
Abstract
The microbial diversity associated with terrestrial groundwater seepage through permafrost soils is tightly coupled to the geochemistry of these fluids. Terrestrial alkaline methane seeps from Lagoon Pingo, Central Spitsbergen (78°N) in Norway, with methane-saturated and oxygen-limited groundwater discharge providing a potential habitat for methanotrophy. Here, we report on the microbial community’s comparative analyses and distribution patterns at two sites close to Lagoon Pingo’s methane emission source. To target methane-oxidizing bacteria from this system, we analysed the microbial community pattern of replicate samples from two sections near the main methane seepage source. DNA extraction, metabarcoding and subsequent sequencing of 16S rRNA genes revealed microbial communities where the major prokaryotic phyla were Pseudomonadota (42–47%), Gemmatimonadota (4–14%) and Actinobacteriota (7–11%). Among the Pseudomonadota, members of the genus Methylobacter were present at relative abundances between 1.6 and 4.7%. Enrichment targeting the methane oxidising bacteria was set up using methane seep sediments as inoculum and methane as the sole carbon and energy source, and this resulted in the isolation of a novel psychrophilic methane oxidizer, LS7-T4AT. The optimum growth temperature for the isolate was 13 °C and the pH optimum was 8.0. The morphology of cells was short rods, and TEM analysis revealed intracytoplasmic membranes arranged in stacks, a distinctive feature for Type I methanotrophs in the family Methylomonadaceae of the class Gammaproteobacteria. The strain belongs to the genus Methylobacter based on high 16S rRNA gene similarity to the psychrophilic species of Methylobacter psychrophilus Z-0021T (98.95%), the psychrophilic strain Methylobacter sp. strain S3L5C (99.00%), and the Arctic mesophilic species of Methylobacter tundripaludum SV96T (99.06%). The genome size of LS7-T4AT was 4,338,157 bp with a G + C content of 47.93%. The average nucleotide identities (ANIb) of strain LS7-T4AT to 10 isolated strains of genus Methylobacter were between 75.54 and 85.51%, lower than the species threshold of 95%. The strain LS7-T4AT represents a novel Arctic species, distinct from other members of the genus Methylobacter, for which the name Methylobacter svalbardensis sp. nov. is proposed. The type of strain is LS7-T4AT (DSMZ:114308, JCM:39463).
Similar content being viewed by others
Explore related subjects
Find the latest articles, discoveries, and news in related topics.Avoid common mistakes on your manuscript.
Introduction
Arctic permafrost is considered critically climate sensitive because temperature increases lead to thaw and higher microbial activity, organic carbon degradation, and increased emissions of the greenhouse gases methane and carbon dioxide (Schuur et al. 2013). In the Arctic, much methane is released into the atmosphere through ice-cored permafrost hills (Hodson et al.2019). These dome-shaped landforms forming on permafrost due to artesian pressure are called open systems pingos (Liestøl, 1977; Gurney 1998; Grosse and Jones 2010). These landforms occur in the lowlands of mountainous cold regions from pressurized water emerging from deep underground (Hammock, et al. 2022). The pingos are ice-cored from the partial or complete freezing of upwelling groundwaters near the land surface (Demidov et al. 2022). When freezing is incomplete, pingos develop springs that discharge biogenic or thermogenic methane (Hodson et al. 2020). Such open system pingos are common in central Svalbard and highly dynamic (Hjelle 1993). They are often found in clusters and are significant sources of groundwater discharge (Gurney 1998). These ubiquitous permafrost-diagnostic landforms’ formations and their internal structure still remain unclear (Hammock et al. 2022). Still, little is known about the origin of the subsurface groundwater and the distribution of methane; however, carbon isotope composition indicates that thermogenic methane mixes with microbial biogenic methane below the permafrost (Hodson et al. 2020). The methane enters the atmosphere by degassing spring water through diffusion and ebullition or by venting directly.
In Adventdalen, four out of six pingo structures described are characterized by uninterrupted annual groundwater discharge, releasing approximately 1040 kg of CH4 into the atmosphere annually (Hodson et al. 2020). Lagoon pingo is the most studied open-pingo system in Adventdalen, making it a model site for understanding such dynamic systems (Orvin 1944; Svensson 1970; Liestol 1976; Yoshikawa 1993; Yoshikawa and Harada 1995; Yoshikawa and Nakamura 1996). Lagoon pingo is the youngest pingo system in Adventdalen, estimated to be about 160 ± 20 years old (Yoshikawa and Nakamura 1996), and is still active. It is situated close to Adventfjorden yet protected from the tides by Moskuslagunen and is composed by three crater ponds discharging groundwater enriched with methane (Hodson et al.2019). In methane-rich environments, two different types of biological methane oxidation occur depending on oxygen availability. Under anaerobic conditions, consortia of anaerobic methane-oxidizing archaea (ANME) and sulfate-reducing bacteria oxidize methane using sulfate as an electron acceptor. In contrast, under aerobic conditions, methane-oxidizing bacteria (MOB) or methanotrophs can utilize methane, either mixotrophically or as their sole source of carbon and energy, with oxygen as an electron acceptor (Knief 2015).
MOB constitute a ubiquitous group of bacteria that share the distinctive ability to metabolize methane (Hanson and Hanson 1996). So far, these bacteria are shown to be phylogenetically affiliated with the phyla of Pseudomonadota (Houghton et al. 2019), Verrucomicrobia (Camp et al. 2009; Islam et al. 2008; Dunfield et al. 2007), and Actinobacteriota (van Spanning et al. 2022). Most methanotrophic bacteria are mesophilic and neutrophilic organisms isolated from diverse extreme environments (Trotsenko and Khmelenina 2002). Psychrophilic, mesophilic and thermophilic methanotrophic bacteria are known to be found in distinct genera of the Gammaproteobacteria class (Knief 2015, 2019). MOBs are critical representatives in the CH4 cycle with a massive influence on CH4 fluxes. They are often found living at the oxic-anoxic interfaces in CH4-emitting ecosystems such as wetlands (Chowdhury and Dick 2013; Danilova et al. 2013; Danilova and Dedysh 2014), lakes (Bowman et al. 1997; Costello and Lindstrom (1999), marine sediments (Deutzmann et al. 2011; Dumont et al. 2011), landfills (Chen et al. 2007), bogs (Dedysh 2002; Dedysh et al. 2000; Belova et al. 2011, 2013) and rice fields (Knief 2015). Many MOBs, especially within Gammaproteobacteria, are obligate methanotrophs oxidizing CH4 for biomass formation and CO2 generation (Knief 2015), while others are facultative methylotrophs, capable of using other carbon and energy sources such as acetate, methanol, or ethanol (i.e. Methylocystis and Methylocella, (Dedysh et al. 2004; Vorobev et al. 2011; Im et al. 2011) and even gases such as H2 and CO2 (Tveit et al. 2019).
Two psychrophilic methanotrophic species, Methylosphaera hansonii (Bowman et al. 1997) and Methylobacter psychrophilus Z-0021 T (Omelchenko et al. 1996), from the family Methylomonadaceae have been characterized and isolated from surface sediments of an Antarctic meromictic lake and from Russian Arctic tundra soil, respectively. Only very few genome sequences of psychrophilic methane oxidizers have been reported. Recently, the genome of the psychrophilic tundra soil strain M. psychrophilus Z-0021T (DSM 9914) was sequenced (Rissanen et al. 2022). An obligate psychrophilic methanotroph in the genus Methylobacter, retrieved from a boreal lake in Finland, has also been characterized by full genome sequencing (Khanongnuch et al. 2022, 2023). Other psychrophilic or psychrotolerant isolates have been reported from tundra soil, permafrost environments, arctic wetlands, saline meromictic lakes, polar lakes, wet plant material, Arctic thermal spring water and sediments in maritime Antarctica (Trotsenko et al. 2005; Bowman et al. 1997; Wartiainen et al. 2006; Oshkin et al. 2016; Mateos-Rivera et al. 2018; Islam et al. 2020; Roldán and Menes 2023).
For the detection and diversity analysis of C1-utilizing bacteria, several functional marker genes are commonly used, such as pmoA (encoding a subunit of the particulate methane monooxygenase, pMMO: a copper-dependent enzyme), mmoX (encoding a subunit of the soluble methane monooxygenase, sMMO: an iron-dependent enzyme), mxaF (encoding the large subunit of PQQ-dependent methanol dehydrogenase, MDH: a calcium-containing enzyme) and cbbL (encoding the large subunit RuBisCo for autotrophic CO2 fixation). The pmoA gene is the most frequently applied phylogenetic marker to distinguish aerobic methanotrophs from other bacteria in different ecosystems (McDonald et al. 2008; Lau et al. 2013).
