
Abstract
The accumulation of pro-inflammatory senescent cells within tissues is a common hallmark of the aging process and many age-related diseases. This modification has been called the senescence-associated secretory phenotype (SASP) and observed in cultured cells and in cells isolated from aged tissues. Currently, there is a debate whether the accumulation of senescent cells within tissues should be attributed to increased generation of senescent cells or to a defect in their elimination from aging tissues. Emerging studies have revealed that senescent cells display an increased expression of several inhibitory immune checkpoint ligands, especially those of the programmed cell death protein-1 (PD-1) ligand-1 (PD-L1) proteins. It is known that the PD-L1 ligands, especially those of cancer cells, target the PD-1 receptor of cytotoxic CD8+ T and natural killer (NK) cells disturbing their functions, e.g., evoking a decline in their cytotoxic activity and promoting their exhaustion and even apoptosis. An increase in the level of the PD-L1 protein in senescent cells was able to suppress their immune surveillance and inhibit their elimination by cytotoxic CD8+ T and NK cells. Senescent cells are known to express ligands for several inhibitory immune checkpoint receptors, i.e., PD-1, LILRB4, NKG2A, TIM-3, and SIRPα receptors. Here, I will briefly describe those pathways and examine whether these inhibitory checkpoints could be involved in the immune evasion of senescent cells with aging and age-related diseases. It seems plausible that an enhanced inhibitory checkpoint signaling can prevent the elimination of senescent cells from tissues and thus promote the aging process.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
A chronic low-grade inflammation and immunosenescence are common immune-related hallmarks of the aging process; this remodelling phenomenon has been called inflammaging (Franceschi et al. 2018). While many age-related and chronic inflammatory diseases display similar properties, it does seem that these immune alterations are clearly augmented in these diseases. Nonetheless, the cellular state of senescence is a common feature encountered in both the aging process and many age-related diseases (Yanai and Fraifeld 2018; Mylonas and O'Loghlen 2022). Senescent cells are pro-inflammatory cells which secrete a number of inflammatory mediators, such as cytokines, chemokines, complements, and colony-stimulating factors (Rodier et al. 2009; Freund et al. 2010; Hernandez-Segura et al. 2018; Sikora et al. 2021). The pro-inflammatory phenotype has been called the senescence-associated secretory phenotype (SASP) (Campisi and Robert 2014). Currently, it seems that there are multiple causes to explain why cells will switch on the senescence phenotype which displays diverse pro-inflammatory properties in a context-dependent manner (Sikora et al. 2021; Wang et al. 2024a). For instance, the inducer of the senescent state and the cell type involved have a crucial role in the heterogeneity of the SASP (Wang et al. 2024a). Recently, several single-cell transcriptome studies have confirmed that many cell types isolated from aged tissues display a pro-inflammatory phenotype thus verifying results observed in cultured cells (Salzer et al. 2018; Benayoun et al. 2019; Zou et al. 2021; Xu et al. 2022). Moreover, the senescence in aged tissues has revealed the presence of a robust heterogeneity within cell populations. The single-cell transcriptomic studies also confirmed that an increased expression of senescence markers in isolated cells was associated with an enhanced expression of SASP factors. These studies verified that senescent cells are an important source of the elevated expression of pro-inflammatory factors in aged tissues.
The chronic inflammatory microenvironment, either associated with aging or as encountered in many inflammatory diseases, stimulates a counteracting immunosuppression in an attempt to maintain tissue homeostasis and thus avoid tissue destruction (Kanterman et al. 2012; Hotchkiss et al. 2013; Wang and DuBois 2015; Salminen 2020). There is abundant evidence that with aging the number of immunosuppressive cells was significantly increased in tissues. For instance, several investigators have revealed that the populations of regulatory T cells (Treg), myeloid-derived suppressor cells (MDSC), and M2 macrophages were augmented in aged tissues (Grizzle et al. 2007; Enioutina et al. 2011; Jackaman et al. 2013; Garg et al. 2014; Ruhland et al. 2016). The counteracting immune responses also involve the activation of inhibitory immune checkpoint pathways in many pathological states. The host tissues can inhibit the cytotoxic effects of CD8+ T and natural killer (NK) cells by increasing the expression of inhibitory checkpoint ligands which suppress the activity of these cytotoxic cells (Buckle and Guillerey 2021; Baldanzi 2022). Currently it is known that there are several inhibitory checkpoint receptor/ligand systems possessing different properties and distributions in many immune cells. The blockade therapies for certain inhibitory checkpoints are clinically used in cancer immunotherapy (Guo et al. 2023). Emerging studies have revealed that senescent cells can evade immune surveillance by activating the expression of the ligands for the programmed cell death protein 1 (PD-1) receptor, i.e., the PD-L1 protein on their cell surface (Onorati et al. 2022; Wang et al. 2022a; Salminen 2024). However, there are several other inhibitory checkpoint receptor/ligand systems which are associated with the immune surveillance of senescent cells. Here, I will briefly introduce those pathways which are known to control the immunosurveillance of senescent cells and examine whether they could be involved in the immune evasion of senescent cells associated with aging.
Accumulation of senescent cells increases with aging and in age-related diseases
Since the characterization of the major properties of senescent cells in cultured cells, there has been mounting interest to determine whether the same molecular markers could be upregulated in aging tissues. There is growing evidence indicating that the aging process is associated with a progressive increase in the expression of common markers of cellular senescence not only in the tissues of mammalian species (Krishnamurthy et al. 2004; Wang et al. 2009; Biran et al. 2017; Yanai and Fraifeld 2018; Idda et al. 2020; Yousefzadeh et al. 2020) but also in zebrafish, Caenorhabditis elegans, and Drosophila (Kishi 2004; Dmitrieva and Burg 2007; Ito and Igaki 2016). Moreover, single-cell transcriptome techniques have been used to confirm the presence of age-related senescence phenotypes in the cells of many murine tissues (Tabula Muris Consortium 2020; Zou et al. 2021). Several new techniques have recently become available to help to reveal in vivo the senescence process. For axample, there are now many reporter models that can monitor the gradual increase of senescence in aging mice (Gorgoulis et al. 2019; Gonzalez-Gualda et al. 2021). It seems that an age-related increase in senescence is a tissue-specific process which is greatly augmented in the mouse models of accelerated aging (Yousefzadeh et al. 2020; Cai et al. 2022). Interestingly, several mouse models of an accelerated aging have demonstrated that the removal of senescent cells from tissues actually attenuate pathological alterations taking place in aging tissues (Baker et al. 2016; Yi et al. 2023). For instance, Baker et al. (2011) developed a transgenic mouse model with an inducible elimination of the p16INK4a-positive cells, a common senescence marker (INK-ATTAC model). They demonstrated that both in wild-type and in the accelerated aging background (the BubR1 progeroid mouse) the depletion of senescent cells attenuated and delayed the appearance of many age-related pathologal changes. Currently there are many drug discovery programs aiming to develop pharmacological medications to eliminate senescent cells, i.e., senolytic therapies (Chaib et al. 2022; Zhang et al. 2023).
Many age-related diseases are associated with an expansion of cellular senescence although it seems that cellular senescence does not play the same role in different diseases. For example, it is known that cellular senescence can possess both tumor-suppressive and tumor-promoting properties in a phase-dependent manner (Sun et al. 2018; Takasugi et al. 2022). Given that senescent cells have pro-inflammatory properties, there is mounting evidence that cellular senescence acts as a driver of several age-related diseases, such as many cardiac diseases (Mehdizadeh et al. 2022), skin pathologies (Chin et al. 2023), atherosclerosis (Wu et al. 2020), and hepatic steatosis (Ogrodnik et al. 2017). The accumulation of fibrotic lesions is a significant pathological characteristic in certain age-related diseases. For example, there is clear evidence that cellular senescence, especially the senescence of stromal fibroblasts, enhanced the pathogenesis of fibrotic diseases, e.g., the clearance of senescent fibroblasts reduced the severity of fibrotic pulmonary lesions in mice (Schafer et al. 2017). Cellular senescence also has a crucial role in chronic kidney disease (Xu et al. 2020), myocardial fibrosis (Osorio et al. 2023), and systemic sclerosis (Tsou et al. 2022). It seems that cellular senescence may have a double-edge role in fibrotic diseases, i.e., it can either prevent or promote fibrosis, a feature also observed in tumor biology. Krizhanovsky et al. (2008) demonstrated that in murine hepatic liver fibrosis the presence of senescent hepatic stellate fibroblasts limited the progression of fibrosis and even facilitated the reversal of this pathological trait.
Currently, it is not known why senescent cells accumulate within tissues during aging and age-related diseases. Eventually, it could be attributable to an increase in the generation of senescent cells or to their reduced removal via impaired immune surveillance. There are diverse experimental insults which can switch cells to adopt an irreversible state of senescence. Interestingly, it is known that senescent cells can induce bystander senescence in the neighboring cells within tissues (Nelson et al. 2012; da Silva et al. 2019). This paracrine senescence is most likely induced by the secretion of SASP factors, such as ROS and cytokines. However, senescent cells possess an increased capacity to secrete extracellular vesicles (EV) which not only target neighboring cells but via the systemic circulation they can expand throughout the whole organism (Nelson et al. 2018; Alibhai et al. 2020; Salminen et al. 2020). Krtolica et al. (2001) demonstrated that senescent fibroblasts promoted the growth of epithelial cells and accordinly stimulated tumorigenesis in mice. Interestingly, there are observations indicating that the EVs secreted from senescent cells are able to enhance immunosuppression in chronic inflammatory conditions (Salminen et al. 2020). It is known that EVs can carry a diverse cargo, e.g., many immunosuppressive miRNAs and other suppressive enhancers, such as ARG1, PGE2, and TGF-β proteins. In fact, the EVs released by tumor cells contain membrane-bound PD-L1 proteins which can act as decoys to confuse the anti-PD-L1 antibodies and thus induce a resistance to immunotherapy (Yin et al. 2021; Chen et al. 2022).
There has been an active debate on the role of the immune system in the elimination of senescent cells, i.e., whether immune cells enhance or prevent the removal of senescent cells. Currently, it is known that there are differences in the clearance process of senescent cells between the microenvironments, such as aging tissues, tumors, and fibrotic lesions (Wang et al. 2022a; Chen et al. 2023; Marin et al. 2023; Lee et al. 2024) (see below). There are several reports indicating that impairments in the function of cytotoxic immune cells, i.e., CD8+ T and NK cells, promote to the accumulation of senescent cells within aged tissues. For instance, Ovadya et al. (2018) demonstrated that deficiency in immunosurveillance activity, evoked either via genetic or pharmacological treatments, disturbed the clearance of senescent cells (SA-β-gal and p16-positive) from tissues in aged mice. The knockout of the perforin gene (Prf−/−), a cytolytic protein expressed in cytotoxic CD8+ T and NK cells, (i) increased cellular senescence in mouse liver, lung, and skin, (ii) enhanced chronic inflammation, and (iii) promoted age-related disorders in several mouse tissues. Moreover, the lack of the Prf gene in progeroid mice shortened their lifespan. They also confirmed that the cytotoxicity of CD8+ T cells was robustly declined in these transgenic mice. These observations indicate that functional deficiencies in surveying immune cells promote the accumulation of senescent cells within aged tissues.
The age-related immunosenescence has been associated with a robust decline in the cytotoxic activity of CD8+ T and NK cells that might impair the elimination of senescent cells (Salminen 2021; Brauning et al. 2022). There is clear evidence that the aging process is associated with activation of the immunosuppressive network intended to inhibit the functions of cytotoxic immune cells (Ruhland et al. 2016; Salminen 2020). Currently, it is known that the age-related immunosuppression can be associated with (i) an increase in the recruitment and differentiation of immunosuppressive cells, such as Tregs, MDSCs, and M2 macrophages, as well as (ii) the activation of inhibitory immune checkpoint signaling, e.g., in the accumulating senescent cells (see below). It is recognized that the immunosuppressive cells secrete anti-inflammatory cytokines, such as TGF-β and IL-10, which inhibit the surveillance activity of CD8+ T and NK cells (Li et al. 2006; Ouyang et al. 2011). For instance, Trzonkowski et al. (2006) demonstrated that Tregs inhibited the cytotoxic activity of human CD8+ T and NK cells. Accordingly, Filippi et al. (2008) revealed that TGF-β exposure suppressed the activation of mouse naïve CD8+ T cells but not that of antigen experienced T cells. Moreover, TGF-β exposure inhibits the cytotoxic activity of NK cells in many experimental models (Viel et al. 2016; Slattery et al. 2021). Nunez et al. (2018) reported that human M2 macrophages reduced the cytotoxic activity of NK cells through the secretion of TGF-β. In their seminal study, Ruhland et al. (2016) demonstrated that senescent stromal cells in mouse skin enhanced inflammation and recruited immunosuppressive Tregs and myeloid-derived suppressor cells (MDSC) into mouse skin. They revealed that senescent stroma in mouse skin robustly decreased immune surveillance of cancer cells and consequently the immunosuppressive environment increased tumorigenesis. Recently, it has been demonstrated that not only immunosuppressive cells but also inhibitory immune checkpoints, such as the PD-1/PD-L1 signaling pathway, can suppress the immune surveillance of senescent cells and thus promote their accumulation with aging (Wang et al. 2022a).
Inhibitory immune checkpoint signaling suppresses immunosurveillance
The immune system drives immune surveillance in the host intended to recognize foreign pathogens as well as aberrant cells, such as malignant and senescent cells. Cytotoxic CD8+ T cells, NK cells and macrophages are the major surveying immune cells which can detect and eliminate not only invading microbes but also abnormal host cells. Antigen presentation via the major histocompatibility complexes (MHC) and the activation of cytotoxic cells are highly regulated processes which are controlled by multilevel regulatory systems (Paul and Lal 2017; Shah et al. 2021). The immune checkpoint receptors fine-tune this process by either stimulating or inhibiting the function of cytotoxic cells. In fact, many checkpoint receptors function as co-receptors which cooperate to maintain the immune balance by regulating the activity of major signaling pathways of cytotoxic cells, such as T cell receptor (TCR) signaling (Baldanzi 2022). For instance, the inhibitory immune checkpoint receptors have an important role in the suppression of autoimmunity disorders, whereas an excessive activity of inhibitory checkpoints prevents the clearance of tumor cells (Toor et al. 2020; Burke et al. 2023).
Inhibitory immune checkpoint receptors
The receptors of the inhibitory immune checkpoints are ligand-activated receptors which are expressed mainly in cytotoxic T and NK cells and macrophages (Ravetch and Lanier 2000; Buckle and Guillerey 2021; Baldanzi 2022; Guo et al. 2023). There are a number of different receptors which possess inhibitory immune activity but I will focus only on those five receptors which are known to act as inhibitory immune checkpoint receptors for the ligand proteins expressed in senescent cells, i.e., programmed cell death protein-1 (PD-1), leukocyte immunoglobulin-like receptor B4 (LILRB4), natural killer cell receptor G2A (NKG2A), T cell immunoglobulin and mucin domain-3 (TIM-3), and signal regulatory protein α (SIRPα). The inhibitory checkpoint receptors are membrane-bound glycoproteins which contain an extracellular Ig-like V domain and a cytoplasmic tail which encompasses the immunoreceptor tyrosine-based inhibitory motifs (ITIM) and tyrosine-based switch motifs (ITSM) (Xu et al. 2021; Baldanzi 2022; Xiang et al. 2024) (Fig. 1). The binding of the ligand to the inhibitory checkpoint receptor induces the phosphorylation of the ITIM or ITSM sites, thus providing the docking sites for the Src-homology-2 (SH2) protein phosphatases, e.g., SHP-1, SHP-2, and SHIP (Xu et al. 2021; Baldanzi 2022; Xiang et al. 2024). Subsequently, these activated phosphatases can dephosphorylate the components of the TCR and costimulatory CD28 pathways, thus suppressing signaling through the TCR axis (Mizuno et al. 2019; Baldanzi 2022). The inhibition of TCR signaling suppresses the activation, proliferation, and cytokine production of CD8+ T cells, thus attenuating immune surveillance and cytotoxic activity. Intriguingly, similar defects in the properties of CD8+ T and NK cells are typical changes encountered in the immunosenescence state which is not only associated with the aging process but also found in tumors and age-related diseases (Barbe-Tuana et al. 2020; Lian et al. 2020; Salminen 2021). Currently, it seems probable that an increase in the expression of ligands for inhibitory checkpoint receptors (discussed below) could reduce the functional efficiency of the immune system and promote immunosenescence. Next, I will briefly describe the properties of certain inhibitory checkpoint pathways and examine whether an increase in the expression of their ligands in senescent cells could impair immunosurveillance and consequently promote the accumulation of senescent cells within aged tissues.

