitin and ubiquitin-like modifiers to evade the host cell's immune response (Lin and Zhong 2015; Tang et al. 2018). Recent research indicated that deubiquitinating enzymes play an essential role in coronavirus pathogenesis, involving the production of non-structural proteins required for the replication process of coronavirus (Clemente et al. 2020). ORF9b interrupts its K63-linked polyubiquitination upon viral stimulation, thereby inhibiting the canonical IκB kinase alpha (IKKα)/β/γ-NF-κB signaling and subsequent interferon production, which contributes mainly to the viral pathogenesis and development of COVID-19 (Wu et al. 2021). These studies indicated that protein ubiquitination is associated with the coronavirus's replication process and the pathogenesis and development of COVID-19, which means both SKA1 and WSB1 modules may play essential roles in the replication process of coronavirus.
Then, we built a logistic regression model only using the expression or DNA methylation values of genes from SKA1 or WSB1 modules. our results showed that the genes in these two modules could be used to distinguish COVID-19 samples from controls. The AUC is 1 and 0.79 for gene expression and DNA methylation of SKA1 module, respectively.
Finally, we screened out two potential marker genes, CENPM and KNL1, from SKA1 module. These two genes are epigenetically activated in COVID-19 samples. Surprisingly, these two genes are significantly overexpressed in HPV-positive CESC and OPSCC tumor samples, as well as HBV-positive LIHC tumor samples. In addition, the expression and DNA methylation profile of CENPM and KNL1 are also significantly associated with the overall survival of HPV- or HBV- positive CESC, OPSCC, or LIHC tumor samples.
To conclude, we identified two functional epigenetic modules, SKA1 and WSB1, and potential biomarkers, CENPM and KNL1, that are associated with the replication process of coronavirus and may be used as potential therapeutic targets for COVID-19 after further verification.
Data Availability
The datasets used in this study can be downloaded from GEO with accession IDs GSE152641 and GSE174818.
References
Balaky STJ, Zaki Abdullah SM, Alexander M et al (2020) A comprehensive review of histopathological findings of infections induced by COVID-19. Cell Mol Biol (noisy-Le-Grand) 66:143–151. https://doi.org/10.14715/cmb/2020.66.7.22
Balnis J, Madrid A, Hogan KJ et al (2021) Blood DNA methylation and COVID-19 outcomes. Clin Epigenetics 13:118. https://doi.org/10.1186/s13148-021-01102-9
Cancer Genome Atlas Research Network, Albert Einstein College of Medicine, Analytical Biological Services et al (2017) Integrated genomic and molecular characterization of cervical cancer. Nature 543:378–384. https://doi.org/10.1038/nature21386
Castro de Moura M, Davalos V, Planas-Serra L et al (2021) Epigenome-wide association study of COVID-19 severity with respiratory failure. EBioMedicine 66:103339. https://doi.org/10.1016/j.ebiom.2021.103339
Clemente V, D’Arcy P, Bazzaro M (2020) Deubiquitinating enzymes in coronaviruses and possible therapeutic opportunities for COVID-19. Int J Mol Sci. https://doi.org/10.3390/ijms21103492
Ding W, Chen G, Shi T (2019) Integrative analysis identifies potential DNA methylation biomarkers for pan-cancer diagnosis and prognosis. Epigenetics 14:67–80. https://doi.org/10.1080/15592294.2019.1568178
Ding W, Chen J, Feng G et al (2020a) DNMIVD: DNA methylation interactive visualization database. Nucleic Acids Res 48:D856–D862. https://doi.org/10.1093/nar/gkz830
Ding W, Feng G, Hu Y et al (2020b) Co-occurrence and mutual exclusivity analysis of DNA methylation reveals distinct subtypes in multiple cancers. Front Cell Dev Biol 8:20. https://doi.org/10.3389/fcell.2020.00020