The genus Methylobacter, belonging to the family Methylomonadaceae (Type Ia), was initially proposed by Bowman and collaborators in 1993 (Bowman et al. 1993). Currently, the genus now contains 8 validly published species (Collins et al. 2015 and 2017). All members of the genus Methylobacter are strictly aerobic, rod-shaped, and capable of aerobically oxidizing methane to carbon dioxide. Moreover, cells assimilate carbon by using the ribulose monophosphate (RuMP) pathway and possess an arranged stack membrane system in its cell compartment (i.e. a Type I intracytoplasmic membrane). Until now, none of the reported species of the genus Methylobacter has been found to maintain soluble methane monooxygenases (sMMO) (Houghton et al. 2019). Methylobacter species generally produce pink, yellow, or white color colonies on solid medium and have been isolated from various environments like wetland soil, sediments, freshwater, and rumen (Wartiainen et al. 2006; Whittenbury et al. 1970; Finn et al. 2012; Khatri et al. 2021).
Here, we report on the microbial community’s comparative analyses and distribution patterns with emphasis on methane oxidizing-bacteria at two different sites close to the methane emission source in Lagoon Pingo. We used sediment samples from Lagoon Pingo, where methane is enriched, and oxygen-limited groundwater is discharged continuously, forming crater ponds (Hodson et al.2019). Initially, molecular community analyses at this site revealed a distinct and unusual methanotrophic community assemblages across hydrological transitions (Fåne 2020). In one of our enrichments, we recovered an extant cold-adapted, obligate psychrophilic bacterium, which was assigned LS7-T4AT. This isolate showed high 16S rRNA gene sequence similarity to members of the genus Methylobacter and low similarity to other methanotrophic genera in the family Methylomonadaceae. For further verification of the taxonomic position of this strain, a polyphasic characterisation and a genomic overview were implemented to give valid evidence of the novelty of this new isolate.
Materials and methods
Site description and sample collection
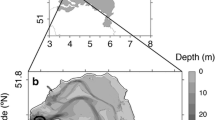
Sediment samples were collected from Lagoon Pingo in Adventdalen Valley, located on the Northern side of Adventdalen River close to Longyearbyen, Svalbard (78°14.403′N, 15°45.281′E) in early August 2019 (Fig. 1a). The pingo is separated from tidal waters by the Moskuslagoon near the coastline. The site is an open pingo system and collapses as a shallow crater lake during summer (Fig. 1b) and builds up as an icy hill during winter and spring (Fig. 1c). The system consists of several elevated mounds with craters spanning 500 m in length, 150 m in width, and up to 10 m in height (Yoshikawa and Nakamura 1996).

Sample site showing the location of Lagoon Pingo indicated with a red circle situated close to Moskuslagoon near Adventdfjorden in Svalbard at 78°N a shows the geographical location of Lagoon Pingo on map taken from toposvalbard b picture taken during summer 2021 showing the Shallow Lake with crater-like structure c image showing elevated Ice-hill structure covered in snow during winters
Lagoon Pingo has three active springs (Hodson et al. 2020). The samples used in this study were collected from the accessible dry sediment area at the rim of the active spring at Lagoon Pingo (Figs. 1b, 2). During autumn, when samples were collected, the structure of the site consisted of a still pond (SP) with methane seeps identified by ebullition to the surface. The main methane spring was situated in the centre, discharged water saturated with methane, and a temperature of 0.5 °C at the surface (Fig. 2).

A schematic drawing of Lagoon Pingo showing the still pond (SP) in light blue and the mini source (MS) in light pink, surrounded by dry sediment indicated with the grey color. Samples from the transect T, which is indicated as a straight line with five methane measurement and sampling locations, was used in this study. The SP consists of a water-logged methane spring at TC1 with a high methane flux indicated by blue bubbles. The diamond shapes indicate where methane flux chambers were located, starting from the centre towards the dry sediment zone. At the locations of the methane flux chambers, sample collection was also performed (Fåne 2020). Samples from the locations of chambers TC2 and TC4, shown in violet diamond shapes, were included in the current study. The temperatures of the collected material at TC2 and TC4, were 10.0 and 5.4 °C, respectively
The sediments used were collected from transect 2 (T) from the methane spring from the middle of the pond into the rim, where an additional “mini methane source” (MS) was located (shown in Fig. 2) resulting in five sampling points labelled as the chamber (C1–C5). Replicate samples were taken from TC2 closer to the main spring from the still pond (SP) with a temperature of 10 °C and from TC4 with small discharges called mini source (MS) where the temperature was 5 °C, at a depth of 10 cm. The recorded pH on the site was 9, which indicates an alkaline environment. Sediment samples collected for nucleic acid extraction were immediately frozen in a portable dry shipper (Air Liquide, Paris, France) on site (< − 150 °C). pH measurements were performed in 1:5 dilution (sediment:MilliQ water). Samples for enrichment were stored cold at 4 °C in a sterile serum vial with thick rubber septum. Samples were kept cold during transportation, until further processing in the laboratory at University of Bergen, Norway.
Methane fluxes
Net methane fluxes were measured using custom-made, static acrylic glass chambers (3603 cm3 inner volume) in combination with a recirculating multiplexer (eosMX-P, Eosense, Dartmouth, Canada) and OA-ICOS ultraportable greenhouse gas analyser (U-GGA-915, Los Gatos Research, San José, USA). Inert, gas-tight perfluoro alkoxy alkanes polyurethane tubing was used as a gas line for the gas transfer between chambers, multiplexer, and greenhouse gas analyser. Before each methane flux measurement, the setup was flushed with ambient air. Depending on the magnitude of methane fluxes, measuring time amounted to 5 min. During each flux measurement, chamber temperature and pressure were monitored using a temperature logger (HOBO MX2201, Onset, Cape Cod, USA) and a manometer (Leo1, Keller AG, Winterthur; Switzerland). The net methane flux estimates were determined by linear regression implemented in the eosAnalyse-AC program (Version 3.7.9, Eosense, Dartmouth, Canada). The accuracy and consistency of the greenhouse gas analyser were periodically checked by referencing ambient air and a standard gas (1000 ppm methane in N2).
DNA extraction, sequencing of 16S rRNA gene, and assignment of taxonomy
Environmental DNA (eDNA) was extracted using the DNeasy PowerSoil Kit (QIAGEN, 12,888–100, Germany) using manufacturers protocol. Extracted DNA was quantified using a high-sensitivity kit of Qubit 2.0 Fluorometer (Invitrogen, Singapore) following the manufacturer’s instructions and then stored at − 20 °C. The eDNA was amplified by targeting the highly conserved V4 -region of the 16S rRNA gene by using nested polymerase chain reaction (PCR) as previously described (Wilson et al. 2017). The 16S rRNA gene amplicon libraries were sequenced on an Illumina MiSeq platform (Norwegian Sequencing Centre, Oslo).
The demultiplexed pairedend fastq sequences were analysed using the DADA2 pipeline (Divisive Amplicon Denoising Algorithm 2, Callahan et al. 2016) using default parameters. Sequence qualities were verified using plotQualityProfile and low-quality reads were removed using filter and Trim function. Primers were removed using the cutadapt function. The core method from DADA2 packages was applied using multithreads to infer the composition of samples. Paired readwere merged to obtain full denoised sequences (merged sequences) using dereplication function. amplicon sequence variants (ASVs) table was made using Seqtab. Chimeric sequences were removed from merged reads using nochim function and taxonomy assigned to the ASVs using the assign taxonomic function in DADA2 package which is based on naïve Bayesian classifier method and SILVA reference database (Quast et al. 2012). ASVs showing bootstrap values above 90 were included in further analyses and subsamples were presented in the pie charts. The statistical analysis used the online MicrobiomeAnalyst platform (Dhariwal et al. 2017).
Enrichment and isolation of aerobic methanotroph
To enrich for isolation of methanotrophic bacteria, 2 g sediment from mini source (MS) was selected and inoculated in 20 mL low-salt mineral media (both LMM: added vitamin solution and LMA: without vitamin solution) in 120 mL serum vial closed with a sterile butyl rubber septum and sealed with aluminium crimps. The pH was adjusted to 8.5, and the substrate for growth was a sterile mixture of methane (80%) and air (20%) in the headspace (purity of methane, 99.5%, Yara Praxair, Oslo, Norway) as previously described for LMM and LMA medium (Islam et al. 2015; Islam et al. 2016). The bottle was incubated at 10 °C for 4 weeks in the dark, without shaking. The gas mixture was substituted every 15 days. When the enrichment culture became visibly turbid, they were checked for cell growth using phase-contrast microscopy (Eclipse E400 microscope, Nikon Corporation, Tokyo, Japan). Two mL of primary enrichment cultures were transferred to fresh LMM and LMA media and re-incubated under the same conditions. To recover a factual aerobic methane oxidiser, the enriched sample was transferred five times in fresh media and incubated with a combination of methane and air. Serial dilutions (10−6 to 10−8) were prepared, and 0.1 mL aliquots were spread onto agar plates (Difco) containing LMA medium. The plates were incubated for 5 weeks at 15 °C in gas-tight jars filled with methane gas and air in a 2:1 mixture. One single colony was then selected, re-streaked onto fresh agar plates, and re-incubated for 5 weeks. After the pure culture was obtained, LMA was used for its routine cultivation at 10 and 15 °C at pH 8.0 for 2 weeks. The purity of the cell culture was again checked by phase-contrast microscopy. A heterotrophic contamination test used glucose (10 mM), yeast extract (5%), and R2A agar plates.