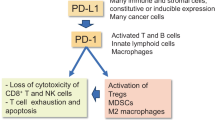
A schematic presentation of the inhibitory immune checkpoint receptors in cytotoxic immune cells and their ligands in senescent cells. CEACAM 1 carcinoembryonic antigen-related cell adhesion molecule 1, Gal galectin, HLA-E human leukocyte antigen-E, HMGB1 high mobility group box 1, ITIM immunoreceptor tyrosine-based inhibitory motif, ITSM immunoreceptor tyrosine-based switch motif, LILRB4 leukocyte immunoglobulin-like receptor B4, NKG2A natural killer cell receptor G2A, PD-1 programmed cell death protein-1, PD-L1 programmed death-ligand 1, SIRPα signal regulatory protein α, TIM-3 T cell immunoglobulin and mucin domain-3
PD-1
The PD-1 receptor (CD279), a member of the immunoglobulin superfamily, is expressed on activated T and B cells, NK cells, and some myeloid cells (Ghosh et al. 2021; Beenen et al. 2022). The two ligands for the PD-1 receptor, PD-L1 (CD274 or B7-H1) and PD-L2 (CD273 or B7-DC), are members of the B7 family of immune regulatory ligand proteins (Collins et al. 2005). The PD-L1 proteins are expressed in several antigen presenting and immunosuppressive cells as well as in many stromal cells, e.g., fibroblasts, pericytes, epithelial, endothelial, and smooth muscle cells (Ghosh et al. 2021, Human Protein Atlas). Moreover, the PD-L1 protein is expressed in senescent cells (Onorati et al. 2022; Wang et al. 2022a) and especially in diverse cancer cells and cancer-associated fibroblasts (CAF) (Jiang et al. 2019; Pei et al. 2023). In tumors, the PD-1/PD-L1 checkpoint signaling has a crucial role in the immune evasion of tumor cells and in the generation of an immunosuppressive microenvironment. The binding of the PD-L1 protein to the PD-1 receptor of CD8+ T cells activates an inhibitory signaling cascade which leads to a loss of cytotoxicity and even promotes the exhaustion of CD8+ T cells. The PD-1/PD-L1 checkpoint axis possesses both beneficial and harmful properties, e.g., it confers immunosuppressive protection in autoimmune diseases, whereas in cancers and fibrotic diseases it provides an immune escape mechanism for abnormal cells to avoid immunosurveillance (Francisco et al. 2010; Toor et al. 2020; Zhao et al. 2023). Currently, the inhibition of the PD-1/PD-L1 signaling with PD-1 and PD-L1 antibodies is a widely used anti-cancer therapy (Tang et al. 2022). However, therapy with PD-1/PD-L1 blockers can induce an immune-based resistance which is due to a loss of PD-L1 expression and defects in antigen presentation and interferon signaling (Vesely et al. 2022). It is known that many combination therapies can improve the treatment response to PD-1/PD-L1 checkpoint inhibitors.
LILRB4
The LILRB4 receptor (ILT3, gp49B) belongs to the leukocyte immunoglobulin-like receptor subfamily B and it contains three inhibitory ITIM domains (Redondo-Garcia et al. 2023; Xiang et al. 2024). The LILRB4 receptor is expressed in T cells, neutrophils, monocytes, macrophages, dendritic cells, microglia, and endothelial cells. The five members of the LILRB family have different specificities for their activating ligands, i.e., the LILRB4 receptor is activated by fibronectin, apolipoprotein E (ApoE), and galectin-8 (Gal-8) (Redondo-Garcia et al. 2023). The activation of LILRB4 signaling enhances the development of an immunosuppressive microenvironment in mouse and human tumors and it facilitates the immune escape of tumor cells (Sharma et al. 2021). Wang et al. (2024b) demonstrated that an exposure to Gal-8 induced the differentiation of mouse monocytes into monocytic MDSCs and subsequently enhanced their expansion through the activation of STAT3 and inhibition of NF-κB signaling. They also revealed that the activation of LILRB4 promoted tumor growth in mice, whereas antibodies blocking the interaction between Gal-8 and LILRB4 receptor suppressed tumor growth in mice. Accordingly, Deng et al. (2018) reported that the signaling via the ApoE/LILRB4/SHP-2/Arginase-1 pathway in mouse myeloid leukemia cells suppressed T cell activity and created an immunosuppressive microenvironment conducive to tumor invasion. Given that the activation of LILRB4 induces immune tolerance, it seems that LILRB4 signaling may well exert beneficial effects in autoimmune diseases, transplantation biology, and maternal–fetal interactions (Xiang et al. 2024).
Fibronectin, a protein abundantly present in the extracellular matrix (ECM), is also a physiological ligand for the immunosuppressive LILRB4 receptor (Itoi et al. 2022; Itagaki et al. 2023; Xiang et al. 2024). In their seminal study, Itoi et al. (2022) demonstrated that fibronectin was tethered with integrins and the LILRB4 receptor on the cell surface of human and mouse macrophages. The terminal 30 kD domain of fibronectin (FN30) interacted with the LILRB4 receptor, whereas the RGD motif could bind the β-integrin protein. Subsequently, they revealed that the cytotoxicity of mouse NK cells was attenuated when the LILRB4 receptor was activated (Itagaki et al. 2023). These experiments indicated that FN30/LILRB4 signaling inhibited the integrin-assisted natural cytotoxicity of NK cells. In addition to some other inhibitory immune checkpoints in NK cells, such as NKG2A (see below), it seems that FN30/LILRB4 signaling can mitigate the cytotoxicity of NK cells and thus inhibit the elimination of cancer cells. Moreover, Paavola et al. (2021) demonstrated that the fibronectin/LILRB4 interaction inhibited the antigen-presentation by human dendritic cells as well as suppressing cytokine production by the Fcγ receptor (FcR)-activated human THP-1 macrophages. These inhibitory effects could be reversed by treatment with an antibody to the LILRB4 receptor. Paavola et al. (2021) also reported that the blockade of LILRB4 signaling robustly increased the secretion of chemokines, such as CCL3, CCL5, and CXCL10, from tumor cells. There is mounting evidence that fibronectin-activated LILRB4 signaling has profound effects on immune responses therefore LILRB4 signaling is a promising target in drug discovery.
NKG2A
The NKG2A receptor is a member of the C-type lectin-like receptor superfamily which includes seven NKG2 receptors, both inhibitory and activating checkpoint receptors (Guo et al. 2023). For instance, the NKG2A receptor is an inhibitory checkpoint in immunosurveillance, whereas the NKG2D receptor is a potent activator of the immune clearance of cancer and senescent cells (Iannello and Raulet 2013; Sagiv et al. 2016). The NKG2A receptor is abundantly expressed on the surface of human NK and CD8+ T cells. Currently, the HLA class I histocompatibility antigen, α chain E (HLA-E) is the sole ligand for the NKG2A receptor (Lee et al. 1998). The expression of the HLA-E protein is enriched on the surfaces of unhealthy cells, such as infected cells as well as tumor and senescent cells. Ljubic et al. (2023) examined the molecular mechanisms in the binding process between the HLA-E ligand and the NKG2A receptor and they revealed that the inhibitory signaling pathways of the NKG2A receptor were mediated via its two ITIM domains. There is clear evidence that the NKG2A/HLA-E complex inhibits the immune surveillance and impairs the clearance of tumor cells by the cytotoxic NK and CD8+ T cells (Andre et al. 2018; Kamiya et al. 2019; Fisher et al. 2022). The blockade of the interaction between the HLA-E and the NKG2A receptor is an important drug discovery target since it can unleash the anti-tumor immunity of cytotoxic NK and CD8+ T cells (Andre et al. 2018; Kamiya et al. 2019). It has been claimed that the immunosuppressive NKG2A/HLA-E axis has an important role in many immune-mediated diseases, such as in tumors, infections, and autoimmune diseases (Wang et al. 2022b).
TIM-3
The TIM-3 receptor, also called hepatitis A virus cellular receptor 2 (HAVCR2), is expressed in T, NK, NKT, and many myeloid cells (Wolf et al. 2020; Buckle and Guillerey 2021). There are several ligands for the TIM-3 receptor, i.e., galectin-9 (Gal-9), phosphatidylserine (PtdSer), high mobility group protein B1 (HMGB1), and carcinoembryonic antigen cell adhesion molecule-1 (CECAM-1) (Wolf et al. 2020; Buckle and Guillerey 2021). The Gal-9 protein is also a ligand for another inhibitory immune checkpoint receptor, i.e., the V-domain immunoglobulin suppressor of T cell activation (VISTA) (Shekari et al. 2023). While the cytoplasmic tail of the TIM-3 receptor lacks the ITIM or ITSM domains, it does contain five conserved tyrosine residues which mediate the downstream inhibitory signaling. The function of TIM-3 is associated with (i) dysfunction and exhaustion of CD8+ T cells by disturbing TCR signaling, (ii) inhibition of the activity of Th1 and Tc1 cells, (iii) induction of antigen-specific tolerance, (iv) suppression of some properties of NK cells, (v) increase in transplantation tolerance, and (vi) inhibition of many activities of the innate immune system (Anderson et al. 2016; Wolf et al. 2020; Buckle and Guillerey 2021). In autoimmune diseases, IFN-β therapy increased the expression of TIM-3 and thus enhanced protective immunity, whereas in chronic viral infections and cancers, TIM-3 expression repressed immunosurveillance and inhibited the immune clearance (Anderson et al. 2016; Wolf et al. 2020). Currently, the TIM-3 receptor is a promising target for immunotherapy, especially in many tumors (Sauer et al. 2023).
The Gal-9 ligand of the TIM-3 receptor is a secreted, β-galactoside-binding protein which also has important intracellular functions linked with its lectin property, e.g., Gal-9 can enhance cytosolic ubiquitination and thus activate AMP kinase (Jia et al. 2020; Wolf et al. 2020). As a ligand for the TIM-3 receptor, Gal-9 was shown to promote immunosuppression, e.g., in human cancers, by triggering an exhaustion of CD8+ T cells and impairing the cytotoxicity of NK cells (Kandel et al. 2021; Yang et al. 2021). Moreover, the Gal-9 protein can enhance the differentiation of immunosuppressive Tregs, whereas it reduces the level of Th17 cells and thus attenuates the severity of mouse collagen-induced arthritis (Seki et al. 2008). The HMGB1 ligand of the TIM-3 receptor is a multifunctional alarmin localized in chromatin structures which is secreted in many pathological states (Tang et al. 2023). When it gains access to the extracellular space, HMGB1 can interact with many immunoreceptors, both immunosuppressive and immunostimulatory. For instance, HMGB1-mediated TIM-3 signaling was able to inhibit the innate immunity responses to nucleic acids induced by Toll-like receptors 3, 7, and 9 in mouse tumors (Chiba et al. 2012). Moreover, it is known that the HMGB1 protein is an important mediator of the senescence phenotype (Davalos et al. 2013). DeKruyff et al. (2010) demonstrated in mice that PtdSer, a membrane phospholipid and a marker of apoptotic cells, is a ligand for the TIM-3 receptor. There are studies indicating that the TIM-3 receptor mediates the phagocytosis of apoptotic cells and consequently induces the cross-presentation of antigens from dying cells (Nakayama et al. 2009; DeKruyff et al. 2010). The membrane-bound CEACAM1 protein is the fourth ligand for the TIM-3 receptor expressed in many immune and non-immune cells (Wolf et al. 2020). CEACAM1 is a multifunctional glycoprotein which has diverse other functions independent of TIM-3 signaling, e.g., it can enhance cell–cell adhesion and control insulin clearance (Kuespert et al. 2006). CEACAM1 also regulates several TIM-3-dependent immune functions. For example, Zhang et al. (2017) demonstrated that CEACAM1 and TIM-3 promoted the exhaustion of CD8+ T cells in colorectal cancer patients. Moreover, the expression of the CEACAM1 protein in activated mouse NK cells inhibited the cytolytic activity of the NKG2D receptor (Hosomi et al. 2013). It seems that it is difficult to develop immunotherapy based on CEACAM1 protein because it is involved in many crucial immune and metabolic functions.
SIRPα
The SIRPα protein is a membrane-bound inhibitory checkpoint receptor which is mainly expressed in myeloid cells, especially in macrophages, although it can also be present in stem cells and neurons (Barclay and Van den Berg 2014; Logtenberg et al. 2020). The SIRPα receptor interacts with the CD47 protein, also called the integrin associated protein (IAP), which is widely expressed in both immune and non-immune cells (Barclay and Van den Berg 2014; Isenberg and Montero 2024). The major function of the SIRPα/CD47 axis is to inhibit the phagocytosis of apoptotic and abnormal cells, i.e., the CD47 protein transmits a “don’t eat me” signal to macrophages via the activation of SIRPα. The cytoplasmic tail of SIRPα contains four conserved tyrosine residues which act as an ITIM domain to block the phagocytosis of apoptotic cells by macrophages (Logtenberg et al. 2020). Tumor cells as well as senescent cells utilize this mechanism to avoid their phagocytosis by macrophages (Jia et al. 2021; Schloesser et al. 2023). The CD47 protein is a potent marker of the “self” and for instance, the Cd47-induced Sirpα signaling in mouse NK cells prevented immunosurveillance and the NK cell-mediated destruction of xenogeneic tissues (Deuse et al. 2021). Moreover, it is known that cancer cells can pack the CD47 protein into extracellular vesicles and thus evade immune surveillance (Shimizu et al. 2021). The activation of SIRPα is reputed to be involved in the pathogenesis of many diseases, such as atherosclerosis, chronic viral infections, and tumor growth (Feng et al. 2023). In addition, the CD47 protein can also serve as a receptor for the matricellular thrombospondin 1 (TSP1) protein which can trigger the CD47-mediated downstream responses thus inducing disturbances in gene expression, mitochondrial function, and many metabolic activities (Roberts and Isenberg 2021). Currently, the downstream signaling mechanisms of the CD47 protein still need to be clarified.
Other inhibitory checkpoint receptors
There are many other inhibitory checkpoint receptors in the immune system (Baldanzi 2022; Guo et al. 2023) although currently there is not enough relevant evidence on whether their ligands are expressed in senescent cells. For instance, many human Siglec receptors contain tyrosine motifs and the ITIM domain and they display many of the properties associated with inhibitory immune checkpoint receptors, e.g., Siglec 5, 7, 10, 15 (Ravetch and Lanier 2000; Guo et al. 2023). It is known that the Siglec/sialic acid axis acts as a glyco-immune checkpoint which can inhibit the functions of many surveying immune cells (Saini et al. 2022) and it has a crucial role in many human diseases (Duan and Paulson 2020). For instance, it is known that cancer cells exploit sialoglycan-Siglec interactions to evade immunosurveillance (van de Wall et al. 2020). However, currently it is not known whether cellular senescence affects the surface sialylation of senescent cells in a way which could provide immune evasion for senescent cells. However, several investigators have reported that the level of many sialoglycans decreases rather than increases with cellular senescence (Tadokoro et al. 2006; Itakura et al. 2016).
Senescent cells evade immune clearance by increasing the expression of ligands for inhibitory immune checkpoint receptors
As described above, the ligands of checkpoint receptors have a crucial role in the immune prevention of host tissues. It seems that cancer cells and senescent cells are able to evade immune surveillance by increasing their expression of the ligand proteins for inhibitory checkpoint receptors. It is known that signaling of many inhibitory checkpoint receptors suppresses the activation of the TCR or B cell receptors (BCR) (Okazaki et al. 2001; Mizuno et al. 2019). There are studies indicating that senescent cells can also increase their antigen presentation by stimulating the expression of proteins of the MHC-I/II machinery (van Tuyn et al. 2017; Chen et al. 2023; Marin et al. 2023; Sinning et al. 2023). For instance, Marin et al. (2023) demonstrated that the cellular senescence induced by three different senescence inducers (genotoxic, CDK4/6 inhibitor, and p53 activator) robustly upregulated antigen presentation via the MHC-I machinery in human IMR-90 fibroblasts and mouse embryonic fibroblasts (MEF) as well as in two human melanoma cell lines. Moreover, tumor senescent cells induced the production of immunogenic peptides which were able to activate CD8+ T-dependent antitumor immunity. However, the immune surveillance performed by CD8+ T and NK cells is dependent on the balance between the function of the stimulatory and inhibitory co-receptors in these cytotoxic cells and it is this balance that determines whether a senescent cell will or will not be eliminated (Chen and Flies 2013; Zhang and Vignali 2016; Baldanzi 2022).
