
Abstract
Aspergillus niger secretes proteins throughout the colony except for the zone that forms asexual spores called conidia. Inactivation of flbA that encodes a regulator of G-protein signaling results in colonies that are unable to reproduce asexually and that secrete proteins throughout the mycelium. In addition, the ΔflbA strain shows cell lysis and has thinner cell walls. Expression analysis showed that 38 predicted transcription factor genes are differentially expressed in strain ΔflbA. Here, the most down-regulated predicted transcription factor gene, called fum21, was inactivated. Growth, conidiation, and protein secretion were not affected in strain Δfum21. Whole genome expression analysis revealed that 63 and 11 genes were down- and up-regulated in Δfum21, respectively, when compared to the wild-type strain. Notably, 24 genes predicted to be involved in secondary metabolism were down-regulated in Δfum21, including 10 out of 12 genes of the fumonisin cluster. This was accompanied by absence of fumonisin production in the deletion strain and a 25% reduction in production of pyranonigrin A. Together, these results link FlbA-mediated sporulation-inhibited secretion with mycotoxin production.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The genus Aspergillus consists of more than 300 species (Samson et al. 2014) that are among the most abundant fungi on the globe. Aspergilli degrade plant waste and as such play a role in carbon cycling in nature. Moreover, the genus includes opportunistic pathogens of plants, animals, and humans (Krijgsheld et al. 2013a). Enzymes secreted by Aspergillus play an important role in degradation of organic material and pathogenicity. The property to secrete high levels and a diversity of enzymes make aspergilli such as Aspergillus niger important cell factories for the production of proteins and metabolites (Meyer et al. 2011; Wösten et al. 2013).
Aspergilli form a mycelium consisting of a network of hyphae that grow at their apex and that branch subapically. The mycelium grows initially vegetative but at a certain moment asexual development is initiated (Krijgsheld et al. 2013a). The resulting conidia are the only spore type that are produced by A. niger. Conidiation starts with the formation of thick aerial hyphae called stalks. When a stalk has reached a certain height, its tip swells to form a vesicle. In many aspergilli, this structure buds resulting in a layer of metulae. The metulae in turn form a layer of phialides, from which chains of conidia develop. These spores are heterogeneous in composition, water dispersal efficiency, and germination rate (Teertstra et al. 2017) and give rise to new mycelia.
The zone forming the asexual conidia within an A. niger colony does not secrete proteins, while non-sporulating zones do release proteins into their environment (Krijgsheld et al. 2013b). Preventing conidiation of the colony by covering it with a polycarbonate membrane does not impact the spatial secretion pattern indicating that the capacity to sporulate but not the conidiation process itself inhibits secretion. FlbA was shown to impact spatial secretion in the colony (Krijgsheld et al. 2013b). This protein regulates the Gα subunit FadA by activating its GTPase activity. By doing so, it promotes asexual development and inhibits vegetative growth and autolysis (Wieser et al. 1994; Yu et al. 1996). Conidiation is abolished in the ΔflbA strain of A. niger and, as a consequence, protein secretion takes place throughout the colony (Krijgsheld et al. 2013b). Moreover, the cell wall of ΔflbA is thinner and intracellular proteins can be found in the culture medium, indicative of cell lysis. The ΔflbA strain therefore has a pleiotropic phenotype. Inactivation of flbA is accompanied by differential expression of 38 transcription factor genes, of which 18 are down-regulated and 20 are up-regulated (Krijgsheld and Wösten 2013). These downstream regulatory genes may impact one or more of the processes affected by FlbA. Here, the role of the most down-regulated predicted transcription factor gene, fum21, was studied. It is shown that fum21 regulates production of the mycotoxins fumonisins. Thus, sporulation-inhibited protein secretion is linked to production of secondary metabolites via FlbA.
Materials and methods
Strains and culture conditions
A. niger strain MA234.1 (transient ku70::amdS) (Park et al. 2016) and its derivatives were grown at 30 °C using minimal medium (MM) (de Vries et al. 2004) containing 25 mM xylose as a carbon source and either (MM-XA) or not (MM-X) containing 1.5% agar. Alternatively, strains were grown on Czapek Yeast Auto-lysate (CYA) agar (Frisvad and Samson 2004), Yeast Extract Sucrose (YES) agar (Frisvad and Samson 2004), or transformation medium (TM; MM containing 0.5% yeast extract, 0.2% casaminoacids, and 25 mM glucose, pH 6)(Kusters-van Someren et al. 1991).