Phylogenetic classification
The cells of strain LS7-T4AT were targeted for the amplification of genes, including 16S rRNA genes, pmoA, mmoX, mxaF, nifH, cbbL and mauA using specific primers (list of primers given in Table S1) and positive amplification products sequenced as described previously (Islam et al. 2020)0.16S rRNA gene sequences and protein sequences of the pmoA gene inferred from PCR products using the ExPASy Translate tool (Artimo et al. 2012) were compared to available sequences from the GenBank database using the NCBI tools of Blastn and Blastp, respectively. Phylogenetic trees of both 16S rRNA and pmoA genes were reconstructed using the neighbor-joining (NJ) and maximum likelihood (ML) in MEGA software version 7.0 (Kumar et al. 2016).
Physiology and TEM characterization
Different organic substrates (glucose, acetate, pyruvate, lactate, malate, succinate, and ethanol) were tested at concentration of 10 mM in fresh LMA (Islam et al. 2008). Growth on methanol, methylamine, formate, and formaldehyde were examined at concentrations from 0.03 to 0.2% (v/v) in an LMA medium. Moreover, growth was tested with nitrogen-free LMA (without KNO3 or NH4Cl) adjusted to pH 8.0 in triplicates, where the only nitrogen source was N2 from the air. The samples were monitored during the incubation time and observed every week for visible growth. After 2 weeks of incubation, growth could be observed. Salt tolerance was determined by adding different concentrations of NaCl (0.1, 0.5, 1.0, 2.0, and 3.0% w/v) to the LMA medium. After 2 weeks of incubation the turbidity of each sample was assessed at 600 nm using a spectrophotometer. The generation time and the growth rate (µ) at 15 °C and pH 8.0 on methane were determined from the exponential growth phase. Growth measurements were recorded after 2 weeks of incubation. To determine optimum temperature for growth, the culture was incubated at 0, 2, 5, 8, 10, 13, 15, 18, 20, 22, 25 and 30 °C (at pH 8.0) with methane as the only available carbon source. The influences of pH on growth were recorded, and antibiotic sensitivity of strain LS7-T4AT was examined at the optimum temperature of 15 °C and pH 8.0 as previously described (Islam et al. 2020). The morphology was studied using phase-contrast microscope, and the internal structures of pure cells were evaluated using transmission electron microscopy (TEM, Hitachi HT7800) as described by Islam et al. (2015).
Metagenome sequencing, assembly, and annotation
Total genomic DNA was extracted from strain LS7-T4AT using GenElute Genomic DNA kit (Sigma), and the metagenome was sequenced using short-read Illumina sequencing platform (Illumina Novoseq 6000 platform: Novogen Co. Ltd., Cambridge, UK). Library preparation, sequencing for short read and annotation was done at Novogene Co. Ltd. To ensure the accuracy and reliability of the subsequent information analysis results, the original data were filtered by the step of quality control using the Novogen compiling pipeline. The genome was assembled using defalt K-mer of three different softwares (1) SOAP denovo version 2.04 (Li et al. 2010) (2) SPAdes (Bankevich et al. 2012a, b) (3) Abyss (Simpson et al. 2009). The assembly results of the three softwares were integrated with CISA software (Lin and Liao 2013) and the assembly result with the least scaffolds was selected. The genome was subjected for prediction of the coding gene using GeneMarkS (Besemer et al. 2001) Transfer RNA (tRNA) genes were predicted by tRNAscan-SE (Lowe et al. 1997). Ribosomal RNA (rRNA) genes were analyzed by the rRNAmmer (Lagesen et al. 2007). Small nuclear RNAs (snRNA) were predicted by Rfam (Gardner et al. 2009). Among several database for gene prediction KEGG is Kyoto Encyclopedia of Genes and Genomes (Kanehisa et al. 2004) and COG Clusters of Orthologous Groups) were used for functional annotation and investigation of the metabolic potential.
Metagenome analyses, genome identity, and phylogeny
The phylogenomic tree was constructed based on the 16 whole genomes from the Methylomonadacecae family, which was created using the automated codon tree method in V-BRC Patric using protein homology groups and coding DNA from single-copy genes (Wattam et al. 2014; Davis et al. 2020). The genome identity analysis was done using ANI, AAI, and GGDC. The average nucleotide identity (ANI) values amid strain LS7-T4AT and other associated species in the genus Methylobacter were calculated using JSpeciesWS (Richter et al. 2016) which is a web server for prokaryotic species circumscription based on pairwise genome comparison (Richter et al. 2016). Additionally, digital DNA-DNA hybridization (dDDH) values between strain LS7-T4AT and other related species in the genus Methylobacter were acquired using the Genome-to-Genome Distance Calculator (GGDC) (Auch et al. 2010) using the method described by Meier-Kolthoff et al. (2013).
Culture deposition and nucleotide sequence submission
The GenBank accession numbers for the sequences of the16S rRNA genes of strain LS7-T4AT is OQ832782. The raw reads of 16S rRNA amplicon Illumina sequence data submitted in sequence read archive (SRA) accession numbers BioProject ID PRJNA1024519 in GenBank and draft genome sequence under the BioProject ID: PRJNA1024098.
Results
Microbial community diversity
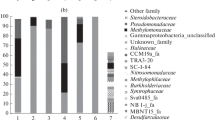
Samples from two locations in Lagoon Pingo, the mini source (MS) and still pond (SP), were used in this study. Each location was characterized by proximity to Lagoon Pingo’s central subsurface water discharge. Both locations were close to the primary water source (MS, 7.61 m and SP, 3.71 m) with differences in temperature (MS, 8.4 °C and SP, 11.4 °C) and methane fluxes (MS, 687.5 and SP, 1.5 nmol m2 s−1), but not water content (both waterlogged). The 16S rRNA genes were amplified and sequenced in DNA samples from four replicates for each location. A total of 2018 bacterial ASV (Amplicon Sequence Variants) were identified in the eight samples. All assigned ASVs belonged to the bacterial domain. The microbial communities were dominated by Pseudomonadota phylum with 47% and 42%, followed by Bacteroidota at 24% and 12%, Actinobacteriota with 11% and 7%, Acidobacteriota with 5% and 7% followed by Gemmatimonadota 4% and 14% relative abundance in SP and MS, respectively, as shown in Fig. 3a. The MS source was dominated by the families of Pseudomonadota phylum Burkholderiaceae followed by Hydrogenophilaceae and the still pond was dominated by Hydrogenophilaceae followed by Flavobacteriaceae (Bacteroidota phylum) (Fig. S1). Further resolution of Pseudomonadota phylum showed the dominance of Gammaproteobacteria, composed of the three common genera Thiobacillus (57.5% and 66.0%), Methylobacter (4.7% and 1.6%), and JTB255_marine_benthic_group (3.0% and 0.9%) in MS and SP respectively Fig. 3b.

Microbial community composition in the two sediments samples mini source (MS) and still pond (SP) from Lagoon Pingo based on high throughput metabarcoding and Illumina sequencing. The pie charts show relative abundance at a the phylum level and b at the genus level, selectively showing distribution within the phylum Pseudomonadota
Enrichment, isolation, and classification
Using sediment samples from the mini source, the enriched cultures grew after 5 weeks of incubation at 10 °C with methane as the only carbon source in LMA medium. Cells observed under phase contrast microscope were dominated by short rod-shaped cells with mucus-like capsules, with few coccoid cells and small thin rod cells after four consecutive transfers. Two distinct types of colonies were found after plating the enrichments on LMA agar plates. One of the colony types consisted of small white colonies about 0.4–0.6 mm in diameter; the other colonies were light pink coloured colonies about 1.8–2.5 mm in diameter. Under the microscope, the white colonies consisted of small rod-shaped cells, and the pink colonies were a mixture of coccoid and rod-shaped cells. Only the pink colonies sustained growth on methane after 5 weeks of incubation (Fig. 4). The pink colonies were used for further physiological and phylogenetic characterisation. The isolate was designated LS7-T4AT, which grew on methane or methanol as the sole carbon and energy source. No growth was found on multi-carbon substrates like glucose, acetate, succinate, malate, lactate, pyruvate ethanol, yeast extract, or R2A agar plates. The growth on different substrates confirmed that the strain was an obligate aerobic MOB that could grow on methanol. The purity of strain LS7-T4AT was confirmed by microscopy in addition to 16S rRNA gene sequencing.