Interestingly, Chen et al. (2023) reported that the p53 activation-induced senescence of mouse hepatic tumors and tumor cells removed their immune resistance and increased their immune clearance. They also revealed that IFNγ signaling promoted the immune surveillance and elimination of senescent tumor cells. Bojko et al. (2019) demonstrated that different chemotherapeutic agents induced diverse senescence phenotypes in respect to different drugs and cancer cell types. It seems that the inducer of senescence state has a crucial role in tumor cell immunogenicity. For instance, Lee et al. (2024) demonstrated that the senescence of human breast cancer cells induced by CDK4/6 inhibitors promoted immunogenic anti-tumor properties, whereas treatment with DNA-damaging agents augmented the pro-tumorigenic microenvironment. This difference was attributable to an increase in the level of SASP factors produced by DNA-damaging treatment. This is an important observation since it is known that pro-inflammatory SASP factors are present in aged tissues and they activate the expression of many inhibitory immune checkpoints, e.g., that of PD-L1 in aged mice, and thus increase the ability of senescent cells to avoid surveillance by cytotoxic immune cells (Wang et al. 2022a). Next, I will examine the expression of the ligands of certain inhibitory immune checkpoint receptors in senescent cells and aged tissues and their effects on the immunosurveillance of senescent cells.
PD-1/PD-L1
There is clear evidence that the expression levels of PD-L1 are increased with aging in many mouse and human tissues, such as cerebellum, cardiac muscle, kidney, liver, lung, and spleen (Benayoun et al. 2019; Garcia et al. 2022; Onorati et al. 2022; Wang et al. 2022a). This is not surprising since it is known that pro-inflammatory SASP mediators are potent inducers of the expression of PD-L1 protein (Antonangeli et al. 2020; Betzler et al. 2020; Rong et al. 2021; Nawas et al. 2023; Salminen 2024). It has been reported that the NF-κB system, especially the non-canonical RelB signaling, as well as JAK-STAT signaling stimulates the expression of the PD-L1 protein (Antonangeli et al. 2020; Onorati et al. 2022; Zhang et al. 2022). Moreover, the NLRP3 inflammasomes, when primed by NF-κB signaling, were observed to be involved in the upregulation of PD-L1 expression and immunosuppression in mouse lymphoma (Lu et al. 2021). Given that the SASP-related inflammatory markers are augmented in many age-related diseases, there are reports indicating that the expression of PD-L1 is upregulated in coronary artery disease (CAD), obstructive lung disease (COPD), and Alzheimer’s disease (AD) (Saresella et al. 2012; Weyand et al. 2018; Polverino et al. 2022). Moreover, it is known that the expression of PD-L1 has an important role in the development of age-related fibrotic lesions in cardiac muscle, kidney, and lungs (Jiang et al. 2022; Onorati et al. 2022; Zhao et al. 2023).
The accumulation of senescent cells seems to be one source of the increased expression of the PD-L1 protein in aged tissues. Onorati et al. (2022) demonstrated that the exposure of cultured human IMR-90 fibroblasts to a wide variety of different inducers of cellular senescence, e.g., replicative exhaustion, etoposide, oncogenic HrasV12, and ionizing radiation, stimulated a robust increase in the expression of the PD-L1 protein in senescent fibroblasts. A cytochemical evaluation of the fibroblasts revealed that there was an intense staining of the PD-L1 protein at the cell-surface of senescent fibroblasts. While it is known that the PD-L1 protein can also accumulate into the nuclei (Hudson et al. 2020), these results clearly indicated that after senescence treatments, the PD-L1 protein was localized to the cell-surface and thus was able to target the PD-1 receptor of immune cells. Interestingly, Onorati et al. (2022) reported that the SASP factors released from cultured senescent fibroblasts were able to induce an upregulation of PD-L1 expression in neighboring cells. They also revealed that secreted factors induced the expression of PD-L1 via the JAK-STAT pathway. Using different imaging techniques, Wang et al. (2022a) demonstrated that with aging the PD-L1-positive cells accumulated within mouse liver and kidney. They also observed that the expression level of PD-L1 correlated with the degree of SASP markers in cells of aged tissues. Interestingly, Wang et al. (2022a) demonstrated that those cells with a high expression level of the PD-L1 protein in cultured mouse pulmonary fibroblasts were protected against the immune surveillance and clearance by cytotoxic CD8+ T cells. This observation indicates that an increase in the expression of the PD-L1 protein in senescent cells was able to reduce the elimination of senescent cells by cytotoxic immune cells.
LILRB4/Fibronectin/Gal-8
Fibronectin is an abundant glycoprotein which binds not only to ECM proteins, such as collagens and some proteoglycans, but also to the cell-membrane bound LILRB4 and integrin receptors (see above). There are many investigations revealing that the aging process affects the properties of fibronectin in aged tissues, i.e., (i) its expression level increases in several cell types in aged tissues (Boyer et al. 1991; Kumazaki et al. 1993; Wicher et al. 2021), (ii) the expression of fibronectin is activated by NF-κB-mediated inflammatory signaling (Qwarnström et al. 1994; Lee et al. 2002), and (iii) the age-related structural alterations occurring in the fibronectin matrix increase the rigidity of the aged ECM (Antia et al. 2008). Interestingly, already two decades before the characterization of the SASP, an increased expression and secretion of fibronectin was identified as a marker of cellular senescence (Shevitz et al. 1986; Smith and Pereira-Smith 1989; Kumazaki et al. 1991). Kumazaki et al. (1993) reported that human fibroblasts in the skin and in human vascular endothelial cells displayed a similar upregulation in the expression of fibronectin both in aged tissues and in cell cultures of replicative senescence. Accordingly, Wang et al. (2015) demonstrated that the replicative senescence of mouse cardiac fibroblasts displayed comparable fibrogenic changes, i.e., an increase in the expression of fibronectin and collagen 1A1 and 3A1, as were evident in the fibroblasts present in the fibrotic myocardium of aged mice. The deposition of fibronectin into tissues in conjunction with pro-inflammatory senescent cells might have a protective role in many inflammatory diseases, e.g., atherosclerosis and autoimmune diseases, since the activation of LILRB4 signaling inhibits NF-κB signaling and moreover, it promotes the activation of immunosuppressive MDSCs (Zhou et al. 2020; Singh et al. 2021; Liu et al. 2023). The pathway of LILRB4, but not those of the integrins, was most probably involved since treatment with the antagonist of the LILRB4 receptor reversed the suppression of T cells induced by human monocytic MDSCs (Singh et al. 2021). Zhou et al. (2020) demonstrated that the activation of LILRB4 signaling protected mouse cardiac muscle from fibrosis and hypertrophy through an inhibition of NF-κB signaling.
Galectins (Gal) are a family of the β-galactoside-binding lectins which have important functions in both intracellular and extracellular spaces. For instance, the Gal-8 protein inhibits the activity of mammalian target of rapamycin (mTOR), an essential regulator of metabolism and physiology (Jia et al. 2018). The Gal-8 protein is highly expressed and secreted in inflammatory conditions and it can regulate many innate and adaptive responses (Tribulatti et al. 2020). Wang et al. (2024b) demonstrated that the Gal-8 protein is a functional ligand for the LILRB4 receptor and it is able to induce the differentiation of M-MDSCs from mouse monocytes by activating the STAT3 pathway and inhibiting NF-κB signaling. The Gal-8 protein is robustly expressed in cancer cells and it is a significant enhancer of the LILRB4-mediated immunosuppressive milieu and the growth of tumors (Wang et al. 2024b). However, there are only a few studies examining the effect of cellular senescence on the expression and secretion of the Gal-8 protein. Wei et al. (2019) revealed that the expression of Gal-8 was robustly increased in senescent human corneal endothelial cells (HCEC) accompanied by senescence markers, such as SA-β-gal and p21. These investigators also revealed that the Gal-8 protein together with matrix metalloproteinase-2 (MMP-2) enhanced the role of senescent HCECs in corneal inflammatory neovascularization (Yu et al. 2019). It seems that in most occasions, the Gal-8 protein as well as the LILRB4 receptor evoked immunosuppression thus promoting cancer growth while alleviating the severity of inflammatory diseases.
NKG2A/HLA-E
Pereira et al. (2019) demonstrated that the cellular senescence induced by replicative senescence, ionising radiation (X-ray), and oncogenic RAS activation stimulated a robust increase in the expression of the HLA-E protein in human fibroblasts and endothelial cells. They also reported that SASP factors, such as cytokines IL-6 and IL-8 as well as chemokine CCL2, induced the expression of HLA-E in human non-senescent fibroblasts. It is known that in human Tera-2 cells, the transcription of the HLA-E gene can be stimulated by IFN-γ signaling via the STAT1 pathway (Gobin and van den Elsen 2000). Pereira et al. (2019) reported that the HLA-E protein targeted the NKG2A receptor on the surface of NK and CD8+ T cells. Subsequently, they revealed that an inhibition of the expression of HLA-E in senescent human fibroblasts enhanced their immune elimination in vitro by NK and CD8+ T cells. Moreover, Pereira et al. (2019) performed histochemical stainings for HLA-E and p16INK4A, a marker of cellular senescence, and they observed that these proteins were co-localized in fibroblasts in the skin samples from aged humans. These results indicate that senescent cells are able to evade immune clearance by NK and CD8+ T cells by stimulating the expression of the HLA-E protein. The HLA-E protein can also be packaged into EVs (Jiang et al. 2021) although currently it is not known whether senescent cells can inhibit cytotoxic cells by secreting the HLA-E protein in EVs, a signaling mechanism known to be activated in senescent cells (Salminen et al. 2020).
TIM-3/HMGB1/Gal-9/CEACAM1
There is abundant evidence that the cellular senescence upregulates the expression of HMGB1, Gal-9, and CEACAM1 in different models. HMGB1 is an alarmin which is secreted from senescent cells and many immune cells during times of tissue stress and inflammation. The HMGB1 protein can bind to certain immunoreceptors, such as TIM-3 and Toll-like receptor 4 (TLR4). Davalos et al. (2013) demonstrated that several treatments that could induce cellular senescence were able to stimulate the secretion of the HMGB1 protein from human fibroblasts. They also observed that senescent cells actively exported nuclear HMGB1 into the extracellular space. It is known that the HMGB1 protein can stimulate inflammatory responses although it seems that it can also inhibit some specific immune responses via the TIM-3 receptor (Chiba et al. 2012; Huang et al. 2022a). As described above, in the cancer microenvironment, the Gal-9 protein inhibits the cytotoxicity of NK and CD8+ T cells via the activation of TIM-3 signaling. Berben et al. (2020) demonstrated that the level of the Gal-9 protein progressively increased with aging in the human blood. Moreover, the Gal-9 protein was an important marker of frailty in old women (Mitchell et al. 2023). Currently, it is not known whether cellular senescence is involved in the increase in the Gal-9 level associated with aging. Tarallo et al. (2024) demonstrated that the temozolomide-induced senescence of mouse melanoma cells robustly increased the expression and secretion of the Gal-9 protein. Interestingly, silencing of mitofusin 1, a mitochondrial fusion protein, reduced the expression of SASP factors, decreased the expression of the Gal-9 protein, promoted immune cell recruitment, and delayed the growth of melanoma. These results indicate that cellular senescence controls the expression level of the Gal-9 protein.
The CEACAM1 protein is upregulated not only in cancer cells but also in diverse types of senescent cells. For instance, Sappino et al. (2012) demonstrated that the inducers of DNA double-strand breaks, such as neocarzinostatin and etoposide, robustly stimulated the expression of CEACAM1 and senescence markers in human epithelial cells and colon cancer cells. They revealed that the upregulation of this protein was mediated through the ATM/p53 signaling pathway. Moreover, they reported that the silencing of CEACAM1 expression impaired the p53-induced cellular senescence. Lee et al. (2024) also demonstrated that two DNA damaging agents, cisplatin and doxorubicin, increased the expression of CEACAM1 and many SASP factors in human breast cancer cells. There are observations indicating that the expression of CEACAM1 promotes vascular aging in humans and mice. For instance, Kleefeldt et al. (2019) reported that the aging process increased the expression of CEACAM1 in human internal thoracic artery and murine aorta. An age-related increase in the expression of CEACAM1 augmented oxidative stress and the expression of TNF-α as well as consequently, it increased collagen accumulation and impaired the integrity of the endothelial barrier. Currently, it is not known whether an increased expression of CEACAM1 is associated with the TIM-3-induced vascular disturbances (Cong et al. 2020).
SIRPα/CD47
Schloesser et al. (2023) demonstrated that an increase in the expression of the CD47 protein was a hallmark of cellular senescence induced by different senescence inducers in many human and mouse fibroblast populations. These results are in agreement with the reports that the expression of CD47 increased with aging in mouse tissues, e.g. in the lungs (Tabula Muris Consortium 2020; Schloesser et al. 2023). This implies that senescent cells upregulate the expression of the marker-of-self, i.e., the CD47 protein, to evade their elimination through phagocytosis. Interestingly, Schloesser et al. (2023) revealed that senescent human fibroblasts were resistant to their efferocytosis by human macrophages. Moreover, they reported that senescent fibroblasts also inhibited the capacity of macrophages to engulf surrounding cell corpses. The suppression of macrophage-induced phagocytosis was dependent on the cell–cell contact with senescent fibroblasts rather than with secreted SASP factors. They also revealed that the inhibition of phagocytic activity of macrophages was attributed to the activation of SHP-1 by means of the ITIM domain in the SIRPα protein. Kojima et al. (2016) reported that exposure to CD47 blocking antibodies restored the phagocytic activity of macrophages and subsequently inhibited the severity of atherosclerosis in mouse models. As described above, the CD47 protein is also a receptor for the TSP1 protein and this regulation mechanism is independent of SIRPα signaling. For instance, TSP1 signaling via the CD47 receptor induces cellular senescence in endothelial cells (Gao et al. 2016; Bitar 2019). Moreover, there are observations indicating that TSP1/CD47 signaling disturbs the normal function of arteries and promotes a vascular aging process. Ghimire et al. (2020) demonstrated that the expression levels of both TSP1 and CD47 significantly increased in human and mouse arteries. They observed that the expression of the CD47 protein reduced the migration of mouse endothelial cell and the formation of capillary tubes. Ghimire et al. (2020) also reported that the angiogenic activity was significantly greater in old CD47-null mice than in their aged wild-type counterparts. The blood flow in the hind limbs was also significantly greater in the CD47-null mice than in wild-type mice. However, it should be emphasized that the TSP1 protein has many other targets which could affect blood flow with aging. In fact, the CD47 protein can act as the ligand for the SIRPα receptor as well as concurrently acting as the receptor of the TSP1 protein.
Cooperation between inhibitory immune checkpoints and immunosuppressive cells
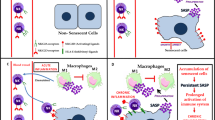
The aging process is associated with a gradual decline and remodelling of the properties of the immune system, i.e., a phenomenon called immunosenescence (Fulop et al. 2018; Salminen 2021; Santoro et al. 2021). However, immunosenescence is not only connected to the aging process since many age-related and chronic inflammatory diseases are associated with a clear immune suppressive state (Kanterman et al. 2012; Barbe-Tuana et al. 2020). Interestingly, an important hallmark of immunosenescence is the decline in the activity of cytotoxic cells, including NK and CD8+ T cells (Martinez-Zamudio et al. 2021; Salminen 2021; Brauning et al. 2022). An accumulation of senescent cells with enriched inhibitory immune checkpoint ligands could represent a mechanism to explain the increase in tissue immunotolerance with aging. Moreover, there is abundant evidence that aged tissues contain an increased number of immunosuppressive cells, such as MDSCs, Tregs, and M2 macrophages (Grizzle et al. 2007; Enioutina et al. 2011; Garg et al. 2014; Ruhland et al. 2016; Salminen 2020, 2022). Currently, it is known that the cooperation between inhibitory immune checkpoint proteins and immunosuppressive cells can inhibit the elimination of senescent cells by cytotoxic NK and CD8+ T cells (Fig. 2).