Huang DW, Sherman BT, Lempicki RA (2009a) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37:1–13. https://doi.org/10.1093/nar/gkn923
Huang DW, Sherman BT, Lempicki RA (2009b) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44–57. https://doi.org/10.1038/nprot.2008.211
Huttlin EL, Ting L, Bruckner RJ et al (2015) The BioPlex network: a systematic exploration of the human interactome. Cell 162:425–440. https://doi.org/10.1016/j.cell.2015.06.043
Jiao Y, Widschwendter M, Teschendorff AE (2014) A systems-level integrative framework for genome-wide DNA methylation and gene expression data identifies differential gene expression modules under epigenetic control. Bioinformatics 30:2360–2366. https://doi.org/10.1093/bioinformatics/btu316
Li T, Wernersson R, Hansen RB et al (2017) A scored human protein-protein interaction network to catalyze genomic interpretation. Nat Methods 14:61–64. https://doi.org/10.1038/nmeth.4083
Lin D, Zhong B (2015) Regulation of cellular innate antiviral signaling by ubiquitin modification. Acta Biochim Biophys Sin (shanghai) 47:149–155. https://doi.org/10.1093/abbs/gmu133
Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. https://doi.org/10.1093/bioinformatics/btp616
Roshandel MR, Nateqi M, Lak R et al (2020) Diagnostic and methodological evaluation of studies on the urinary shedding of SARS-CoV-2, compared to stool and serum: a systematic review and meta-analysis. Cell Mol Biol (noisy-Le-Grand) 66:148–156
Tang Q, Wu P, Chen H, Li G (2018) Pleiotropic roles of the ubiquitin-proteasome system during viral propagation. Life Sci 207:350–354. https://doi.org/10.1016/j.lfs.2018.06.014
Tang G, Cho M, Wang X (2022) OncoDB: an interactive online database for analysis of gene expression and viral infection in cancer. Nucleic Acids Res 50:D1334–D1339. https://doi.org/10.1093/nar/gkab970
Teschendorff AE, Gao Y, Jones A et al (2016) DNA methylation outliers in normal breast tissue identify field defects that are enriched in cancer. Nat Commun 7:10478. https://doi.org/10.1038/ncomms10478
Thair SA, He YD, Hasin-Brumshtein Y et al (2021) Transcriptomic similarities and differences in host response between SARS-CoV-2 and other viral infections. iScience 24:101947. https://doi.org/10.1016/j.isci.2020.101947
Wang X, Li Y, Hu H et al (2020) Comprehensive analysis of gene expression and DNA methylation data identifies potential biomarkers and functional epigenetic modules for lung adenocarcinoma. Genet Mol Biol 43:e20190164. https://doi.org/10.1590/1678-4685-GMB-2019-0164
Wu J, Shi Y, Pan X et al (2021) SARS-CoV-2 ORF9b inhibits RIG-I-MAVS antiviral signaling by interrupting K63-linked ubiquitination of NEMO. Cell Rep 34:108761. https://doi.org/10.1016/j.celrep.2021.108761
Xiao Y, Najeeb RM, Ma D et al (2019) Upregulation of CENPM promotes hepatocarcinogenesis through mutiple mechanisms. J Exp Clin Cancer Res 38:458. https://doi.org/10.1186/s13046-019-1444-0
Zhu Y, Cao X, Lu Y et al (2020) Lymphocyte cell population as a potential hematological index for early diagnosis of COVID-19. Cell Mol Biol (noisy-Le-Grand) 66:202–206
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
LYH conceived the study, LL, LLH, XLQ and RM analyzed the data, LL, LYH and GYL wrote the manuscript, and all authors read and approved the final version.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Ethical Approval
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, L., Hu, L., Qiao, X. et al. Integrative Analysis of DNA Methylation and Gene Expression Data Identifies Potential Biomarkers and Functional Epigenetic Modules for SARS-CoV-2. Biochem Genet 61, 2318–2329 (2023). https://doi.org/10.1007/s10528-023-10373-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10528-023-10373-1