Morphological characteristics of the strain LS7-T4AT a image of LMA agar plate showing light pink colonies Indicated with large arrow heads) and small white colonies (indicated with small arrow heads). The light pink colonies were examined under microscope and micrographs shown in b and phase- contrast micrograph of live cells showed rod-shaped bacteria. c Electron micrograph of a cross-section showing the internal characteristic of the cells (i– iii). White arrow (i) indicates the intracytoplasmic membranes (ICM) arranged in stacks, black arrow (ii) shows the cell wall (CW) and white arrow (iii) point to inclusion bodies observed as tiny white sacs under the microscope. Scale bar is 500 nm
Phylogenetic analysis revealed that the 16S rRNA genes sequence cluster within the genus Methylobacter and the nearest cultivated species is Methylobacter tundripaludum SV96T, with a sequence identity at 99.06%. The second closest match was Methylobacter S3L5C with a sequence identity of 99.00% (Fig. 5). The pmoA gene (particulate methane monooxygenase subunit A), used as a biomarker gene for defining methanotrophic bacteria, was aligned and based on their gene sequences a phylogenetic tree was constructed (Fig. 6).

Neighbour-Joining (NJ) phylogenetic tree of strain LS7-T4AT from Lagoon Pingo (showed in bold red) based on the analyses of the 16S rRNA gene using the Kimura 2-parameter model showing phylogenetic relationship related to strains from genus Methylobacter and other cultured and uncultured Type Ia and Type Ib genera from Methylomonadaceae and Methylococcaceae family. The Type IIb methanotroph, Methylocapsa acidiphila B2T (AJ278726) of the family Beijerinkiaceae, was used as an outgroup. The number displayed at the branches refer to NJ-bootstrap values

Neighbour-joining (NJ) phylogenetic tree derived using the pmoA gene amino acid sequences, based on Dayhoff matrix-based model showing the position of the strain LS7-T4A (showed in bold red) and other related Type Ia and Type Ib methanotrophs. The Type IIb methanotroph, Methylocapsa acidiphila B2T (AJ278727) of the family Beijerinkiaceae, was used as an outgroup. The number displayed at the branches refers to NJ-bootstrap values
Physiological and TEM features
The LS7-T4AT isolate had an optimum growth temperature of 13 °C at pH 8.0 (Table S2). The growth rate declined after 13 °C, and no growth was observed at 25 °C (Fig. S2). The optimal pH was pH 8.0, and no growth was recorded at pH 6.0 and 9.5. Growth was inhibited when NaCl concentrations exceeded 0.5% (w/v). Growth was not achieved under aerobic conditions in the absence of methane or under anaerobic conditions in the presence of methane. Multi-carbon substrates prevented the growth of strain LS7-T4AT. Moreover, the strain was able to grow with low methanol concentrations, between 0.05 and 0.5%. The generation time and growth rate (µ) when growing on methane was 19 h and 0.016 h−1, respectively. All antibiotics tested inhibited the growth of LS7-T4AT. Ammonia and nitrate compounds were used by cells as nitrogen sources. Vitamins were not found to be required for growth. The strain was able to grow on nitrogen-free LMA and LMM, indicating the ability to fix atmospheric N2. Still optimal growth was observed on LMA (containing NH4Cl) compared to LMM (containing KNO3). These observations were also supported by positive amplification of the nifH gene (Fig. S3). The strain was non-motile, and the cells multiply by binary fission. No flagella were visible by transmission electron microscopy. The ultrathin sections in TEM analysis showed the presence of extensive intracytoplasmic membranes, close-packed in vesicular disks (as stacked), which is a typical feature of the family Methylomonadaceae (Fig. 4c).
Genome features of methylobacter strain LS7-T4AT
To better understand the genome feature and metabolism of strain LS7-T4AT the genome was sequenced, and draft genome assembled. The metagenome-assembled genome was constructed using 220 contigs giving a total size of 4,338,157 bp. The genome indicated 99.7% completeness with only 0.845% contamination. The GC content was 47.93%, and the genome included one rRNA operon, 41 tRNAs, and 4271 total number of genes. Genome features are summarized in Table 1.
Of the predicted genes, 84.85% were assigned to Clusters of Orthologous Groups (COGs). The genome of strain LS7-T4AT had one complete sequence of 16S rRNA gene sequence, with 1530 bp on scaffold no.112 and the pmoA gene (particulate methane monooxygenase subunit A), was complete 744 bp) and present in the genome on scaffold no 17 (Fig. 7).

Gene organization of particulate methane monooxygenase gene pmoCAB in strain LS7-T4AT compared to four other species of the genus Methylobacter
The average nucleotide identity (ANI) values were determined using online resources JspeciesWS (Richter et al. 2016) were 85.51% with M. tundripaludum SV96T and 75.63% Methylobacter S3L5C. The dDDH values were 31.70% with M. tundripaludum SV96T and 22.40% Methylobacter S3L5C (Table 2). The percent identity of the sequence was 97.57% with M. tundripaludum SV96T and 97.98% with M. psychrophilus. ANI values below 95–96% for Bacteria and Archaea can be considered as a novel species (Yoon et al. 2017).
To validate the novelty of the LS7-T4AT strain in the genus Methylobacter, the genomic tree with reported Methylobacter strains was constructed using BV-BRC Patric phylogenomic function (Davis et al. 2020). The phylogenomic analysis of the LS7-T4AT showed that strain M. tundripaludum SV96T and Ca. Methylobacter oryzae were the closest relatives, followed by strain Methylobacter S3L5C (Fig. 8).

Phylogenetic tree of strains from Methylomonadacacae family relative to isolated strain LS7-T4AT (in bold red) constructed by the Codon Tree method used on BV-BRC online service. The tree was based on the total number of 16 genomes, 100 aligned proteins, 45,521 amino acids and 136,563 aligned nucleotides and 100 CDS from the genomes
Predicted metabolic potential
Methanotrophy
The draft genome analysis of strain Methylobacter LS7-T4AT revealed that genes for pmoCAB of the particulate membrane-bound methane monooxygenase (pMMO), the first step of converting methane to methanol were detected. We did, however, not detect the soluble methane monooxygenase (sMMO) coding gene clusters. The enzymes in subsequent steps of the CH4 oxidation pathways transforming methanol to formaldehyde were also revealed (Fig. 9). The genome contained genes encoding subunits of the Ca-dependent methanol dehydrogenase (mxaFI) but lacked genes encoding the lanthanide-containing pyrroloquinoline quinone (PQQ) dependent methanol dehydrogenase (xoxF).

Cellular putative metabolic pathways reconstructed from the genome of Methylobacter LS7-T4AT. The carbon pathways are shown in circles and the genes involved in methane oxidation are shown in yellow boxes pMMO (particulate methane monooxygenase), MDH (methanol dehydrogenase), fae (5,6,7,8-tetrahydromethanopterin hydro-lyase), fdh (formate dehydrogenase). The genes shown in the green boxes represents electron transport chain complexes (I, II, III, IV, V). The cellular transporter genes on the right side in purple boxes are involved in nitrate assimilation and GS-GOGAT ammonia assimilation pathway nasA (assimilatory nitrate reductase), narHG (dissimilatory nitrate reductase), nirBD (nitrite reductase), AMT (ammonia transporter), GS-GOGAT (glutamine synthetase-glutamine2-oxoglutarate aminotransferase). Followed by the sulfur assimilating gene cys (cysteine synthase) and pst (inorganic phosphate transport genes) into the cell
Screening the genome revealed the presence of genes encoding the enzymes for a complete tetrahydromethanopterin (H4MPT) C-transfer pathway for formaldehyde oxidation to formate. This included genes for the formaldehyde-activating enzyme (fae), the NAD(P)-dependent methylene tetrahydromethanopterine dehydrogenase (mtdB), methenyl-H4MPT cyclohydrolase (mch), and formylmethanofuran dehydrogenase subunits BCA (fwdBCA). We also detected the genes encoding the major subunit (fdhF and fdoG) and delta subunit (fdsD) of format dehydrogenase, responsible for oxidation formate to CO2.
Carbon fixation
The strain LS7-T4AT contained a complete set of genes for the ribulose monophosphate pathway for carbon fixation from formaldehyde. Hexose phosphates are initial products formed by the condensation of formaldehyde and ribulose-5-phosphate. The key enzymes of the RuMP pathway are hexose phosphate synthase, encoded by hxlA, and phosphohexulose isomerase, encoded by hxlB which both were found in the genome of strain LS7-T4AT. The strain will thus likely assimilate carbon through RuMP pathway as shown in Fig. 9. The genome lacked key enzyme serine-glyoxylate aminotransferase encoded by gene sga for serine pathway and genes encoding the ribulose- 1,5-bisphosphate carboxylase/oxygenase /RuBisCO).