Cooperation between inhibitory immune checkpoints and immunosuppressive cells. Senescent cells suppress the functions of cytotoxic cells via inhibitory checkpoint ligands. Senescent cells also secrete chemokines which recruit immunosuppressive cells into senescent tissues. Senescent cells can also induce the differentiation of immunosuppressive cells via the PD-1/PD-L1 signaling. On the other hand, immunosuppressive cells secrete anti-inflammatory cytokines which enhance the senescence process itself and stimulate the expression of inhibitory checkpoint ligands in senescent cells. MDSC myeloid-derived suppressor cell, NK natural killer cell. Other abbreviations are as in Fig. 1
Chemokines are well characterized SASP factors secreted by pro-inflammatory senescent cells (Rodier et al. 2009; Freund et al. 2010; Acosta et al. 2013). For instance, cultured senescent cells secrete many members of two families of chemokines, i.e., CCL2,3,5,8 and CXCL1,2,3,5,10. Moreover, granulocyte–macrophage colony-stimulating factor (GM-CSF) is highly secreted by human senescent fibroblasts (Freund et al. 2010). Moreover, single-cell studies have revealed that fibroblasts isolated from aged tissues abundantly express and secrete chemokines (Salzer et al. 2018; Sole-Boldo et al. 2020). The GM-CSF and chemokines secreted from senescent cells induce haematopoiesis in the bone marrow producing myeloid cells which consequently can be recruited into inflamed tissues. For instance, GM-CSF and certain chemokines coordinate the development of immunosuppressive MDSC via myelopoiesis (Li et al. 2022). Accordingly, Hotta et al. (2019) reported that in mice GM-CSF therapy increased the expansion of Tregs. Moreover, it is known that chemokine exposure can augment the polarization of human and mouse M2 macrophages in inflammatory conditions (Mantovani et al. 2004). These studies indicated that senescent cells by secreting GM-CSF and chemokines can recruit and even stimulate the generation of immunosuppressive cells (Fig. 2).
Given that inhibitory immune checkpoints and Tregs have a crucial role in the maintenance of peripheral tolerance, it is not surprising that they are able to cooperate with each other. Francisco et al. (2009) demonstrated that PD-L1 signaling, stimulated by either PD-L1-positive immune cells or PD-L1-bound beads, was able to convert mouse naïve CD4+ T cells into induced Tregs (iTreg). They also reported that iTreg cells suppressed the activity of CD4+ T effector cells. DiDomenico et al. (2018) revealed that mouse PD-L1 ligands stimulated the expansion of iTregs by binding to the PD-1 receptor of the CD4+ T cells. They also reported that the PD-L1-induced Tregs inhibited the proliferation of naïve CD4+ T cells. Moreover, DiDomenico et al. (2018) demonstrated that the blockade of the PD-1 receptor suppressed the expansion of iTregs in a murine glioma model. It is also known that activation of the PD-1/PD-L1 checkpoint pathway stimulated the polarization of macrophages toward the immune suppressive M2 phenotype. Wei et al. (2021) reported that when human THP-1 cells were treated with human recombinant PD-L1 protein, this promoted their polarization toward the M2 macrophage phenotype. The M2 polarization was induced through the ERK/AKT/mTOR signaling pathway in human THP-1 cells. They also revealed that treatment with the PD-1 antagonist, nivolumab, inhibited the PD-L1-induced M2 polarization. There are also indications that the NF-κB-mediated increase in the expression of the PD-1 protein in MDSCs increased their proliferation in the tumor milieu (Nam et al. 2019). It is known that immunosuppressive cells are potent inhibitors of the cytotoxic NK and CD8+ T cells by secreting TGF-β cytokine, a mediator of interactions between immunosuppressive cells (Thomas and Massague 2005; Ralainirina et al. 2007; Flavell et al. 2010). Flavell et al. (2010) have reviewed the role of TGF-β in the suppression of cytotoxicity induced by NK and CD8+ T cells. It seems that the immunosuppressive microenvironment induced by MDSCs, Tregs, and M2 macrophages effectively suppresses the clearance of senescent cells from aged tissues (Fig. 2).
Immunosuppressive cells, either recruited or polarized in aging tissues, secrete anti-inflammatory cytokines, such as TGF-β and IL-10, which can promote the senescence process through two different mechanisms, (i) they can enhance the senescence process itself, or (ii) they can stimulate the expression of inhibitory checkpoint ligands in senescent cells and thus prevent their elimination. Several factors in the senescence microenvironment can activate the latent TGF-β protein, such as reactive oxygen species (ROS) and integrin β3 protein (Frippiat et al. 2001; Rapisarda et al. 2017). Lyu et al. (2018) demonstrated that the ROS-induced TGF-β signaling altered the epigenetic H4K20me3 status by increasing the expression of miR-29 which suppressed the expression of SUV4-20 h and consequently reduced the methylation status of the H4K20me site in mouse embryonic fibroblasts. The loss of H4K20me3 reduced the integrity of DNA and caused a premature fibroblast senescence. They also reported that an increase in TGF/miR-29 signaling promoted the aging process in mouse cardiac muscle. Several other studies have revealed that exposure to TGF-β increased cellular senescence in several experimental models (Tominaga and Suzuki 2019; Matsuda et al. 2023). Cellular senescence is not the only response induced by TGF-β exposure since TGF-β is a robust enhancer of age-related tissue fibrosis (Ren et al. 2023). Moreover, the IL-10 cytokine can also trigger cellular senescence via STAT3 signaling, e.g., in hepatic stellate cells (Huang et al. 2020). These examples indicate that immunosuppressive cells are potent enhancers of cellular senescence both as encountered in aging and in many age-related inflammatory diseases.
Immunosuppressive cells not only enhance cellular senescence but they are also able to stimulate the expression of many inhibitory immune checkpoint proteins, thus promoting immunosenescence in aged tissues (Fig. 2). For instance, Park et al. (2016) demonstrated that TGF-β1 treatment increased the expression of the PD-1 receptors in activated human CD4+ and CD8+ T cells. TGF-β exposure activated the SMAD3 signaling pathway which increased the transcription of the PDCD1 gene. Park et al. (2016) also confirmed that an increase in the expression of PD-1 decreased the function of T cells. Accordingly, Shiri et al. (2024) demonstrated that the IL-10 cytokines secreted by mouse Tregs stimulated the expression of the PD-L1 protein on myeloid cells and subsequently inhibited the immunosurveillance of cancer cells by cytotoxic immune cells. Currently, a dual blockade of the TGF-β and PD-L1 proteins has been used for therapeutic purposes (Gulley et al. 2022). There are several investigations indicating that when human fibroblasts were exposed to TGF-β this stimulated the expression of fibronectin, a major ligand for the LILRB4 receptor (Varga et al. 1987; Roberts et al. 1988). On the other hand, it seems that exposure to TGF-β can inhibit the expression of activating immune checkpoint receptors. Castriconi et al. (2003) demonstrated that the treatment of human NK cells with TGF-β1 inhibited the surface expression of NKp30 and NKG2D receptors, which are major activating checkpoint receptors involved in the immunosurveillance by NK cells. They also confirmed that TGF-β1 exposure prevented the elimination of inducible dendritic cells (iDC). This suggests that TGF-β and probably other immunosuppressive mediators can promote the development of immunosuppressive states in two ways, i.e., either by activating inhibitory checkpoints or inhibiting activating checkpoints.
In conclusion, it seems that the inhibitory checkpoint receptors and their ligands of immune and non-immune cells cooperate with recruited and tissue-resident immunosuppressive cells in aged tissues and in age-related diseases. This immunosuppressive network prevents the elimination of senescent cells from aged tissues. Paradoxically, it seems that pro-inflammatory senescent cells promote tissue inflammation in aged tissues, whereas increased expression of the ligands for inhibitory checkpoint receptors in senescent cells prevents excessive devastating inflammation with aging thus maintaining the chronic low-grade inflammatory state.
Blockade of inhibitory immune checkpoints could provide a novel senolytic therapy
Given that there are studies indicating that the clearance of senescent cells can delay the aging-associated disorders (Baker et al. 2011; Chaib et al. 2022), there has been intensive research to discover either chemical drugs or forms of immunotherapy to eliminate senescent cells from aging tissues (Sikora et al. 2019; Chaib et al. 2022; Krzystyniak et al. 2022). Immunotherapies based on the blockade of inhibitory checkpoints are in clinical use for the treatment of diverse cancers (Shiravand et al. 2022). Antibodies to the PD-1, PD-L1, and CTLA-4 proteins have demonstrated good efficacy in certain cancer types but unfortunately not in all cancers. Currently there are also some small molecule inhibitors and peptides under examination which have been claimed to block the properties of certain inhibitory checkpoint proteins (Fuchs et al. 2024). There are also some preclinical ongoing investigations examining the pros and cons of blockade of immune checkpoints in cardiovascular diseases (Suda et al. 2023), infectious diseases (Wykes and Lewin 2018), and idiopathic pulmonary fibrosis (Tan et al. 2024). Several cancer studies have compared the efficacy of the PD-1/PD-L1 blockade therapy between young and old patients. However, a meta-analysis did not find any differences in treatment efficacy between young and elderly patients (Elias et al. 2018).
The earlier experiments on the elimination of senescent cells used genetic techniques for the removal of senescent cells (Baker et al. 2011). In fact, there are rather few studies which have examined whether the blockade of the PD-1/PD-L1 axis could eliminate senescent cells in tissues, whereas many senolytic trials have utilized chemical approaches (Chaib et al. 2022). Wang et al (2022a) demonstrated that a blockade of the PD-1/PD-L1 axis enhanced immunosurveillance of senescent cells in mouse kidney, liver, and lung and moreover, it also reduced some age-related disorders. They used the anti-PD-1 therapy protocol in aged mice and thus it seems that this treatment prevented the PD-L1-induced exhaustion of CD8+ T cells. Currently, it is known that there are some adverse events, such as systemic inflammatory syndromes, associated with the use of immune checkpoint blockade therapies (Baldini et al. 2020; Zheng 2023). For instance, Baldini et al. (2020) reported that an anti-PD-L1 therapy for solid tumors induced immune-related adverse events more frequently in the elderly than in young patients. This could be attributed to an increase in the expression of the PD-L1 receptor in aged tissues and its wide distribution in the body. Nonetheless, there are many ongoing experiments with inhibitors of other inhibitory checkpoints. For instance, Kojima et al. (2016) demonstrated that the blocking antibodies of the CD47 protein attenuated the severity of atherosclerosis in mouse models by improving the phagocytosis capacity of immune cells and enhanced the clearance of abnormal cells from mouse vascular tissues.
Conclusions
Given that the aging process is associated with a gradual increase in the inflammatory state, it is not surprising that as a counteracting effect there is an activation of immunosuppressive mechanisms, i.e., an increase in the expression of several inhibitory immune checkpoint proteins as well as an upregulation of immunosuppressive cells in tissues, both via recruitment into tissues and alterations in the polarization of tisssue-resident immune cells. Moreover, there are some other mechanisms to counteract increasing inflammation with aging. For instance, an increase in the aging-linked activation of NF-κB system (Salminen et al. 2008a) can be suppressed, e.g., by stimulating the expression of heat-shock proteins HSP70 and HSP90 (Salminen et al. 2008b; van Eden et al. 2017) and inducing an endotoxin tolerance state (Chan et al. 2005). Moreover, it is known that there are many metabolic immune checkpoints, such as the adenosine-induced immunosuppression (Ohta 2016). Currently, the role of cellular senescence in physiology and pathology is under debate; while it seems that it has beneficial effects during embryogenesis and development, however in some pathological conditions, such as in cancer and wound healing, it can evoke both beneficial and harmful responses in a context-dependent manner (Huang et al. 2022b). Currently, it is not known whether the upregulation of inhibitory checkpoint expression is also a context-dependent process with aging. The age-related cellular senescence seems to be a paradoxical process since senescent cells possess both pro-inflammatory and immunosuppressive properties. However, it is known that many signaling pathways which stimulate the expression of the PD-L1 ligand are well-known enhancers of the aging process, such as epigenetic regulation as well as signaling via the mTOR-related, cGAS-STING, and AhR pathways (Salminen 2024). Moreover, the anti-aging treatments with metformin and rapamycin reduce the expression levels of PD-L1 ligand protein. It does seem that inhibitory immune checkpoint signaling has an important role in the aging process.
Data availability
No datasets were generated or analysed during the current study.