Energy conservation and respiration
The strain LS7-T4AT is obligate aerobe and uses O2 as a terminal electron acceptor. Energy conservation is through oxidative phosphorylation. the respiratory complexes comprised of nadh-quinone oxidoreductase (electron transport chain (ETC) complex I, characterised by the genes nuo BCDHJKNM were found, in addition to succinate dehydrogenase (ETC complex II, with the gene sdh) and cytochrome c oxidase (ETC complex III with the genes coxA and coxB) followed by F-type ATPase (ETC complex V, genes atpABCDEFGHIK) as shown in Fig. 9. This validates a complete aerobic respiration chain present in our isolate.
Nitrogen, sulfate, and phosphate metabolism
Genome analyses of the LS7-T4AT strain indicated that it has the potential for using ammonia as a nitrogen source. The genes for the membrane bound ammonium transporter (AMT) was found in addition to the genes glnA (glutamine synthetase) and GDH2 (glutamate dehydrogenase), demonstrating its assimilation of ammonia through the glutamin synthetase/ glutamate synthetase (GS/GOGAT) system and providing available nitrogen for cellular anabolism. Potential for nitrate and nitrite assimilation was found by genes encoding ABC-type nitrate transporter (NasA) together with the membrane bound nitrate reductase (and NarHG, large and small subunits) and conversion of nitrite to ammonium by assimilatory nitrite reductase (NirBD). The molecular marker gene for denitrification NirK was not found. The strain also possesses genes for the nitrogen fixation process. Nitrogenases genes nifHDK (alpha and beta chain), nifE (co-factor synthase protein), nifN (iron protein), and nifW (nitrogenase-stabilizing/protective protein) were found. These include sulfate adenylyltransferase subunit 2 (cysD), bifunctional enzyme (CysN/CysC), phosphoadenosine phosphosulfate reductase (cycH), sulfite reductase (NADPH) hemoprotein beta-component (cycI), and tRNA 2-thiouridine synthesizing protein A (Sir). For dissimilatory sulfate reduction and oxidation, encoded by tRNA 2-thiouridine synthesizing protein (DsrHCEF) was present but lacked specific genes like sat AprAB and DsrAB.
The genome showed potential for an inorganic phosphate transport system (PstI) to incorporate inorganic phosphate. This transport system is comprised of a periplasmic substrate-binding protein (pstS), a membrane-bound protein (pstA and pstC), and a protein that releases free Pi in the cytoplasm (pstB).
Discussion
Several pingos situated along the Adventdalen Valley in Svalbard are formed by a combination of climate, geology, and hydrology. One of these pingos is the Lagoon Pingo, which boasts an active spring and a dynamic ecosystem that experiences yearly freeze–thaw cycles and erosion throughout the year. Lagoon Pingo is shaped by groundwater-rich methane fluids pushing upwards through the continuous permafrost, making it a methane source, and enables methane release. During winter the Lagoon Pingo builds up as several dome shaped landforms with icy layers on top. During the summer, the ice melts, the domes collapses and a several crater lakes are generated. Due to the varying moisture levels, sediment grains and methane availability in these pingo crater lakes unique microbial habitats establishes.
In Lagoon Pingo methane fluxes were found to vary between − 0.5 and 1650 nmol m2 s−1 (Nagel 2020). In our study, two sample sites along a transect (T) were collected, one from the mini source (MS) which had a subsurface water discharge with an elevated methane flux (687.5 nmol m2 s−1) and one from the still pond (SP), containing water covered locations with no water movements, which exhibited a relatively low methane flux of 1.5 nmol m2/s. Previously a high number of pmoA genes relative to the copy number of 16S rRNA genes were found in the still pond was (Nagel 2020), suggesting the potential for high methanotrophic activity which matches our observation of a low methane flux. When analysing the microbial community diversity at these two sites, we found that the alpha diversity was highest in the MS, and the overall bacterial communities were highly diverse within the bacterial domain. Dominant ASVs were affiliated to the phyla Pseudomonadota, Bacteriodota, Gemmatimonadota, and Actinomycetota. The same taxa have also been reported as dominant in studies done across the Arctic and Antarctic marine sediments in addition to studies from lake sediments of the Tibetan Plateau (Xiong et al. 2012; Carr et al. 2015; Müller et al. 2018). The Lagoon Pingo samples were dominated by family Hydrogenophilaceae, represented by Thiobacillus. Thiobacillus is a genus with known chemolithotrophic or mixotrophic bacteria that uses various inorganic electron donors like reduced sulfur compounds and has the ability for carbon fixation by the Calvin-Benson cycle (Hayashi et al. 1999; Orlygsson and Kristjansson 2014). Thiobacillus genera are abundant, indicating high sulfur activity and potential denitrification chances at this site.
Within the phylum Pseudomonadota, the Gammaproteobacteria was the most dominant class in Lagoon Pingo. We observed high abundance of the gammaproteobacterial methanotrophic family Methylococcaceae which represented up to 2.5% of the community in MS and only < 1% in the still pond. Among the genera within this family, Methylobacter dominated. This genus has shown to be present in many soils on Svalbard, close to Lagoon Pingo (Wartiainen et al. 2003; Høj. et al. 2006; Tveit et al. 2014; Fåne 2020).
Strain LS7-T4AT was successfully isolated and classified in this study, and classified as a Methylobacter sp. Its identity was confirmed by 16S rRNA gene- and the pmoA gene sequencing, molecular marker genes that can be used for the classification of methanotrophic taxa (Knief 2015). Phylogenetic analysis of the pmoA gene revealed that our isolate clusters within Methylobacter, indicating that this is a new species within this genus, and further genome analyses revealed clustering with Methylobacter sp. isolated from Arctic ecosystems (Fig. 8). The most closely related strain to our isolate is M. psychrophilus Z-0021 (Omelchenko et al. 1996) and M. tundripaludum SV96T isolated from High Arctic wetland soil, Ny-Ålesund, Svalbard, Norway and was first described in 2006 (Wartiainen et al. 2006), and the genome sequencing of this strain was completed in 2011 (Svenning et al. 2011). This species was identified to have a significant role in the biogeochemistry of Arctic wetland soils emitting methane (Tveit et al. 2023).
Our strain LS7-T4AT has an optimal growth temperature of 13 °C and a maximum growth temperature at 22 °C, which differs from the M. tundripaludum SV96T, a psychrotolerant strain with optimal growth at 23 °C and maximum growth temperature at 30 °C. Very few psychrophilic methanotrophs, which thrive in low-temperature environments, have been isolated and characterised (Table 3). However, Methylosphaera hansonii and Methylobacter psychrophilus are two true psychrophilic methanotrophs that have been successfully isolated. M. hansonii was found in the surface sediments of an Antarctic meromictic lake (Bowman et al. 1997), while M. psychrophilus was isolated from Russian Arctic tundra soil (Omelchenko et al. 1996). Recently, a study conducted in boreal lake ecosystems in Finland reported that the isolate Methylobacter sp S3L5C, which based on its characterization and genomic data, is also a psychrophilic methanotroph (Khanongnuch et al. 2022). The dominance of Methylobacter sp. in the oxic-anoxic transition zone from boreal and subarctic lakes, ponds, and wetlands is reported through several different studies (Smith et al. 2018; Rissanen et al. 2018; Rissanen et al. 2021; Cabrol et al. 2020) confirming that this is a ubiquitous genus in low temperature environments.
The genome of Methylobacter LS7-T4AT sp. has genes encoding enzymes required for aerobic methane metabolism. Compared with other species, the features of LS7-T4AT are distinguishable, and the average nucleotide identity showed that our strain is a new addition to the genus Methylobacter. The GC content also has differences between 0.7 and 5 when compared with other species in the same genus. Methanotrophs obtain energy from oxidation of C1 substrates to CO2 and can obtain energy in the form of ATP from oxidative phosphorylation. Our isolate uses methane as a substrate for growth catalyzed by the pMMO enzyme. Type I methanotrophs also have xoxF-type pyrroloquinoline (PQQ) dependent methanol dehydrogenase (MDH) genes (Chu and Lidstrom 2016) which were absent in our strain. The genome carries the genes necessary for the synthesis of methanofuran (MFR), and tetrahydromethanepterin (THPMT), which were absent in the recently described Ca. Methylobacter titanis sp. nov (Roldán and Menes 2023). Conversion of formate to CO2 is the final methane oxidation stage catalyzed by formate dehydrogenase, which we also found in the genome of our isolate.
The isolate LS7-T4AT uses the RuMP pathway for carbon fixation as most type Ia methanotrophs in the genus Methylobacter sp (Collins et al. 2017), but it lacks the enzymes for the serine pathway. Recent studies about Ca. Methylobacter favarea B2 (Hogendoorn et al. 2021) and Ca. Methylobacter titanis sp. nov (Roldán and Menes, 2023) revealed almost complete serine pathways along with RuMP, which is not typically seen within the genus Methylobacter (Chistoserdova et al. 2009). Like most Methylobacter species, the genome of our strain lacks RuBisCo (1,5-bisphosphate carboxylase/oxygenase), which was found in the Methylococcus capsulatus strain bath (a member of the type Ib Methylococcaceae) and some Verrucomicrobial methane oxidizers (Henard et al. 2021; Khadem et al. 2011).