References
Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, Athineos D, Kang TW, Lasitschka F, Andrulis M, Pascual G, Morris KJ, Khan S, Jin H, Dharmalingam G, Snijders AP, Carroll T, Capper D, Pritchard C, Inman GJ, Longerich T, Sansom OJ, Benitah SA, Zender L, Gil J (2013) A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol 15:978–990. https://doi.org/10.1038/ncb2784
Alibhai FJ, Lim F, Yeganeh A, DiStefano PV, Binesh-Marvasti T, Belfiore A, Wlodarek L, Gustafson D, Millar S, Li SH, Weisel RD, Fish JE, Li RK (2020) Cellular senescence contributes to age-dependent changes in circulating extracellular vesicle cargo and function. Aging Cell 19:e13103. https://doi.org/10.1111/acel.13103
Anderson AC, Joller N, Kuchroo VK (2016) Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity 44:989–1004. https://doi.org/10.1016/j.immuni.2016.05.001
Andre P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, Arnoux T, Blery M, Bonnafous C, Gauthier L, Morel A, Rossi B, Remark R, Breso V, Bonnet E, Habif G, Guia S, Lalanne AI, Hoffmann C, Lantz O, Fayette J, Boyer-Chammard A, Zerbib R, Dodion P, Ghadially H, Jure-Kunkel M, Morel Y, Herbst R, Narni-Mancinelli E, Cohen RB, Vivier E (2018) Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell 175:1731-1743.e13. https://doi.org/10.1016/j.cell.2018.10.014
Antia M, Baneyx G, Kubow KE, Vogel V (2008) Fibronectin in aging extracellular matrix fibrils is progressively unfolded by cells and elicits an enhanced rigidity response. Faraday Discuss 139:229–249. https://doi.org/10.1039/b718714a. (discussion 309-25, 419-20)
Antonangeli F, Natalini A, Garassino MC, Sica A, Santoni A, Di Rosa F (2020) Regulation of PD-L1 expression by NF-κB in cancer. Front Immunol 11:584626. https://doi.org/10.3389/fimmu.2020.584626
Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM (2011) Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479:232–236. https://doi.org/10.1038/nature10600
Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, Khazaie K, Miller JD, van Deursen JM (2016) Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 530:184–189. https://doi.org/10.1038/nature16932
Baldanzi G (2022) Immune checkpoint receptors signaling in T cells. Int J Mol Sci 23:3529. https://doi.org/10.3390/ijms23073529
Baldini C, Martin Romano P, Voisin AL, Danlos FX, Champiat S, Laghouati S, Kfoury M, Vincent H, Postel-Vinay S, Varga A, Vuagnat P, Ribrag V, Mezquita L, Besse B, Hollebecque A, Lambotte O, Michot JM, Soria JC, Massard C, Marabelle A (2020) Impact of aging on immune-related adverse events generated by anti-programmed death (ligand)PD-(L)1 therapies. Eur J Cancer 129:71–79. https://doi.org/10.1016/j.ejca.2020.01.013
Barbe-Tuana F, Funchal G, Schmitz CRR, Maurmann RM, Bauer ME (2020) The interplay between immunosenescence and age-related diseases. Semin Immunopathol 42:545–557. https://doi.org/10.1007/s00281-020-00806-z
Barclay AN, Van den Berg TK (2014) The interaction between signal regulatory protein alpha (SIRPα) and CD47: structure, function, and therapeutic target. Annu Rev Immunol 32:25–50. https://doi.org/10.1146/annurev-immunol-032713-120142
Beenen AC, Sauerer T, Schaft N, Dörrie J (2022) Beyond cancer: regulation and function of PD-L1 in health and immune-related diseases. Int J Mol Sci 23:8599. https://doi.org/10.3390/ijms23158599
Benayoun BA, Pollina EA, Singh PP, Mahmoudi S, Harel I, Casey KM, Dulken BW, Kundaje A, Brunet A (2019) Remodeling of epigenome and transcriptome landscapes with aging in mice reveals widespread induction of inflammatory responses. Genome Res 29:697–709. https://doi.org/10.1101/gr.240093.118
Berben L, Floris G, Kenis C, Dalmasso B, Smeets A, Vos H, Neven P, Antoranz Martinez A, Laenen A, Wildiers H, Hatse S (2020) Age-related remodelling of the blood immunological portrait and the local tumor immune response in patients with luminal breast cancer. Clin Transl Immunology 9:e1184. https://doi.org/10.1002/cti2.1184
Betzler AC, Theodoraki MN, Schuler PJ, Döscher J, Laban S, Hoffmann TK, Brunner C (2020) NF-κB and its role in checkpoint control. Int J Mol Sci 21:3949. https://doi.org/10.3390/ijms21113949
Biran A, Zada L, Abou Karam P, Vadai E, Roitman L, Ovadya Y, Porat Z, Krizhanovsky V (2017) Quantitative identification of senescent cells in aging and disease. Aging Cell 16:661–671. https://doi.org/10.1111/acel.12592
Bitar MS (2019) Diabetes impairs angiogenesis and induces endothelial cell senescence by up-regulating thrombospondin-CD47-dependent signaling. Int J Mol Sci 20:673. https://doi.org/10.3390/ijms20030673
Bojko A, Czarnecka-Herok J, Charzynska A, Dabrowski M, Sikora E (2019) Diversity of the senescence phenotype of cancer cells treated with chemotherapeutic agents. Cells 8:1501. https://doi.org/10.3390/cells8121501
Boyer B, Kern P, Fourtanier A, Labat-Robert J (1991) Age-dependent variations of the biosyntheses of fibronectin and fibrous collagens in mouse skin. Exp Gerontol 26:375–383. https://doi.org/10.1016/0531-5565(91)90049-r
Brauning A, Rae M, Zhu G, Fulton E, Admasu TD, Stolzing A, Sharma A (2022) Aging of the immune system: focus on natural killer cells phenotype and functions. Cells 11:1017. https://doi.org/10.3390/cells11061017
Buckle I, Guillerey C (2021) Inhibitory receptors and immune checkpoints regulating natural killer cell responses to cancer. Cancers (basel) 13:4263. https://doi.org/10.3390/cancers13174263
Burke KP, Patterson DG, Liang D, Sharpe AH (2023) Immune checkpoint receptors in autoimmunity. Curr Opin Immunol 80:102283. https://doi.org/10.1016/j.coi.2023.102283
Cai N, Wu Y, Huang Y (2022) Induction of accelerated aging in a mouse model. Cells 11:1418. https://doi.org/10.3390/cells11091418
Campisi J, Robert L (2014) Cell senescence: role in aging and age-related diseases. Interdiscip Top Gerontol 39:45–61. https://doi.org/10.1159/000358899
Castriconi R, Cantoni C, Della Chiesa M, Vitale M, Marcenaro E, Conte R, Biassoni R, Bottino C, Moretta L, Moretta A (2003) Transforming growth factor β1 inhibits expression of NKp30 and NKG2D receptors: consequences for the NK-mediated killing of dendritic cells. Proc Natl Acad Sci U S A 100:4120–4125. https://doi.org/10.1073/pnas.0730640100
Chaib S, Tchkonia T, Kirkland JL (2022) Cellular senescence and senolytics: the path to the clinic. Nat Med 28:1556–1568. https://doi.org/10.1038/s41591-022-01923-y
Chan C, Li L, McCall CE, Yoza BK (2005) Endotoxin tolerance disrupts chromatin remodeling and NF-κB transactivation at the IL-1β promoter. J Immunol 175:461–468. https://doi.org/10.4049/jimmunol.175.1.461
Chen L, Flies DB (2013) Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol 13:227–242. https://doi.org/10.1038/nri3405
Chen J, Yang J, Wang W, Guo D, Zhang C, Wang S, Lu X, Huang X, Wang P, Zhang G, Zhang J, Wang J, Cai Z (2022) Tumor extracellular vesicles mediate anti-PD-L1 therapy resistance by decoying anti-PD-L1. Cell Mol Immunol 19:1290–1301. https://doi.org/10.1038/s41423-022-00926-6
Chen HA, Ho YJ, Mezzadra R, Adrover JM, Smolkin R, Zhu C, Woess K, Bernstein N, Schmitt G, Fong L, Luan W, Wuest A, Tian S, Li X, Broderick C, Hendrickson RC, Egeblad M, Chen Z, Alonso-Curbelo D, Lowe SW (2023) Senescence rewires microenvironment sensing to facilitate antitumor immunity. Cancer Discov 13:432–453. https://doi.org/10.1158/2159-8290.CD-22-0528
Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, Fujioka Y, Ohba Y, Gorman JV, Colgan JD, Hirashima M, Uede T, Takaoka A, Yagita H, Jinushi M (2012) Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol 13:832–842. https://doi.org/10.1038/ni.2376
Chin T, Lee XE, Ng PY, Lee Y, Dreesen O (2023) The role of cellular senescence in skin aging and age-related skin pathologies. Front Physiol 14:1297637. https://doi.org/10.3389/fphys.2023.1297637
Collins M, Ling V, Carreno BM (2005) The B7 family of immune-regulatory ligands. Genome Biol 6:223. https://doi.org/10.1186/gb-2005-6-6-223
Cong Y, Wang X, Wang S, Qiao G, Li Y, Cao J, Jiang W, Cui Y (2020) Tim-3 promotes tube formation and decreases tight junction formation in vascular endothelial cells. Biosci Rep 40(10):BSR20202130. https://doi.org/10.1042/BSR20202130
da Silva PFL, Ogrodnik M, Kucheryavenko O, Glibert J, Miwa S, Cameron K, Ishaq A, Saretzki G, Nagaraja-Grellscheid S, Nelson G, von Zglinicki T (2019) The bystander effect contributes to the accumulation of senescent cells in vivo. Aging Cell 18:e12848. https://doi.org/10.1111/acel.12848
Davalos AR, Kawahara M, Malhotra GK, Schaum N, Huang J, Ved U, Beausejour CM, Coppe JP, Rodier F, Campisi J (2013) p53-dependent release of alarmin HMGB1 is a central mediator of senescent phenotypes. J Cell Biol 201:613–629. https://doi.org/10.1083/jcb.201206006
DeKruyff RH, Bu X, Ballesteros A, Santiago C, Chim YL, Lee HH, Karisola P, Pichavant M, Kaplan GG, Umetsu DT, Freeman GJ, Casasnovas JM (2010) T cell/transmembrane, Ig, and mucin-3 allelic variants differentially recognize phosphatidylserine and mediate phagocytosis of apoptotic cells. J Immunol 184:1918–1930. https://doi.org/10.4049/jimmunol.0903059
Deng M, Gui X, Kim J, Xie L, Chen W, Li Z, He L, Chen Y, Chen H, Luo W, Lu Z, Xie J, Churchill H, Xu Y, Zhou Z, Wu G, Yu C, John S, Hirayasu K, Nguyen N, Liu X, Huang F, Li L, Deng H, Tang H, Sadek AH, Zhang L, Huang T, Zou Y, Chen B, Zhu H, Arase H, Xia N, Jiang Y, Collins R, You MJ, Homsi J, Unni N, Lewis C, Chen GQ, Fu YX, Liao XC, An Z, Zheng J, Zhang N, Zhang CC (2018) LILRB4 signalling in leukaemia cells mediates T cell suppression and tumour infiltration. Nature 562:605–609. https://doi.org/10.1038/s41586-018-0615-z
Deuse T, Hu X, Agbor-Enoh S, Jang MK, Alawi M, Saygi C, Gravina A, Tediashvili G, Nguyen VQ, Liu Y, Valantine H, Lanier LL, Schrepfer S (2021) The SIRPα-CD47 immune checkpoint in NK cells. J Exp Med 218:e20200839. https://doi.org/10.1084/jem.20200839
DiDomenico J, Lamano JB, Oyon D, Li Y, Veliceasa D, Kaur G, Ampie L, Choy W, Lamano JB, Bloch O (2018) The immune checkpoint protein PD-L1 induces and maintains regulatory T cells in glioblastoma. Oncoimmunology 7:e1448329. https://doi.org/10.1080/2162402X.2018.1448329
Dmitrieva NI, Burg MB (2007) High NaCl promotes cellular senescence. Cell Cycle 6:3108–3113. https://doi.org/10.4161/cc.6.24.5084
Duan S, Paulson JC (2020) Siglecs as immune cell checkpoints in disease. Annu Rev Immunol 38:365–395. https://doi.org/10.1146/annurev-immunol-102419-035900
Elias R, Giobbie-Hurder A, McCleary NJ, Ott P, Hodi FS, Rahma O (2018) Efficacy of PD-1 & PD-L1 inhibitors in older adults: a meta-analysis. J Immunother Cancer 6:26. https://doi.org/10.1186/s40425-018-0336-8
Enioutina EY, Bareyan D, Daynes RA (2011) A role for immature myeloid cells in immune senescence. J Immunol 186:697–707. https://doi.org/10.4049/jimmunol.1002987
Feng Y, Huang C, Wang Y, Chen J (2023) SIRPα: A key player in innate immunity. Eur J Immunol 53:e2350375. https://doi.org/10.1002/eji.202350375
Filippi CM, Juedes AE, Oldham JE, Ling E, Togher L, Peng Y, Flavell RA, von Herrath MG (2008) Transforming growth factor-β suppresses the activation of CD8+ T-cells when naive but promotes their survival and function once antigen experienced: a two-faced impact on autoimmunity. Diabetes 57:2684–2692. https://doi.org/10.2337/db08-0609
Fisher JG, Doyle ADP, Graham LV, Khakoo SI, Blunt MD (2022) Disruption of the NKG2A:HLA-E immune checkpoint axis to enhance NK cell activation against cancer. Vaccines (basel) 10:1993. https://doi.org/10.3390/vaccines10121993
Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limon P (2010) The polarization of immune cells in the tumour environment by TGFβ. Nat Rev Immunol 10:554–567. https://doi.org/10.1038/nri2808
Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A (2018) Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat Rev Endocrinol 14:576–590. https://doi.org/10.1038/s41574-018-0059-4
Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, Sharpe AH (2009) PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med 206:3015–3029. https://doi.org/10.1084/jem.20090847
Francisco LM, Sage PT, Sharpe AH (2010) The PD-1 pathway in tolerance and autoimmunity. Immunol Rev 236:219–242. https://doi.org/10.1111/j.1600-065X.2010.00923.x
Freund A, Orjalo AV, Desprez PY, Campisi J (2010) Inflammatory networks during cellular senescence: causes and consequences. Trends Mol Med 16:238–246. https://doi.org/10.1016/j.molmed.2010.03.003
Frippiat C, Chen QM, Zdanov S, Magalhaes JP, Remacle J, Toussaint O (2001) Subcytotoxic H2O2 stress triggers a release of transforming growth factor-β1, which induces biomarkers of cellular senescence of human diploid fibroblasts. J Biol Chem 276:2531–2537. https://doi.org/10.1074/jbc.M006809200
Fuchs N, Zhang L, Calvo-Barreiro L, Kuncewicz K, Gabr M (2024) Inhibitors of immune checkpoints: Small molecule- and peptide-based approaches. J Pers Med 14:68. https://doi.org/10.3390/jpm14010068
Fulop T, Larbi A, Dupuis G, Le Page A, Frost EH, Cohen AA, Witkowski JM, Franceschi C (2018) Immunosenescence and inflamm-aging as two sides of the same coin: Friends or foes? Front Immunol 8:1960. https://doi.org/10.3389/fimmu.2017.01960
Gao Q, Chen K, Gao L, Zheng Y, Yang YG (2016) Thrombospondin-1 signaling through CD47 inhibits cell cycle progression and induces senescence in endothelial cells. Cell Death Dis 7:e2368. https://doi.org/10.1038/cddis.2016.155
Garcia MG, Deng Y, Murray C, Reyes RM, Padron A, Bai H, Kancharla A, Gupta H, Shen-Orr S, Curiel TJ (2022) Immune checkpoint expression and relationships to anti-PD-L1 immune checkpoint blockade cancer immunotherapy efficacy in aged versus young mice. Aging Cancer 3:68–83. https://doi.org/10.1002/aac2.12045
Garg SK, Delaney C, Toubai T, Ghosh A, Reddy P, Banerjee R, Yung R (2014) Aging is associated with increased regulatory T-cell function. Aging Cell 13:441–448. https://doi.org/10.1111/acel.12191
Ghimire K, Li Y, Chiba T, Julovi SM, Li J, Ross MA, Straub AC, O’Connell PJ, Rüegg C, Pagano PJ, Isenberg JS, Rogers NM (2020) CD47 promotes age-associated deterioration in angiogenesis, blood flow and glucose homeostasis. Cells 9:1695. https://doi.org/10.3390/cells9071695