In this study, we have isolated a psychrophilic methane oxidizer belonging to the genus Methylobacter in the family Type Ia Methylomonadaceae. Relative to M. tundripaludum SV96T, M. psychrophilus Z-0021T, Methylobacter sp. S3L5C, ‘Ca. Methylobacter titanis, the Methylobacter sp. LS7-T4AT presented in this paper is likely to be distinct species compared to commonly used ANI and dDDH thresholds to distinguish separate species (95% ANI and 70% dDDH; Table S2). Strain LS7-T4AT might have an important role in the biological methane sinks of terrestrial methane seeps such as Lagoon Pingo in Svalbard. Our knowledge of the cold-adapted methane oxidizing bacteria in the open-system pingos is still very limited, yet the results from this work together with the recovered aerobic methanotroph isolate, indicates that the microbial community is important in the methane mitigation in these systems.
Description of Methylobacter svalbardensis sp. nov
Methylobacter svalbardensis (sval.bar.den’sis. N.L. gen pl. n). The local name of a Norwegian archipelago in the Arctic Ocean refers to “the land with the cold coasts.”.
The strain has the following properties: Gram-stain-negative, strictly aerobic and coccoid to rod-shaped cells with a size of 0.8–1.2 × 1.6–2.2 µm. Some cells are motile. Reproduce by binary fission. Colonies are pigmented, light pink, circular and smooth colonies on agar with 1.8 to 2.5 mm in diameter. It is a psychrophilic and obligately methylotrophic strain utilizing methane and methanol via RuMP pathway. Cells do not grow on methylamine, formate, and formaldehyde. Utilise nitrate as a nitrogen source. Vitamins are not required for its growth. Cells contain pMMO and MDH but not sMMO; the genes xoxF, mauA and cbbL are not found in the genome. Contains a nifH gene. Growth occurs at 1–22 °C (optimum 10 to 13 °C), at pH 6.4 to 9.3 (optimum pH 7.5 to 8.0). Does not grow on glucose, acetate, succinate, malate, lactate, pyruvate ethanol, methylamine, yeast extract or R2A agar plates. Phylogenetically, strain LS7-T4AT belongs to the genus Methylobacter of the family Methylomonadaceae Type Ia. The closest present species are M. psychrophilus Z-0021T (98.95%) and M. tundripaludum SV96T (99.06%). DNA Genome sequencing of strain LS7-T4AT unveiled a genome length of 4.3 Mbp of 226 contigs with 4272 annotated genes. The G + C content of the DNA is 47.93 mol % from genome. The type of strain LS7-T4AT (DSMZ: 114308; JCM: 39463 ) was isolated from terrestrial methane seep sediments located in Svalbard, Norway.
Data availability
The GenBank accession numbers of the16S rRNA genes sequence is OQ832782. The 16S rRNA amplicon Illumina sequence GenBank accession numbers SAMN37693933, SAMN37693934, SAMN37693935, SAMN37693936, SAMN37693937, SAMN37693938, SAMN37693939, SAMN37693940 and draft genome sequence under the BioProject ID: PRJNA1024098 (all sequences will be publicly available after publication). The culture of our isolate is deposited and available in DSMZ with assigned accession number 114308 and JCM with the accession number from 39463.
References
Auch AF, Klenk HP, Göker M (2010) Standard operating procedure for calculating genome-to-genome distances based on high-scoring segment pairs. Stand Genomic Sci 2:142–148. https://doi.org/10.4056/sigs.541628
Bankevich A, Nurk S et al (2012a) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19(5):455–477
Bankevich A et al (2012b) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19(5):455–477. https://doi.org/10.1089/cmb.2012.0021
Belova SE, Baani M et al (2011) Acetate utilization as a survival strategy of peat-inhabiting Methylocystis spp. Environ Microbiol Rep 3(1):36–46. https://doi.org/10.1111/j.1758-2229.2010.00180.x
Belova SE, Kulichevskaya IS, Bodelier PL, Dedysh SN (2013) Methylocystis bryophila sp. nov., a facultatively methanotrophic bacterium from acidic Sphagnum peat, and emended description of the genus Methylocystis (ex Whittenbury et al. 1970) Bowman et al. 1993. Int J Syst Evolut Microbiolo 63(Pt_3):1096–1104. https://doi.org/10.1099/ijs.0.043505-0
Besemer J, Lomsadze A, Borodovsky M (2001) GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res 29(12):2607–2618. https://doi.org/10.1093/nar/29.12.2607
Bowman JP et al (1993) Characterization of the methanotrophic bacterial community present in a trichloroethylene-contaminated subsurface groundwater site. Appl Environ Microbiol 59(8):2380–2387. https://doi.org/10.1099/00207713-43-4-735
Bowman JP, Mccammon SA, Skerratt JH (1997) Methylosphaera hansonii gen nov, sp nov, a psychrophilic, group I methanotroph from Antarctic marine-salinity, meromictic lakes. Microbiology-UK 143:1451–1459. https://doi.org/10.1099/00221287-143-4-1451
Cabrol L, Thalasso F et al (2020) Anaerobic oxidation of methane and associated microbiome in anoxic water of Northwestern Siberian lakes. Sci Total Environ 736:139588. https://doi.org/10.1016/j.scitotenv.2020.139588
Callahan BJ et al (2016) DADA2: high-resolution sample inference from Illumina amplicon data. Nat Method 13(7):581–583. https://doi.org/10.1038/nmeth.3869
Carr SA et al (2015) Abundant Atribacteria in deep marine sediment from the Adélie Basin, Antarctica. Front Microbiol 6:145828. https://doi.org/10.3389/fmicb.2015.00872
Chen Y, Dumont MG, Cébron A, Murrell JC (2007) Identification of active methanotrophs in a landfill cover soil through detection of expression of 16S rRNA and functional genes. Environ Microbiol 9(11):2855–2869. https://doi.org/10.1111/j.1462-2920.2007.01401.x
Chistoserdova L, Kalyuzhnaya MG, Lidstrom ME (2009) The expanding world of methylotrophic metabolism. Annu Rev Microbiol 63:477–499. https://doi.org/10.1146/annurev.micro.091208.073600
Chowdhury TR, Dick RP (2013) Ecology of aerobic methanotrophs in controlling methane fluxes from wetlands. Appl Soil Ecol 65:8–22. https://doi.org/10.1016/j.apsoil.2012.12.014
Chu F, Lidstrom ME (2016) XoxF acts as the predominant methanol dehydrogenase in the type I methanotroph Methylomicrobium buryatense. J Bacteriol 198(8):1317–1325. https://doi.org/10.1128/jb.00959-15
Collins DA, Akberdin IR, Kalyuzhnaya MG (2015) Methylobacter. Bergey’s Man of Syst Archaea Bact. https://doi.org/10.1002/9781118960608.gbm01179.pub2
Collins DA, Akberdin IR, and Kalyuzhnaya M (2017). Genus Methylobacter. Bergey’s Manual of Systematics of Archaea and Bacteria.
Costello AM, Lidstrom ME (1999) Molecular characterization of functional and phylogenetic genes from natural populations of methanotrophs in lake sediments. Applied and environmental microbiology 65(11):5066–5074. https://doi.org/10.1128/AEM.65.11.5066-5074.1999
Danilova OV, Dedysh SN (2014) Abundance and diversity of methanotrophic Gammaproteobacteria in northern wetlands. Microbiology 83:67–76. https://doi.org/10.1134/s0026261714020040
Danilova OV et al (2013) Methylomonas paludis sp nov., the first acid-tolerant member of the genus Methylomonas, from an acidic wetland. Int J Syst Evol Microbiol 63:2282–2289. https://doi.org/10.1099/ijs.0.045658-0
Davis JJ et al (2020) The PATRIC Bioinformatics Resource Center: expanding data and analysis capabilities. Nucleic Acids Res 48(D1):D606–D612. https://doi.org/10.1093/nar/gkz943
Dedysh SN (2002) Methanotrophic bacteria of acidic Sphagnum peat bogs. Microbiology 71:638–650. https://doi.org/10.1023/A:1021467520274
Dedysh SN et al (2000) Methylocella palustris gen. nov., sp. nov., a new methane-oxidizing acidophilic bacterium from peat bogs, representing a novel subtype of serine-pathway methanotrophs. Int J Syst Evolut Microbiol 50(3):955–969. https://doi.org/10.1099/00207713-50-3-955
Dedysh SN et al (2004) Methylocella tundrae sp. nov., a novel methanotrophic bacterium from acidic tundra peatlands. Int J Syst Evol Microbiol 54:151–156. https://doi.org/10.1099/ijs.0.02805-0
Demidov V, Demidov N, Verkulich S, Wetterich S (2022) Distribution of pingos on Svalbard. Geomorphology 412:108326. https://doi.org/10.1016/j.geomorph.2022.108326
Den Camp Op et al (2009) Environmental, genomic, and taxonomic perspectives on methanotrophic Verrucomicrobia. Environ Microbiol Rep 1:293–306. https://doi.org/10.1111/j.1758-2229.2009.00022.x
Deutzmann JS, Wörner S, Schink B (2011) Activity and diversity of methanotrophic bacteria at methane seeps in eastern Lake Constance sediments. Appl Environ Microbiol 77(8):2573–2581. https://doi.org/10.1128/AEM.02776-10
Dhariwal A et al (2017) MicrobiomeAnalyst: a web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acid Res 45(W1):W180–W188. https://doi.org/10.1093/nar/gkx295
Dumont MG, Pommerenke B, Casper P, Conrad R (2011) DNA-, rRNA-and mRNA-based stable isotope probing of aerobic methanotrophs in lake sediment. Environ Microbiol 13(5):1153–1167. https://doi.org/10.1111/j.1462-2920.2010.02415.x
Dunfield PF et al (2007) Methane oxidation by an extremely acidophilic bacterium of the phylum Verrucomicrobia. Nature 450(7171):879–882. https://doi.org/10.1038/nature06411
Fåne, P. M. S. (2020). Novel methanotrophic community assemblages in a terrestrial methane seep in Svalbard (Master’s thesis, UiT Norges arktiske universitet). https://hdl.handle.net/10037/22057
Finn D, Ouwerkerk D and Klieve A (2012). Methanotrophs from natural ecosystems as biocontrol agents for ruminant methane emissions.