Ghosh C, Luong G, Sun Y (2021) A snapshot of the PD-1/PD-L1 pathway. J Cancer 12:2735–2746. https://doi.org/10.7150/jca.57334
Gobin SJ, van den Elsen PJ (2000) Transcriptional regulation of the MHC class Ib genes HLA-E, HLA-F, and HLA-G. Hum Immunol 61:1102–1107. https://doi.org/10.1016/s0198-8859(00)00198-1
Gonzalez-Gualda E, Baker AG, Fruk L, Munoz-Espín D (2021) A guide to assessing cellular senescence in vitro and in vivo. FEBS J 288:56–80. https://doi.org/10.1111/febs.15570
Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, Campisi J, Collado M, Evangelou K, Ferbeyre G, Gil J, Hara E, Krizhanovsky V, Jurk D, Maier AB, Narita M, Niedernhofer L, Passos JF, Robbins PD, Schmitt CA, Sedivy J, Vougas K, von Zglinicki T, Zhou D, Serrano M, Demaria M (2019) Cellular senescence: defining a path forward. Cell 179:813–827. https://doi.org/10.1016/j.cell.2019.10.005
Grizzle WE, Xu X, Zhang S, Stockard CR, Liu C, Yu S, Wang J, Mountz JD, Zhang HG (2007) Age-related increase of tumor susceptibility is associated with myeloid-derived suppressor cell mediated suppression of T cell cytotoxicity in recombinant inbred BXD12 mice. Mech Ageing Dev 128:672–680. https://doi.org/10.1016/j.mad.2007.10.003
Gulley JL, Schlom J, Barcellos-Hoff MH, Wang XJ, Seoane J, Audhuy F, Lan Y, Dussault I, Moustakas A (2022) Dual inhibition of TGF-β and PD-L1: a novel approach to cancer treatment. Mol Oncol 16:2117–2134. https://doi.org/10.1002/1878-0261.13146
Guo Z, Zhang R, Yang AG, Zheng G (2023) Diversity of immune checkpoints in cancer immunotherapy. Front Immunol 14:1121285. https://doi.org/10.3389/fimmu.2023.1121285
Hernandez-Segura A, Nehme J, Demaria M (2018) Hallmarks of cellular senescence. Trends Cell Biol 28:436–453. https://doi.org/10.1016/j.tcb.2018.02.001
Hosomi S, Chen Z, Baker K, Chen L, Huang YH, Olszak T, Zeissig S, Wang JH, Mandelboim O, Beauchemin N, Lanier LL, Blumberg RS (2013) CEACAM1 on activated NK cells inhibits NKG2D-mediated cytolytic function and signaling. Eur J Immunol 43:2473–2483. https://doi.org/10.1002/eji.201242676
Hotchkiss RS, Monneret G, Payen D (2013) Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol 13:862–874. https://doi.org/10.1038/nri3552
Hotta M, Yoshimura H, Satake A, Tsubokura Y, Ito T, Nomura S (2019) GM-CSF therapy inhibits chronic graft-versus-host disease via expansion of regulatory T cells. Eur J Immunol 49:179–191. https://doi.org/10.1002/eji.201847684
Huang YH, Chen MH, Guo QL, Chen ZX, Chen QD, Wang XZ (2020) Interleukin-10 induces senescence of activated hepatic stellate cells via STAT3-p53 pathway to attenuate liver fibrosis. Cell Signal 66:109445. https://doi.org/10.1016/j.cellsig.2019.109445
Huang S, Liu D, Sun J, Zhang H, Zhang J, Wang Q, Gan L, Qu G, Qiu J, Deng J, Jiang J, Zeng L (2022a) Tim-3 regulates sepsis-induced immunosuppression by inhibiting the NF-κB signaling pathway in CD4 T cells. Mol Ther 30:1227–1238. https://doi.org/10.1016/j.ymthe.2021.12.013
Huang W, Hickson LJ, Eirin A, Kirkland JL, Lerman LO (2022b) Cellular senescence: the good, the bad and the unknown. Nat Rev Nephrol 18:611–627. https://doi.org/10.1038/s41581-022-00601-z
Hudson K, Cross N, Jordan-Mahy N, Leyland R (2020) The extrinsic and intrinsic roles of PD-L1 and its receptor PD-1: Implications for immunotherapy treatment. Front Immunol 11:568931. https://doi.org/10.3389/fimmu.2020.568931
Iannello A, Raulet DH (2013) Immune surveillance of unhealthy cells by natural killer cells. Cold Spring Harb Symp Quant Biol 78:249–257. https://doi.org/10.1101/sqb.2013.78.020255
Idda ML, McClusky WG, Lodde V, Munk R, Abdelmohsen K, Rossi M, Gorospe M (2020) Survey of senescent cell markers with age in human tissues. Aging 12:4052–4066. https://doi.org/10.18632/aging.102903
Isenberg JS, Montero E (2024) Tolerating CD47. Clin Transl Med 14:e1584. https://doi.org/10.1002/ctm2.1584
Itagaki F, Nakatsuka K, Sakai H, Endo S, Su MT, Takai T (2023) Fibronectin on target cells attenuates natural cytotoxicity of NK cells via myeloid immune checkpoint ILT3/LILRB4/gp49B. Int Immunol 35:339–348. https://doi.org/10.1093/intimm/dxad012
Itakura Y, Sasaki N, Kami D, Gojo S, Umezawa A, Toyoda M (2016) N- and O-glycan cell surface protein modifications associated with cellular senescence and human aging. Cell Biosci 6:14. https://doi.org/10.1186/s13578-016-0079-5
Ito T, Igaki T (2016) Dissecting cellular senescence and SASP in Drosophila. Inflamm Regen 36:25. https://doi.org/10.1186/s41232-016-0031-4
Itoi S, Takahashi N, Saito H, Miyata Y, Su MT, Kezuka D, Itagaki F, Endo S, Fujii H, Harigae H, Sakamoto Y, Takai T (2022) Myeloid immune checkpoint ILT3/LILRB4/gp49B can co-tether fibronectin with integrin on macrophages. Int Immunol 34:435–444. https://doi.org/10.1093/intimm/dxac023
Jackaman C, Radley-Crabb HG, Soffe Z, Shavlakadze T, Grounds MD, Nelson DJ (2013) Targeting macrophages rescues age-related immune deficiencies in C57BL/6J geriatric mice. Aging Cell 12:345–357. https://doi.org/10.1111/acel.12062
Jia J, Abudu YP, Claude-Taupin A, Gu Y, Kumar S, Choi SW, Peters R, Mudd MH, Allers L, Salemi M, Phinney B, Johansen T, Deretic V (2018) Galectins control mTOR in response to endomembrane damage. Mol Cell 70:120-135.e8. https://doi.org/10.1016/j.molcel.2018.03.009
Jia J, Bissa B, Brecht L, Allers L, Choi SW, Gu Y, Zbinden M, Burge MR, Timmins G, Hallows K, Behrends C, Deretic V (2020) AMPK, a regulator of metabolism and autophagy, is activated by lysosomal damage via a novel galectin-directed ubiquitin signal transduction system. Mol Cell 77:951-969.e9. https://doi.org/10.1016/j.molcel.2019.12.028
Jia X, Yan B, Tian X, Liu Q, Jin J, Shi J, Hou Y (2021) CD47/SIRPα pathway mediates cancer immune escape and immunotherapy. Int J Biol Sci 17:3281–3287. https://doi.org/10.7150/ijbs.60782
Jiang X, Wang J, Deng X, Xiong F, Ge J, Xiang B, Wu X, Ma J, Zhou M, Li X, Li Y, Li G, Xiong W, Guo C, Zeng Z (2019) Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol Cancer 18:10. https://doi.org/10.1186/s12943-018-0928-4
Jiang L, Fei H, Jin X, Liu X, Yang C, Li C, Chen J, Yang A, Zhu J, Wang H, Fei X, Zhang S (2021) Extracellular vesicle-mediated secretion of HLA-E by trophoblasts maintains pregnancy by regulating the metabolism of decidual NK cells. Int J Biol Sci 17:4377–4395. https://doi.org/10.7150/ijbs.63390
Jiang A, Liu N, Wang J, Zheng X, Ren M, Zhang W, Yao Y (2022) The role of PD-1/PD-L1 axis in idiopathic pulmonary fibrosis: friend or foe? Front Immunol 13:1022228. https://doi.org/10.3389/fimmu.2022.1022228
Kamiya T, Seow SV, Wong D, Robinson M, Campana D (2019) Blocking expression of inhibitory receptor NKG2A overcomes tumor resistance to NK cells. J Clin Invest 129:2094–2106. https://doi.org/10.1172/JCI123955
Kandel S, Adhikary P, Li G, Cheng K (2021) The TIM3/Gal9 signaling pathway: an emerging target for cancer immunotherapy. Cancer Lett 510:67–78. https://doi.org/10.1016/j.canlet.2021.04.011
Kanterman J, Sade-Feldman M, Baniyash M (2012) New insights into chronic inflammation-induced immunosuppression. Semin Cancer Biol 22:307–318. https://doi.org/10.1016/j.semcancer.2012.02.008
Kishi S (2004) Functional aging and gradual senescence in zebrafish. Ann N Y Acad Sci 1019:521–526. https://doi.org/10.1196/annals.1297.097
Kleefeldt F, Bömmel H, Broede B, Thomsen M, Pfeiffer V, Wörsdörfer P, Karnati S, Wagner N, Rueckschloss U, Ergun S (2019) Aging-related carcinoembryonic antigen-related cell adhesion molecule 1 signaling promotes vascular dysfunction. Aging Cell 18:e13025. https://doi.org/10.1111/acel.13025
Kojima Y, Volkmer JP, McKenna K, Civelek M, Lusis AJ, Miller CL, Direnzo D, Nanda V, Ye J, Connolly AJ, Schadt EE, Quertermous T, Betancur P, Maegdefessel L, Matic LP, Hedin U, Weissman IL, Leeper NJ (2016) CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature 536:86–90. https://doi.org/10.1038/nature18935
Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, Su L, Sharpless NE (2004) Ink4a/Arf expression is a biomarker of aging. J Clin Invest 114:1299–1307. https://doi.org/10.1172/JCI22475
Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Yee H, Zender L, Lowe SW (2008) Senescence of activated stellate cells limits liver fibrosis. Cell 134:657–667. https://doi.org/10.1016/j.cell.2008.06.049
Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J (2001) Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci U S A 98:12072–12077. https://doi.org/10.1073/pnas.211053698
Krzystyniak A, Wesierska M, Petrazzo G, Gadecka A, Dudkowska M, Bielak-Zmijewska A, Mosieniak G, Figiel I, Wlodarczyk J, Sikora E (2022) Combination of dasatinib and quercetin improves cognitive abilities in aged male Wistar rats, alleviates inflammation and changes hippocampal synaptic plasticity and histone H3 methylation profile. Aging 14:572–595. https://doi.org/10.18632/aging.203835
Kuespert K, Pils S, Hauck CR (2006) CEACAMs: their role in physiology and pathophysiology. Curr Opin Cell Biol 18:565–571. https://doi.org/10.1016/j.ceb.2006.08.008
Kumazaki T, Robetorye RS, Robetorye SC, Smith JR (1991) Fibronectin expression increases during in vitro cellular senescence: correlation with increased cell area. Exp Cell Res 195:13–19. https://doi.org/10.1016/0014-4827(91)90494-f
Kumazaki T, Kobayashi M, Mitsui Y (1993) Enhanced expression of fibronectin during in vivo cellular aging of human vascular endothelial cells and skin fibroblasts. Exp Cell Res 205:396–402. https://doi.org/10.1006/excr.1993.1103
Lee N, Llano M, Carretero M, Ishitani A, Navarro F, Lopez-Botet M, Geraghty DE (1998) HLA-E is a major ligand for the natural killer inhibitory receptor CD94/NKG2A. Proc Natl Acad Sci U S A 95:5199–5204. https://doi.org/10.1073/pnas.95.9.5199
Lee BH, Park SY, Kang KB, Park RW, Kim IS (2002) NF-κB activates fibronectin gene expression in rat hepatocytes. Biochem Biophys Res Commun 297:1218–1224. https://doi.org/10.1016/s0006-291x(02)02356-2
Lee DH, Imran M, Choi JH, Park YJ, Kim YH, Min S, Park TJ, Choi YW (2024) CDK4/6 inhibitors induce breast cancer senescence with enhanced anti-tumor immunogenic properties compared with DNA-damaging agents. Mol Oncol 18:216–232. https://doi.org/10.1002/1878-0261.13541
Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA (2006) Transforming growth factor-β regulation of immune responses. Annu Rev Immunol 24:99–146. https://doi.org/10.1146/annurev.immunol.24.021605.090737
Li R, Mukherjee MB, Lin J (2022) Coordinated regulation of myeloid-derived suppressor cells by cytokines and chemokines. Cancers (basel) 14:1236. https://doi.org/10.3390/cancers14051236
Lian J, Yue Y, Yu W, Zhang Y (2020) Immunosenescence: a key player in cancer development. J Hematol Oncol 13:151. https://doi.org/10.1186/s13045-020-00986-z
Liu H, Yang J, Zhang J, Zhang P, Zhang M, Yang C, Liu L, Huang C, Wang W, Zhai Y, Yang J (2023) Molecular regulatory mechanism of LILRB4 in the immune response. Cent Eur J Immunol 48:43–47. https://doi.org/10.5114/ceji.2023.125238
Ljubic M, Prasnikar E, Perdih A, Borisek J (2023) All-atom simulations reveal the intricacies of signal transduction upon binding of the HLA-E ligand to the transmembrane inhibitory CD94/NKG2A receptor. J Chem Inf Model 63:3486–3499. https://doi.org/10.1021/acs.jcim.3c00249
Logtenberg MEW, Scheeren FA, Schumacher TN (2020) The CD47-SIRPα immune checkpoint. Immunity 52:742–752. https://doi.org/10.1016/j.immuni.2020.04.011
Lu F, Zhao Y, Pang Y, Ji M, Sun Y, Wang H, Zou J, Wang Y, Li G, Sun T, Li J, Ma D, Ye J, Ji C (2021) NLRP3 inflammasome upregulates PD-L1 expression and contributes to immune suppression in lymphoma. Cancer Lett 497:178–189. https://doi.org/10.1016/j.canlet.2020.10.024
Lyu G, Guan Y, Zhang C, Zong L, Sun L, Huang X, Huang L, Zhang L, Tian XL, Zhou Z, Tao W (2018) TGF-β signaling alters H4K20me3 status via miR-29 and contributes to cellular senescence and cardiac aging. Nat Commun 9:2560. https://doi.org/10.1038/s41467-018-04994-z
Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M (2004) The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol 25:677–686. https://doi.org/10.1016/j.it.2004.09.015
Marin I, Boix O, Garcia-Garijo A, Sirois I, Caballe A, Zarzuela E, Ruano I, Attolini CS, Prats N, Lopez-Dominguez JA, Kovatcheva M, Garralda E, Munoz J, Caron E, Abad M, Gros A, Pietrocola F, Serrano M (2023) Cellular senescence is immunogenic and promotes antitumor immunity. Cancer Discov 13:410–431. https://doi.org/10.1158/2159-8290.CD-22-0523
Martinez-Zamudio RI, Dewald HK, Vasilopoulos T, Gittens-Williams L, Fitzgerald-Bocarsly P, Herbig U (2021) Senescence-associated β-galactosidase reveals the abundance of senescent CD8+ T cells in aging humans. Aging Cell 20:e13344. https://doi.org/10.1111/acel.13344
Matsuda S, Revandkar A, Dubash TD, Ravi A, Wittner BS, Lin M, Morris R, Burr R, Guo H, Seeger K, Szabolcs A, Che D, Nieman L, Getz GA, Ting DT, Lawrence MS, Gainor J, Haber DA, Maheswaran S (2023) TGF-β in the microenvironment induces a physiologically occurring immune-suppressive senescent state. Cell Rep 42:112129. https://doi.org/10.1016/j.celrep.2023.112129
Mehdizadeh M, Aguilar M, Thorin E, Ferbeyre G, Nattel S (2022) The role of cellular senescence in cardiac disease: basic biology and clinical relevance. Nat Rev Cardiol 19:250–264. https://doi.org/10.1038/s41569-021-00624-2
Mitchell A, Malmgren L, Bartosch P, McGuigan FE, Akesson KE (2023) Pro-inflammatory proteins associated with frailty and its progression-a longitudinal study in community-dwelling women. J Bone Miner Res 38:1076–1091. https://doi.org/10.1002/jbmr.4861
Mizuno R, Sugiura D, Shimizu K, Maruhashi T, Watada M, Okazaki IM, Okazaki T (2019) PD-1 primarily targets TCR signal in the inhibition of functional T cell activation. Front Immunol 10:630. https://doi.org/10.3389/fimmu.2019.00630
Mylonas A, O’Loghlen A (2022) Cellular senescence and ageing: mechanisms and interventions. Front Aging 3:866718. https://doi.org/10.3389/fragi.2022.866718
Nakayama M, Akiba H, Takeda K, Kojima Y, Hashiguchi M, Azuma M, Yagita H, Okumura K (2009) Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood 113:3821–3830. https://doi.org/10.1182/blood-2008-10-185884
Nam S, Lee A, Lim J, Lim JS (2019) Analysis of the expression and regulation of PD-1 protein on the surface of myeloid-derived suppressor cells (MDSCs). Biomol Ther (seoul) 27:63–70. https://doi.org/10.4062/biomolther.2018.201
Nawas AF, Solmonson A, Gao B, DeBerardinis RJ, Minna JD, Conacci-Sorrell M, Mendelson CR (2023) IL-1β mediates the induction of immune checkpoint regulators IDO1 and PD-L1 in lung adenocarcinoma cells. Cell Commun Signal 21:331. https://doi.org/10.1186/s12964-023-01348-1
Nelson G, Wordsworth J, Wang C, Jurk D, Lawless C, Martin-Ruiz C, von Zglinicki T (2012) A senescent cell bystander effect: senescence-induced senescence. Aging Cell 11:345–349. https://doi.org/10.1111/j.1474-9726.2012.00795.x
Nelson G, Kucheryavenko O, Wordsworth J, von Zglinicki T (2018) The senescent bystander effect is caused by ROS-activated NF-κB signalling. Mech Ageing Dev 170:30–36. https://doi.org/10.1016/j.mad.2017.08.005
Nunez SY, Ziblat A, Secchiari F, Torres NI, Sierra JM, Raffo Iraolagoitia XL, Araya RE, Domaica CI, Fuertes MB, Zwirner NW (2018) Human M2 macrophages limit NK cell effector functions through secretion of TGF-β and engagement of CD85j. J Immunol 200:1008–1015. https://doi.org/10.4049/jimmunol.1700737
Ogrodnik M, Miwa S, Tchkonia T, Tiniakos D, Wilson CL, Lahat A, Day CP, Burt A, Palmer A, Anstee QM, Grellscheid SN, Hoeijmakers JHJ, Barnhoorn S, Mann DA, Bird TG, Vermeij WP, Kirkland JL, Passos JF, von Zglinicki T, Jurk D (2017) Cellular senescence drives age-dependent hepatic steatosis. Nat Commun 8:15691. https://doi.org/10.1038/ncomms15691
Ohta A (2016) A metabolic immune checkpoint: adenosine in tumor microenvironment. Front Immunol 7:109. https://doi.org/10.3389/fimmu.2016.00109
Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T (2001) PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc Natl Acad Sci U S A 98:13866–13871. https://doi.org/10.1073/pnas.231486598
Onorati A, Havas AP, Lin B, Rajagopal J, Sen P, Adams PD, Dou Z (2022) Upregulation of PD-L1 in senescence and aging. Mol Cell Biol 42:e0017122. https://doi.org/10.1128/mcb.00171-22
Osorio JM, Espinoza-Perez C, Rimassa-Tare C, Machuca V, Bustos JO, Vallejos M, Vargas H, Diaz-Araya G (2023) Senescent cardiac fibroblasts: a key role in cardiac fibrosis. Biochim Biophys Acta Mol Basis Dis 1869:166642. https://doi.org/10.1016/j.bbadis.2023.166642
Ouyang W, Rutz S, Crellin NK, Valdez PA, Hymowitz SG (2011) Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu Rev Immunol 29:71–109. https://doi.org/10.1146/annurev-immunol-031210-101312
Ovadya Y, Landsberger T, Leins H, Vadai E, Gal H, Biran A, Yosef R, Sagiv A, Agrawal A, Shapira A, Windheim J, Tsoory M, Schirmbeck R, Amit I, Geiger H, Krizhanovsky V (2018) Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat Commun 9:5435. https://doi.org/10.1038/s41467-018-07825-3
Paavola KJ, Roda JM, Lin VY, Chen P, O’Hollaren KP, Ventura R, Crawley SC, Li B, Chen HH, Malmersjö S, Sharkov NA, Horner G, Guo W, Kutach AK, Mondal K, Zhang Z, Lichtman JS, Song C, Rivera LB, Liu W, Luo J, Wang Y, Solloway MJ, Allan BB, Kekatpure A, Starck SR, Haldankar R, Fan B, Chu C, Tang J, Molgora M, Colonna M, Kaplan DD, Hsu JY (2021) The fibronectin-ILT3 interaction functions as a stromal checkpoint that suppresses myeloid cells. Cancer Immunol Res 9:1283–1297. https://doi.org/10.1158/2326-6066.CIR-21-0240
Park BV, Freeman ZT, Ghasemzadeh A, Chattergoon MA, Rutebemberwa A, Steigner J, Winter ME, Huynh TV, Sebald SM, Lee SJ, Pan F, Pardoll DM, Cox AL (2016) TGFβ1-mediated SMAD3 enhances PD-1 expression on antigen-specific T cells in cancer. Cancer Discov 6:1366–1381. https://doi.org/10.1158/2159-8290.CD-15-1347
Paul S, Lal G (2017) The molecular mechanism of natural killer cells function and its importance in cancer immunotherapy. Front Immunol 8:1124. https://doi.org/10.3389/fimmu.2017.01124
Pei L, Liu Y, Liu L, Gao S, Gao X, Feng Y, Sun Z, Zhang Y, Wang C (2023) Roles of cancer-associated fibroblasts (CAFs) in anti- PD-1/PD-L1 immunotherapy for solid cancers. Mol Cancer 22:29. https://doi.org/10.1186/s12943-023-01731-z
Pereira BI, Devine OP, Vukmanovic-Stejic M, Chambers ES, Subramanian P, Patel N, Virasami A, Sebire NJ, Kinsler V, Valdovinos A, LeSaux CJ, Passos JF, Antoniou A, Rustin MHA, Campisi J, Akbar AN (2019) Senescent cells evade immune clearance via HLA-E-mediated NK and CD8+ T cell inhibition. Nat Commun 10:2387. https://doi.org/10.1038/s41467-019-10335-5
Polverino F, Mirra D, Yang CX, Esposito R, Spaziano G, Rojas-Quintero J, Sgambato M, Piegari E, Cozzolino A, Cione E, Gallelli L, Capuozzo A, Santoriello C, Berrino L, de-Torres JP, HackettPolverino TLM, D’Agostiono B (2022) Similar programmed death ligand 1 (PD-L1) expression profile in patients with mild COPD and lung cancer. Sci Rep 12:22402. https://doi.org/10.1038/s41598-022-26650-9
Qwarnström EE, Ostberg CO, Turk GL, Richardson CA, Bomsztyk K (1994) Fibronectin attachment activates the NF-κB p50/p65 heterodimer in fibroblasts and smooth muscle cells. J Biol Chem 269:30765–30768
Ralainirina N, Poli A, Michel T, Poos L, Andres E, Hentges F, Zimmer J (2007) Control of NK cell functions by CD4+CD25+ regulatory T cells. J Leukoc Biol 81:144–153. https://doi.org/10.1189/jlb.0606409
Rapisarda V, Borghesan M, Miguela V, Encheva V, Snijders AP, Lujambio A, O’Loghlen A (2017) Integrin β3 regulates cellular senescence by activating the TGF-β pathway. Cell Rep 18:2480–2493. https://doi.org/10.1016/j.celrep.2017.02.012
Ravetch JV, Lanier LL (2000) Immune inhibitory receptors. Science 290:84–89. https://doi.org/10.1126/science.290.5489.84
Redondo-Garcia S, Barritt C, Papagregoriou C, Yeboah M, Frendeus B, Cragg MS, Roghanian A (2023) Human leukocyte immunoglobulin-like receptors in health and disease. Front Immunol 14:1282874. https://doi.org/10.3389/fimmu.2023.1282874
Ren LL, Miao H, Wang YN, Liu F, Li P, Zhao YY (2023) TGF-β as a master regulator of aging-associated tissue fibrosis. Aging Dis 14:1633
Roberts DD, Isenberg JS (2021) CD47 and thrombospondin-1 regulation of mitochondria, metabolism, and diabetes. Am J Physiol Cell Physiol 321:C201–C213. https://doi.org/10.1152/ajpcell.00175.2021
Roberts CJ, Birkenmeier TM, McQuillan JJ, Akiyama SK, Yamada SS, Chen WT, Yamada KM, McDonald JA (1988) Transforming growth factor β stimulates the expression of fibronectin and of both subunits of the human fibronectin receptor by cultured human lung fibroblasts. J Biol Chem 263:4586–4592
Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J (2009) Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol 11:973–979. https://doi.org/10.1038/ncb1909
Rong QX, Wang F, Guo ZX, Hu Y, An SN, Luo M, Zhang H, Wu SC, Huang HQ, Fu LW (2021) GM-CSF mediates immune evasion via upregulation of PD-L1 expression in extranodal natural killer/T cell lymphoma. Mol Cancer 20:80. https://doi.org/10.1186/s12943-021-01374-y
Ruhland MK, Loza AJ, Capietto AH, Luo X, Knolhoff BL, Flanagan KC, Belt BA, Alspach E, Leahy K, Luo J, Schaffer A, Edwards JR, Longmore G, Faccio R, DeNardo DG, Stewart SA (2016) Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat Commun 7:11762. https://doi.org/10.1038/ncomms11762
Sagiv A, Burton DG, Moshayev Z, Vadai E, Wensveen F, Ben-Dor S, Golani O, Polic B, Krizhanovsky V (2016) NKG2D ligands mediate immunosurveillance of senescent cells. Aging 8:328–344. https://doi.org/10.18632/aging.100897
Saini P, Adeniji OS, Abdel-Mohsen M (2022) Inhibitory siglec-sialic acid interactions in balancing immunological activation and tolerance during viral infections. EBioMedicine 86:104354. https://doi.org/10.1016/j.ebiom.2022.104354
Salminen A (2020) Activation of immunosuppressive network in the aging process. Ageing Res Rev 57:100998. https://doi.org/10.1016/j.arr.2019.100998
Salminen A (2021) Immunosuppressive network promotes immunosenescence associated with aging and chronic inflammatory conditions. J Mol Med (berl) 99:1553–1569. https://doi.org/10.1007/s00109-021-02123-w
Salminen A (2022) Clinical perspectives on the age-related increase of immunosuppressive activity. J Mol Med (berl) 100:697–712. https://doi.org/10.1007/s00109-022-02193-4
Salminen A (2024) The role of the immunosuppressive PD-1/PD-L1 checkpoint pathway in the aging process and age-related diseases. J Mol Med (berl) 102:733–750. https://doi.org/10.1007/s00109-024-02444-6
Salminen A, Huuskonen J, Ojala J, Kauppinen A, Kaarniranta K, Suuronen T (2008a) Activation of innate immunity system during aging: NF-κB signaling is the molecular culprit of inflamm-aging. Ageing Res Rev 7:83–105. https://doi.org/10.1016/j.arr.2007.09.002
Salminen A, Paimela T, Suuronen T, Kaarniranta K (2008b) Innate immunity meets with cellular stress at the IKK complex: regulation of the IKK complex by HSP70 and HSP90. Immunol Lett 117:9–15. https://doi.org/10.1016/j.imlet.2007.12.017
Salminen A, Kaarniranta K, Kauppinen A (2020) Exosomal vesicles enhance immunosuppression in chronic inflammation: Impact in cellular senescence and the aging process. Cell Signal 75:109771. https://doi.org/10.1016/j.cellsig.2020.109771
Salzer MC, Lafzi A, Berenguer-Llergo A, Youssif C, Castellanos A, Solanas G, Peixoto FO, Stephan-Otto Attolini C, Prats N, Aguilera M, Martin-Caballero J, Heyn H, Benitah SA (2018) Identity noise and adipogenic traits characterize dermal fibroblast aging. Cell 175:1575-1590.e22. https://doi.org/10.1016/j.cell.2018.10.012
Santoro A, Bientinesi E, Monti D (2021) Immunosenescence and inflammaging in the aging process: age-related diseases or longevity? Ageing Res Rev 71:101422. https://doi.org/10.1016/j.arr.2021.101422
Sappino AP, Buser R, Seguin Q, Fernet M, Lesne L, Gumy-Pause F, Reith W, Favaudon V, Mandriota SJ (2012) The CEACAM1 tumor suppressor is an ATM and p53-regulated gene required for the induction of cellular senescence by DNA damage. Oncogenesis 1:e7. https://doi.org/10.1038/oncsis.2012.7
Saresella M, Calabrese E, Marventano I, Piancone F, Gatti A, Farina E, Alberoni M, Clerici M (2012) A potential role for the PD1/PD-L1 pathway in the neuroinflammation of Alzheimer’s disease. Neurobiol Aging 33:624.e11–22. https://doi.org/10.1016/j.neurobiolaging.2011.03.004
Sauer N, Janicka N, Szlasa W, Skinderowicz B, Kolodzinska K, Dwernicka W, Oslizlo M, Kulbacka J, Novickij V, Karlowicz-Bodalska K (2023) TIM-3 as a promising target for cancer immunotherapy in a wide range of tumors. Cancer Immunol Immunother 72:3405–3425. https://doi.org/10.1007/s00262-023-03516-1
Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, Oberg AL, Birch J, Salmonowicz H, Zhu Y, Mazula DL, Brooks RW, Fuhrmann-Stroissnigg H, Pirtskhalava T, Prakash YS, Tchkonia T, Robbins PD, Aubry MC, Passos JF, Kirkland JL, Tschumperlin DJ, Kita H, LeBrasseur NK (2017) Cellular senescence mediates fibrotic pulmonary disease. Nat Commun 8:14532. https://doi.org/10.1038/ncomms14532
Schloesser D, Lindenthal L, Sauer J, Chung KJ, Chavakis T, Griesser E, Baskaran P, Maier-Habelsberger U, Fundel-Clemens K, Schlotthauer I, Watson CK, Swee LK, Igney F, Park JE, Huber-Lang MS, Thomas MJ, El Kasmi KC, Murray PJ (2023) Senescent cells suppress macrophage-mediated corpse removal via upregulation of the CD47-QPCT/L axis. J Cell Biol 222:e202207097. https://doi.org/10.1083/jcb.202207097
Seki M, Oomizu S, Sakata KM, Sakata A, Arikawa T, Watanabe K, Ito K, Takeshita K, Niki T, Saita N, Nishi N, Yamauchi A, Katoh S, Matsukawa A, Kuchroo V, Hirashima M (2008) Galectin-9 suppresses the generation of Th17, promotes the induction of regulatory T cells, and regulates experimental autoimmune arthritis. Clin Immunol 127:78–88. https://doi.org/10.1016/j.clim.2008.01.006
Shah K, Al-Haidari A, Sun J, Kazi JU (2021) T cell receptor (TCR) signaling in health and disease. Signal Transduct Target Ther 6:412. https://doi.org/10.1038/s41392-021-00823-w
Sharma N, Atolagbe OT, Ge Z, Allison JP (2021) LILRB4 suppresses immunity in solid tumors and is a potential target for immunotherapy. J Exp Med 218:e20201811. https://doi.org/10.1084/jem.20201811
Shekari N, Shanehbandi D, Kazemi T, Zarredar H, Baradaran B, Jalali SA (2023) VISTA and its ligands: the next generation of promising therapeutic targets in immunotherapy. Cancer Cell Int 23:265. https://doi.org/10.1186/s12935-023-03116-0
Shevitz J, Jenkins CS, Hatcher VB (1986) Fibronectin synthesis and degradation in human fibroblasts with aging. Mech Ageing Dev 35:221–232. https://doi.org/10.1016/0047-6374(86)90125-9
Shimizu A, Sawada K, Kobayashi M, Yamamoto M, Yagi T, Kinose Y, Kodama M, Hashimoto K, Kimura T (2021) Exosomal CD47 plays an essential role in immune evasion in ovarian cancer. Mol Cancer Res 19:1583–1595. https://doi.org/10.1158/1541-7786.MCR-20-0956
Shiravand Y, Khodadadi F, Kashani SMA, Hosseini-Fard SR, Hosseini S, Sadeghirad H, Ladwa R, O’Byrne K, Kulasinghe A (2022) Immune checkpoint inhibitors in cancer therapy. Curr Oncol 29:3044–3060. https://doi.org/10.3390/curroncol29050247
Shiri AM, Zhang T, Bedke T, Zazara DE, Zhao L, Lücke J, Sabihi M, Fazio A, Zhang S, Tauriello DVF, Batlle E, Steglich B, Kempski J, Agalioti T, Nawrocki M, Xu Y, Riecken K, Liebold I, Brockmann L, Konczalla L, Bosurgi L, Mercanoglu B, Seeger P, Küsters N, Lykoudis PM, Heumann A, Arck PC, Fehse B, Busch P, Grotelüschen R, Mann O, Izbicki JR, Hackert T, Flavell RA, Gagliani N, Giannou AD, Huber S (2024) IL-10 dampens antitumor immunity and promotes liver metastasis via PD-L1 induction. J Hepatol 80:634–644. https://doi.org/10.1016/j.jhep.2023.12.015
Sikora E, Bielak-Zmijewska A, Mosieniak G (2019) Targeting normal and cancer senescent cells as a strategy of senotherapy. Ageing Res Rev 55:100941. https://doi.org/10.1016/j.arr.2019.100941
Sikora E, Bielak-Zmijewska A, Mosieniak G (2021) A common signature of cellular senescence; does it exist? Ageing Res Rev 71:101458. https://doi.org/10.1016/j.arr.2021.101458