Gardner PP et al (2009). Rfam: updates to the RNA families database. Nucleic acids research, 37(suppl_1), D136-D140. https://doi.org/10.1093/nar/gkn766
Grosse G, Jones BM (2010) Spatial distribution of pingos in Northern Asia. The Cryosphere Discuss 4(3):1781–1837
Gurney SD (1998) Aspects of the genesis and geomorphology of pingos: perennial permafrost mounds. Prog Phys Geogr 22(3):307–324. https://doi.org/10.1177/030913339802200301
Hammock CP et al (2022) Seismic and electrical geophysical characterization of an incipient coastal open-system Pingo Lagoon Pingo Svalbard. Earth Space Sci 9(3):e2021EA002093. https://doi.org/10.1029/2021EA002093
Hanson RS, Hanson TE (1996) Methanotrophic bacteria. Microbiol Rev 60:439–471. https://doi.org/10.1128/mr.60.2.439-471.1996
Hayashi NR et al (1999) Hydrogenophilus thermoluteolus gen. nov., sp. nov., a thermophilic, facultatively chemolithoautotrophic, hydrogen-oxidizing bacterium. Int J Syst Evolut Microbiol 49(2):783–786. https://doi.org/10.1099/00207713-49-2-783
Henard CA et al (2021) Ribulose-1, 5-bisphosphate carboxylase/oxygenase (RubisCO) is essential for growth of the methanotroph Methylococcus capsulatus strain Bath. Appl Environ Microbiol 87(18):e00881-e921. https://doi.org/10.1128/AEM.00881-21
Hjelle A (1993). Geology of Svalbard.
Hodson AJ et al (2019) Seasonal dynamics of methane and carbon dioxide evasion from an open system pingo: Lagoon pingo, Svalbard. Frontiers Earth Sci. https://doi.org/10.3389/feart.2019.00030
Hodson AJ et al (2020) Sub-permafrost methane seepage from open-system pingos in Svalbard. Cryosphere 14(11):3829–3842
Hogendoorn C et al (2021) Draft genome of a novel methanotrophic Methylobacter sp. from the volcanic soils of Pantelleria Island. Antonie Van Leeuwenhoek 114:313–324
Høj L, Rusten M, Haugen LE, Olsen RA, Torsvik VL (2006) Effects of water regime on archaeal community composition in Arctic soils. Environ Microbiol 8(6):984–996. https://doi.org/10.1111/j.1462-2920.2006.00982.x
Houghton KM, Carere CR, Stott MB, McDonald IR (2019) Thermophilic methanotrophs In hot pursuit. FEMS Microbiol Ecol 95(9):fiz125. https://doi.org/10.1093/femsec/fiz125
Im J, Lee SW, Yoon S, DiSpirito AA, Semrau JD (2011) Characterization of a novel facultative Methylocystis species capable of growth on methane, acetate and ethanol. Environ Microbiol Reports 3(2):174–181. https://doi.org/10.1111/j.1758-2229.2010.00204.x
Islam T et al (2008) Methane oxidation at 55oC and pH 2 by a thermoacidophilic bacterium belonging to the Verrucomicrobia phylum. Proc Natl Acad Sci USA 105:300–304. https://doi.org/10.1073/pnas.0704162105
Islam T et al (2015) Novel Methanotrophs of the Family Methylococcaceae from Different Geographical Regions and Habitats. Microorganisms 3:484–499. https://doi.org/10.3390/microorganisms3030484
Islam T et al (2016) Acid-tolerant moderately thermophilic methanotrophs of the class Gammaproteobacteria isolated from tropical topsoil with methane seeps. Front Microbiol 7:851. https://doi.org/10.3389/fmicb.2016.00851
Islam T et al (2020) A novel moderately thermophilic methanotroph isolated from an alkaline thermal spring in the Ethiopian Rift Valley. Microorganisms 8:250. https://doi.org/10.3390/microorganisms8020250
Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M (2004) The KEGG resource for deciphering the genome. Nucleic Acid Res 32(Suppl_1):D277–D280
Khadem AF et al (2011) Autotrophic methanotrophy in Verrucomicrobia: Methylacidiphilum fumariolicum SolV uses the Calvin–Benson–Bassham cycle for carbon dioxide fixation. J Bacteriol 193(17):4438–4446. https://doi.org/10.1128/jb.00407-11
Khanongnuch R, Mangayil R, Svenning MM, Rissanen AJ (2022) Characterization and genome analysis of a psychrophilic methanotroph representing a ubiquitous Methylobacter spp. cluster in boreal lake ecosystems. ISME Commun 2(1):85. https://doi.org/10.1038/s43705-022-00172-x
Khanongnuch R, Mangayil R, Rissanen AJ (2023) Conversion of methane to organic acids is a widely found trait among gammaproteobacterial methanotrophs of freshwater lake and pond ecosystems. Microbiol Spectr 11(6):e01742-e1823. https://doi.org/10.1128/spectrum.01742-23
Khatri K, Mohite J, Pandit P, Bahulikar RA, Rahalkar MC (2021) Isolation, description and genome analysis of a putative novel Methylobacter Species (‘Ca. Methylobacter coli’) isolated from the faeces of a blackbuck (Indian antelope). Microbiol Res 12(2):513–523. https://doi.org/10.3390/microbiolres1202003
Knief C (2015) Diversity and habitat preferences of cultivated and uncultivated aerobic methanotrophic bacteria evaluated based on pmoA as molecular marker. Front Microbiol 6:1346. https://doi.org/10.3389/fmicb.2015.01346
Knief C (2019) Diversity of methane-cycling microorganisms in soils and their relation to oxygen. Curr Issue Mol Biol 33(1):23–56. https://doi.org/10.21775/cimb.033.023
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874. https://doi.org/10.1093/molbev/msw054
Lagesen K, Hallin P, Rødland EA, Stærfeldt HH, Rognes T, Ussery DW (2007) RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35(9):3100–3108. https://doi.org/10.1093/nar/gkm160
Lau E, Fisher MC, Steudler PA, Cavanaugh CM (2013) The methanol dehydrogenase gene, mxaF, as a functional and phylogenetic marker for proteobacterial methanotrophs in natural environments. PLoS ONE 8(2):e56993. https://doi.org/10.1371/journal.pone.0056993
Li R et al (2010) De novo assembly of human genomes with massively parallel short read sequencing. Genome Res 20(2):265–272. https://doi.org/10.1101/gr.097261.109
Liestol O (1976) Pingos, springs and permafrost in Spitsbergen. Norsk Polarinstitutt Arbok 1975:7–29
Lin SH, Liao YC (2013) CISA: contig integrator for sequence assembly of bacterial genomes. PLoS ONE 8(3):e60843
Lowe TM, Eddy SR (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acid Res 25(5):955–964. https://doi.org/10.1093/nar/25.5.955
Mateos-Rivera A et al (2018) Activity and diversity of methane-oxidizing bacteria along a Norwegian sub-Arctic glacier forefield. FEMS Microbiol Ecol 94(5):fiy059. https://doi.org/10.1093/femsec/fiy059
McDonald IR, Bodrossy L, Chen Y, Murrell JC (2008) Molecular ecology techniques for the study of aerobic methanotrophs. Appl Environ Microbiol 74(5):1305–1315. https://doi.org/10.1128/AEM.02233-07
Meier-Kolthoff JP et al (2013) Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform 14:1–14. https://doi.org/10.1186/1471-2105-14-60
MüllerO SL, Bratbak G, Paulsen ML (2018) Bacterial response to permafrost derived organic matter input in an Arctic fjord. Front Mar Sci 5:263. https://doi.org/10.3389/fmars.2018.00263
Nagel F (2020). Linking methane fluxes and oxidation rates to methane oxidizing bacteria in an Arctic terrestrial methane seep, Svalbard (Master’s thesis, UiT Norges arktiske universitet)
Omelchenko MV et al (1996) A novel psychrophilic methanotroph of the genus Methylobacter. Microbiology 65:339–343
Orlygsson J, Kristjansson JK (2014) The family Hydrogenophilaceae. The Prokaryotes. https://doi.org/10.1007/978-3-642-30197-1_244
Orvin AK (1944) Outline of the geological history of Spitsbergen. Skrifter Svalbard Og Ishavet 78:1–24