Singh L, Muise ES, Bhattacharya A, Grein J, Javaid S, Stivers P, Zhang J, Qu Y, Joyce-Shaikh B, Loboda A, Zhang C, Meehl M, Chiang DY, Ranganath SH, Rosenzweig M, Brandish PE (2021) ILT3 (LILRB4) promotes the immunosuppressive function of tumor-educated human monocytic myeloid-derived suppressor cells. Mol Cancer Res 19:702–716. https://doi.org/10.1158/1541-7786.MCR-20-0622
Sinning J, Funk ND, Soerensen-Zender I, Wulfmeyer VC, Liao CM, Haller H, Hinze C, Schmidt-Ott KM, Melk A, Schmitt R (2023) The aging kidney is characterized by tubuloinflammaging, a phenotype associated with MHC-II gene expression. Front Immunol 14:1222339. https://doi.org/10.3389/fimmu.2023.1222339
Slattery K, Woods E, Zaiatz-Bittencourt V, Marks S, Chew S, Conroy M, Goggin C, MacEochagain C, Kennedy J, Lucas S, Finlay DK, Gardiner CM (2021) TGFβ drives NK cell metabolic dysfunction in human metastatic breast cancer. J Immunother Cancer 9:e002044. https://doi.org/10.1136/jitc-2020-002044
Smith JR, Pereira-Smith OM (1989) Altered gene expression during cellular aging. Genome 31:386–389. https://doi.org/10.1139/g89-058
Sole-Boldo L, Raddatz G, Schütz S, Mallm JP, Rippe K, Lonsdorf AS, Rodriguez-Paredes M, Lyko F (2020) Single-cell transcriptomes of the human skin reveal age-related loss of fibroblast priming. Commun Biol 3:188. https://doi.org/10.1038/s42003-020-0922-4
Suda M, Paul KH, Minamino T, Miller JD, Lerman A, Ellison-Hughes GM, Tchkonia T, Kirkland JL (2023) Senescent cells: a therapeutic target in cardiovascular diseases. Cells 12:1296. https://doi.org/10.3390/cells12091296
Sun Y, Coppe JP, Lam EW (2018) Cellular senescence: The sought or the unwanted? Trends Mol Med 24:871–885. https://doi.org/10.1016/j.molmed.2018.08.002
Tabula Muris Consortium (2020) A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature 583:590–595. https://doi.org/10.1038/s41586-020-2496-1
Tadokoro T, Yamamoto K, Kuwahara I, Fujisawa H, Ikekita M, Taniguchi A, Sato T, Furukawa K (2006) Preferential reduction of the alpha-2-6-sialylation from cell surface N-glycans of human diploid fibroblastic cells by in vitro aging. Glycoconj J 23:443–452. https://doi.org/10.1007/s10719-006-7152-y
Takasugi M, Yoshida Y, Ohtani N (2022) Cellular senescence and the tumour microenvironment. Mol Oncol 16:3333–3351. https://doi.org/10.1002/1878-0261.13268
Tan J, Xue Q, Hu X, Yang J (2024) Inhibitor of PD-1/PD-L1: a new approach may be beneficial for the treatment of idiopathic pulmonary fibrosis. J Transl Med 22:95. https://doi.org/10.1186/s12967-024-04884-7
Tang Q, Chen Y, Li X, Long S, Shi Y, Yu Y, Wu W, Han L, Wang S (2022) The role of PD-1/PD-L1 and application of immune-checkpoint inhibitors in human cancers. Front Immunol 13:964442. https://doi.org/10.3389/fimmu.2022.964442
Tang D, Kang R, Zeh HJ, Lotze MT (2023) The multifunctional protein HMGB1: 50 years of discovery. Nat Rev Immunol 23:824–841. https://doi.org/10.1038/s41577-023-00894-6
Tarallo D, Martinez J, Leyva A, Monaco A, Perroni C, Tassano M, Gambini JP, Cappetta M, Duran R, Moreno M, Quijano C (2024) Mitofusin 1 silencing decreases the senescent associated secretory phenotype, promotes immune cell recruitment and delays melanoma tumor growth after chemotherapy. Sci Rep 14:909. https://doi.org/10.1038/s41598-024-51427-7
Thomas DA, Massague J (2005) TGF-β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 8:369–380. https://doi.org/10.1016/j.ccr.2005.10.012
Tominaga K, Suzuki HI (2019) TGF-β signaling in cellular senescence and aging-related pathology. Int J Mol Sci 20:5002. https://doi.org/10.3390/ijms20205002
Toor SM, Sasidharan Nair V, Decock J, Elkord E (2020) Immune checkpoints in the tumor microenvironment. Semin Cancer Biol 65:1–12. https://doi.org/10.1016/j.semcancer.2019.06.021
Tribulatti MV, Carabelli J, Prato CA, Campetella O (2020) Galectin-8 in the onset of the immune response and inflammation. Glycobiology 30:134–142. https://doi.org/10.1093/glycob/cwz077
Trzonkowski P, Szmit E, Mysliwska J, Mysliwski A (2006) CD4+CD25+ T regulatory cells inhibit cytotoxic activity of CTL and NK cells in humans-impact of immunosenescence. Clin Immunol 119:307–316. https://doi.org/10.1016/j.clim.2006.02.002
Tsou PS, Shi B, Varga J (2022) Role of cellular senescence in the pathogenesis of systemic sclerosis. Curr Opin Rheumatol 34:343–350. https://doi.org/10.1097/BOR.0000000000000898
van de Wall S, Santegoets KCM, van Houtum EJH, Büll C, Adema GJ (2020) Sialoglycans and siglecs can shape the tumor immune microenvironment. Trends Immunol 41:274–285. https://doi.org/10.1016/j.it.2020.02.001
van Eden W, Jansen MAA, Ludwig I, van Kooten P, van der Zee R, Broere F (2017) The enigma of heat shock proteins in immune tolerance. Front Immunol 8:1599. https://doi.org/10.3389/fimmu.2017.01599
van Tuyn J, Jaber-Hijazi F, MacKenzie D, Cole JJ, Mann E, Pawlikowski JS, Rai TS, Nelson DM, McBryan T, Ivanov A, Blyth K, Wu H, Milling S, Adams PD (2017) Oncogene-expressing senescent melanocytes up-regulate MHC class II, a candidate melanoma suppressor function. J Invest Dermatol 137:2197–2207. https://doi.org/10.1016/j.jid.2017.05.030
Varga J, Rosenbloom J, Jimenez SA (1987) Transforming growth factor β (TGF β) causes a persistent increase in steady-state amounts of type I and type III collagen and fibronectin mRNAs in normal human dermal fibroblasts. Biochem J 247:597–604. https://doi.org/10.1042/bj2470597
Vesely MD, Zhang T, Chen L (2022) Resistance mechanisms to anti-PD cancer immunotherapy. Annu Rev Immunol 40:45–74. https://doi.org/10.1146/annurev-immunol-070621-030155
Viel S, Marcais A, Guimaraes FS, Loftus R, Rabilloud J, Grau M, Degouve S, Djebali S, Sanlaville A, Charrier E, Bienvenu J, Marie JC, Caux C, Marvel J, Town L, Huntington ND, Bartholin L, Finlay D, Smyth MJ, Walzer T (2016) TGF-β inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci Signal 9(415):ra19. https://doi.org/10.1126/scisignal.aad1884
Wang D, DuBois RN (2015) Immunosuppression associated with chronic inflammation in the tumor microenvironment. Carcinogenesis 36:1085–1093. https://doi.org/10.1093/carcin/bgv123
Wang C, Jurk D, Maddick M, Nelson G, Martin-Ruiz C, von Zglinicki T (2009) DNA damage response and cellular senescence in tissues of aging mice. Aging Cell 8:311–323. https://doi.org/10.1111/j.1474-9726.2009.00481.x
Wang X, Guo Z, Ding Z, Khaidakov M, Lin J, Xu Z, Sharma SG, Jiwani S, Mehta JL (2015) Endothelin-1 upregulation mediates aging-related cardiac fibrosis. J Mol Cell Cardiol 80:101–109. https://doi.org/10.1016/j.yjmcc.2015.01.001
Wang TW, Johmura Y, Suzuki N, Omori S, Migita T, Yamaguchi K, Hatakeyama S, Yamazaki S, Shimizu E, Imoto S, Furukawa Y, Yoshimura A, Nakanishi M (2022a) Blocking PD-L1-PD-1 improves senescence surveillance and ageing phenotypes. Nature 611:358–364. https://doi.org/10.1038/s41586-022-05388-4
Wang X, Xiong H, Ning Z (2022b) Implications of NKG2A in immunity and immune-mediated diseases. Front Immunol 13:960852. https://doi.org/10.3389/fimmu.2022.960852
Wang B, Han J, Elisseeff JH, Demaria M (2024a) The senescence-associated secretory phenotype and its physiological and pathological implications. Nat Rev Mol Cell Biol. https://doi.org/10.1038/s41580-024-00727-x
Wang Y, Sun Y, Deng S, Liu J, Yu J, Chi H, Han X, Zhang Y, Shi J, Wang Y, Quan Y, Li H, Xu J (2024b) Discovery of galectin-8 as an LILRB4 ligand driving M-MDSCs defines a class of antibodies to fight solid tumors. Cell Rep Med 5:101374. https://doi.org/10.1016/j.xcrm.2023.101374
Wei X, Luo D, Yan Y, Yu H, Sun L, Wang C, Song F, Ge H, Qian H, Li X, Tang X, Liu P (2019) Kojic acid inhibits senescence of human corneal endothelial cells via NF-κB and p21 signaling pathways. Exp Eye Res 180:174–183. https://doi.org/10.1016/j.exer.2018.12.020
Wei Y, Liang M, Xiong L, Su N, Gao X, Jiang Z (2021) PD-L1 induces macrophage polarization toward the M2 phenotype via Erk/Akt/mTOR. Exp Cell Res 402:112575. https://doi.org/10.1016/j.yexcr.2021.112575
Weyand CM, Berry GJ, Goronzy JJ (2018) The immunoinhibitory PD-1/PD-L1 pathway in inflammatory blood vessel disease. J Leukoc Biol 103:565–575. https://doi.org/10.1189/jlb.3MA0717-283
Wicher SA, Roos BB, Teske JJ, Fang YH, Pabelick C, Prakash YS (2021) Aging increases senescence, calcium signaling, and extracellular matrix deposition in human airway smooth muscle. PLoS ONE 16:e0254710. https://doi.org/10.1371/journal.pone.0254710
Wolf Y, Anderson AC, Kuchroo VK (2020) TIM3 comes of age as an inhibitory receptor. Nat Rev Immunol 20:173–185. https://doi.org/10.1038/s41577-019-0224-6
Wu CM, Zheng L, Wang Q, Hu YW (2020) The emerging role of cell senescence in atherosclerosis. Clin Chem Lab Med 59:27–38. https://doi.org/10.1515/cclm-2020-0601
Wykes MN, Lewin SR (2018) Immune checkpoint blockade in infectious diseases. Nat Rev Immunol 18:91–104. https://doi.org/10.1038/nri.2017.112
Xiang Z, Yin X, Wei L, Peng M, Zhu Q, Lu X, Guo J, Zhang J, Li X, Zou Y (2024) LILRB4 checkpoint for immunotherapy: Structure, mechanism and disease targets. Biomolecules 14:187. https://doi.org/10.3390/biom14020187
Xu J, Zhou L, Liu Y (2020) Cellular senescence in kidney fibrosis: Pathologic significance and therapeutic strategies. Front Pharmacol 11:601325. https://doi.org/10.3389/fphar.2020.601325
Xu X, Masubuchi T, Cai Q, Zhao Y, Hui E (2021) Molecular features underlying differential SHP1/SHP2 binding of immune checkpoint receptors. Elife 10:e74276. https://doi.org/10.7554/eLife.74276
Xu P, Wang M, Song WM, Wang Q, Yuan GC, Sudmant PH, Zare H, Tu Z, Orr ME, Zhang B (2022) The landscape of human tissue and cell type specific expression and co-regulation of senescence genes. Mol Neurodegener 17:5. https://doi.org/10.1186/s13024-021-00507-7
Yanai H, Fraifeld VE (2018) The role of cellular senescence in aging through the prism of Koch-like criteria. Ageing Res Rev 41:18–33. https://doi.org/10.1016/j.arr.2017.10.004
Yang R, Sun L, Li CF, Wang YH, Yao J, Li H, Yan M, Chang WC, Hsu JM, Cha JH, Hsu JL, Chou CW, Sun X, Deng Y, Chou CK, Yu D, Hung MC (2021) Galectin-9 interacts with PD-1 and TIM-3 to regulate T cell death and is a target for cancer immunotherapy. Nat Commun 12:832. https://doi.org/10.1038/s41467-021-21099-2
Yi Z, Ren L, Wei Y, Chen S, Zhao J, Zhu J, Wu J (2023) Generation of a p21 reporter mouse and its use to identify and eliminate p21high cells in vivo. Int J Mol Sci 24:5565. https://doi.org/10.3390/ijms24065565
Yin Z, Yu M, Ma T, Zhang C, Huang S, Karimzadeh MR, Momtazi-Borojeni AA, Chen S (2021) Mechanisms underlying low-clinical responses to PD-1/PD-L1 blocking antibodies in immunotherapy of cancer: a key role of exosomal PD-L1. J Immunother Cancer 9:e001698. https://doi.org/10.1136/jitc-2020-001698
Yousefzadeh MJ, Zhao J, Bukata C, Wade EA, McGowan SJ, Angelini LA, Bank MP, Gurkar AU, McGuckian CA, Calubag MF, Kato JI, Burd CE, Robbins PD, Niedernhofer LJ (2020) Tissue specificity of senescent cell accumulation during physiologic and accelerated aging of mice. Aging Cell 19:e13094. https://doi.org/10.1111/acel.13094
Yu H, Sun L, Cui J, Li Y, Yan Y, Wei X, Wang C, Song F, Jiang W, Liu Y, Ge H, Qian H, Li X, Tang X, Liu P (2019) Three kinds of corneal host cells contribute differently to corneal neovascularization. EBioMedicine 44:542–553. https://doi.org/10.1016/j.ebiom.2019.05.026
Zhang Q, Vignali DA (2016) Co-stimulatory and co-inhibitory pathways in autoimmunity. Immunity 44:1034–1051. https://doi.org/10.1016/j.immuni.2016.04.017
Zhang Y, Cai P, Li L, Shi L, Chang P, Liang T, Yang Q, Liu Y, Wang L, Hu L (2017) Co-expression of TIM-3 and CEACAM1 promotes T cell exhaustion in colorectal cancer patients. Int Immunopharmacol 43:210–218. https://doi.org/10.1016/j.intimp.2016.12.024
Zhang Y, Zhu S, Du Y, Xu F, Sun W, Xu Z, Wang X, Qian P, Zhang Q, Feng J, Xu Y (2022) RelB upregulates PD-L1 and exacerbates prostate cancer immune evasion. J Exp Clin Cancer Res 41:66. https://doi.org/10.1186/s13046-022-02243-2
Zhang L, Pitcher LE, Prahalad V, Niedernhofer LJ, Robbins PD (2023) Targeting cellular senescence with senotherapeutics: senolytics and senomorphics. FEBS J 290:1362–1383. https://doi.org/10.1111/febs.16350
Zhao Y, Qu Y, Hao C, Yao W (2023) PD-1/PD-L1 axis in organ fibrosis. Front Immunol 14:1145682. https://doi.org/10.3389/fimmu.2023.1145682
Zheng M (2023) Systemic inflammation shapes clinical outcomes in response to immune checkpoint blockade treatment: moving toward optimizing antitumor immunity. J Immunother Cancer 11:e006462. https://doi.org/10.1136/jitc-2022-006462
Zhou H, Li N, Yuan Y, Jin YG, Wu Q, Yan L, Bian ZY, Deng W, Shen DF, Li H, Tang QZ (2020) Leukocyte immunoglobulin-like receptor B4 protects against cardiac hypertrophy via SHP-2-dependent inhibition of the NF-κB pathway. J Mol Med (berl) 98:691–705. https://doi.org/10.1007/s00109-020-01896-w
Zou Z, Long X, Zhao Q, Zheng Y, Song M, Ma S, Jing Y, Wang S, He Y, Esteban CR, Yu N, Huang J, Chan P, Chen T, Izpisua Belmonte JC, Zhang W, Qu J, Liu GH (2021) A single-cell transcriptomic atlas of human skin aging. Dev Cell 56:383-397.e8. https://doi.org/10.1016/j.devcel.2020.11.002
Acknowledgements
The author thanks Dr. Ewen MacDonald for checking the language of the manuscript.
Funding
Open access funding provided by University of Eastern Finland (including Kuopio University Hospital). The author declares that no funds, grants, or other forms of support were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
This article is the sole work of the author.
Corresponding author
Ethics declarations
Conflict of interest
The author declares that no conflict of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publish
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Salminen, A. Inhibitory immune checkpoints suppress the surveillance of senescent cells promoting their accumulation with aging and in age-related diseases. Biogerontology (2024). https://doi.org/10.1007/s10522-024-10114-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10522-024-10114-w