Oshkin IY et al (2016) Methylovulum psychrotolerans sp nov: a cold-adapted methanotroph from low-temperature terrestrial environments, and emended description of the genus Methylovulum. Int J Syst Evol Microbiol 66:2417–2423. https://doi.org/10.1099/ijsem.0.001046
Quast C et al (2012) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acid Res 41(D1):D590–D596. https://doi.org/10.1093/nar/gks1219
Richter M et al (2016) JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 32(6):929–931. https://doi.org/10.1093/bioinformatics/btv681
Rissanen AJ et al (2018) Gammaproteobacterial methanotrophs dominate methanotrophy in aerobic and anaerobic layers of boreal lake waters. Aquat Microb Ecol 81(3):257–276. https://doi.org/10.3354/ame01874
Rissanen AJ et al (2021) Vertical stratification patterns of methanotrophs and their genetic controllers in water columns of oxygen-stratified boreal lakes. FEMS Microbiol Ecol 97(2):fiaa252. https://doi.org/10.1093/femsec/fiaa252
Rissanen AJ et al (2022) Draft genome sequence data of a psychrophilic tundra soil methanotroph, Methylobacter psychrophilus Z-0021 (DSM 9914). Data Brief 45:108689. https://doi.org/10.1016/j.dib.2022.108689
Roldán DM, Menes RJ (2023) Characterisation of ‘Candidatus Methylobacter titanis’ sp. nov., a putative novel species of Methylobacter clade 2 and their distribution in sediments of freshwater lakes in maritime Antarctica. Antonie Van Leeuwenhoek 116(7):721–738. https://doi.org/10.1007/s10482-023-01840-1
Schuur EA et al (2013) Expert assessment of vulnerability of permafrost carbon to climate change. Clim Ch 119:359–374. https://doi.org/10.1007/s10584-013-0730-7
Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I (2009) ABySS: a parallel assembler for short read sequence data. Genome Res 19(6):1117–1123. https://doi.org/10.1101/gr.089532.108
Smith GJ et al (2018) Members of the genus Methylobacter are inferred to account for the majority of aerobic methane oxidation in oxic soils from a freshwater wetland. Mbio 9(6):10–1128. https://doi.org/10.1128/mbio.00815-18
Spanning V et al (2022) Methanotrophy by a Mycobacterium species that dominates a cave microbial ecosystem. Nat Microbiol 7(12):2089–2100. https://doi.org/10.1038/s41564-022-01252-3
Svenning MM et al (2011) Genome sequence of the Arctic methanotroph Methylobactertundripaludum. SV96. https://doi.org/10.1128/jb.05380-11
Svensson H (1969) Pingos i ytre dalen av Adventdalen. Norsk Polarinstitutt Arbok 1970:168–174
Trotsenko YA, Khmelenina VN (2002) Biology of extremophilic and extremotolerant methanotrophs. Archives of Microbiology 177:123–131. https://doi.org/10.1007/s00203-001-0368-0
Trotsenko YA, Khmelenina VN (2005) Aerobic methanotrophic bacteria of cold ecosystems. FEMS Microbiol Ecol 53:15–26. https://doi.org/10.1016/j.femsec.2005.02.010
Tveit AT, Urich T, Svenning MM (2014) Metatranscriptomic analysis of arctic peat soil microbiota. Appl Environ Microbiol 80(18):5761–5772. https://doi.org/10.1128/AEM.01030-14
Tveit AT, Hestnes AG, Robinson SL, Schintlmeister A, Dedysh SN, Jehmlich N, von Bergen M, Herbold C, Wagner M, Richter A, Svenning MM (2019) Widespread soil bacterium that oxidizes atmospheric methane. Proc Natl Acad Sci 116(17):8515–8524. https://doi.org/10.1073/pnas.1817812116
Tveit AT et al (2023) Thermal acclimation of methanotrophs from the genus Methylobacter. ISME J 17(4):502–513. https://doi.org/10.1038/s41396-023-01363-7
Vorobev AV et al (2011) Methyloferula stellata gen. nov., sp nov., an acidophilic, obligately methanotrophic bacterium that possesses only a soluble methane monooxygenase. Int J Syst Evol Microbiol 61:2456–2463. https://doi.org/10.1099/ijs.0.028118-0
Wartiainen I, Hestnes AG, Svenning MM (2003) Methanotrophic diversity in high arctic wetlands on the islands of Svalbard (Norway)-denaturing gradient gel electrophoresis analysis of soil DNA and enrichment cultures. Can J Microbiol 49(10):602–612. https://doi.org/10.1139/w03-080
Wartiainen I, Hestnes AG, Mcdonald IR, Svenning MM (2006) Methylobacter tundripaludum sp nov., a methane-oxidizing bacterium from Arctic wetland soil on the Svalbard islands, Norway (78 degrees N). Int J Syst Evol Microbiol 56:109–113. https://doi.org/10.1099/ijs.0.63728-0
Wattam AR et al (2014) PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acid Res 42(D1):D581–D591. https://doi.org/10.1093/nar/gkt1099
Whittenbury R, Phillips KC, Wilkinso JF (1970) Enrichment, isolation and some properties of methane-utilizing bacteria. J Gen Microbiol 61:205–218. https://doi.org/10.1099/00221287-61-2-205
Wilson B et al (2017) Changes in marine prokaryote composition with season and depth over an Arctic polar year. Front Mar Sci 95. https://doi.org/10.3389/fmars.2017.00095
Xiong J, Liu Y, Lin X, Zhang H, Zeng J, Hou J, Yang Y, Yao T, Knight R, Chu H (2012) Geographic distance and pH drive bacterial distribution in alkaline lake sediments across Tibetan Plateau. Environ Microbiol 14(9):2457–2466. https://doi.org/10.1111/j.1462-2920.2012.02799.x
Yoon SH, Ha SM, Lim J, Kwon S, Chun J (2017) A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek 110:1281–1286. https://doi.org/10.1007/s10482-017-0844-4
Yoshikawa K (1993) Notes on open-system pingo ice, Adventdalen, Spitsbergen. Permafrost Periglac Process 4(4):327–334. https://doi.org/10.1002/ppp.3430040405
Yoshikawa K, Harada K (1995) Observations on nearshore pingo growth, Adventdalen. Spitsbergen Permafr Periglac Process 6(4):361–372. https://doi.org/10.1002/ppp.3430060407
Yoshikawa K, Nakamura T (1996) Pingo growth ages in the delta area, Adventdalen. Spitsbergen Polar Rec 32(183):347–352. https://doi.org/10.1017/S0032247400067565
Acknowledgements
This experiment was supported by Norwegian Research Council Grant through CLIMAGAS (ID: 294764) project led by AH and LØ at UNIS and UiB. The authors would like to thank Tilman Scmider (TS) for performing the methane flux rates in the field. All experiments were conducted at the Department of Biological Science (BIO) in Marine Microbiology group at UiB. The electron microscopy imaging was performed at the Molecular Imaging Center (MIC). Department of Biomedicine, University of Bergen. Norway.
Funding
Open access funding provided by University of Bergen (incl Haukeland University Hospital).
Author information
Authors and Affiliations
Contributions
LØ, SP, AT and TJ designed the project. AT and AH collected sediment samples from Lagoon pingo. SP and TJ did experiments of cultivation and isolation. SP performed DNA extraction, sequencing, culture deposition in DSMZ and JCM, TEM microscopy. LØ, SP, TJ and AT contributed to analysis and SP, LØ, TJ wrote the manuscript. All authors reviewed the manuscript and approved for submission.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Patil, S.K., Islam, T., Tveit, A. et al. Targeting methanotrophs and isolation of a novel psychrophilic Methylobacter species from a terrestrial Arctic alkaline methane seep in Lagoon Pingo, Central Spitsbergen (78° N). Antonie van Leeuwenhoek 117, 60 (2024). https://doi.org/10.1007/s10482-024-01953-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10482-024-01953-1