Conidia were harvested from 3-day-old MM-XA cultures. Liquid cultures inoculated with 5 × 108 spores were pre-grown for 16 h at 200 rpm in 300 ml TM in 500 ml Erlenmeyer flasks. After 16 h, 10 g wet weight mycelium was harvested, washed with 0.9% NaCl, and transferred to a 1 l Erlenmeyer flask containing 150 ml MM-X. The culture was shaken at 250 rpm and 30 °C for 4 h (RNA sequencing) or 24 h (SDS-PAGE).
For growth on agar media, strains were inoculated directly on the medium or grown in a layer of 1.25% agarose in between two perforated polycarbonate membranes (pores of 0.1 µm, diameter 76 mm; Profiltra, Almere, The Netherlands) (Wösten et al. 1991). These sandwiched cultures were inoculated in the center of the agarose layer by placing a 2 µl drop containing 104 conidia. The upper polycarbonate membrane was placed on top of the agarose layer 24 h after inoculation to prevent spreading of conidia.
Inactivation and complementation constructs of fum21
For the construction of the fum21 deletion strain, 5′ and 3′ flanks of the gene were amplified from genomic DNA by PCR using primer pairs 1/2 and 3/4, respectively (Supplemental Table 1). The hygromycin resistance gene hph was amplified from vector pAN7.1 (Punt et al. 1987) using primer pair 5/6 (Supplemental Table 1). Split marker fragments of this selection marker were created by fusion PCR (Arenthorst et al. 2015) using primer pairs 1/7 and 4/8 (Supplemental Table 1) for the 5′ and 3′ fragment, respectively.
For the construction of the fum21 complementation construct, the hygromycin resistance cassette contained in pAN7.1 was amplified using primers 9 and 10 (Supplemental Table 1) that contain XbaI sites at their 5′ ends. The amplified 3 kb fragment that had a shorter promoter and terminator region as compared to pAN7.1 was cloned in the XbaI site of pUC20, resulting in vector pWR11. Gene fum21 with 1048 bp promoter and 444 bp terminator sequence was amplified from MA234.1 genomic DNA using Phusion polymerase (Thermo Fisher Scientific, Waltham, MA, USA) and primer pair 11/12 (Supplemental Table 1). The amplified fragment was inserted in the SmaI site of pWR11 by using the InFusion® HD Cloning Kit (Clontech, Mountain View, CA, USA). This resulted in vector pDA1 containing the hygromycin resistance cassette and gene fum21.
Transformation of A. niger
Transformation of A. niger was performed according to de Bekker et al. (2009). For inactivation of fum21, the split marker fragments were transformed to strain MA234.1. Transformants were purified twice on MM-XA containing hygromycin. Deletion of fum21 was verified by Southern blot analysis using HindIII digested genomic DNA (Supplemental Fig. 1). For complementation, the disrupted kusA gene in the Δfum21 strain was first restored by selecting AmdS loop-out strains on 5′ fluoroacetamide (Carvalho et al. 2010) to facilitate ectopic integration. Since the fum21 deletion strain was already hygromycin resistant, the resulting Δfum21 kusA + strain was complemented by co-transforming vectors pDA1 and pMA357. The latter pJet1.2 (Thermo Fisher Scientific) based vector contains the amdS gene and 3′ regulatory sequences of Aspergillus nidulans under control of the gpdA promoter. The amdS expression cassette (Meyer et al. 2007) was PCR amplified with primer pair 13/14 (Supplemental Table 1). Selection was done on MM containing 0.95 M sucrose, 15 mM CsCl, and 10 mM acetamide as sole nitrogen source. Integration of the wild-type copy of the gene was confirmed by Southern blot analysis (Supplemental Fig. 1).
RNA sequencing and analysis
Mycelium of biological duplicates of liquid cultures pre-grown in TM and transferred to MM-X was frozen in liquid nitrogen and ground for 1 min at 25 Hz with a Tissue Lyzer II (Qiagen, Venlo, The Netherlands). Samples were taken up in 1 ml TRIzol reagent (Invitrogen, Bleiswijk, The Netherlands) and incubated for 5 min at room temperature (RT). 0.2 ml chloroform was added and samples were centrifuged for 15 min at 4 °C and 12,000×g after 2 min incubation at RT. Total RNA was precipitated from the resulting aqueous phase by addition of 0.5 ml isopropanol, incubation at RT for 10 min, and centrifugation for 10 min at 4 °C and 12,000×g. RNA was washed with 1 ml 75% ethanol, left to dry, and resuspended in RNAse-free water. RNA was purified using the NucleoSpin® RNA kit (Macherey-Nagel, Düren, Germany). Concentration and purity of RNA was checked using the Nanodrop ND-1000 (Thermo Fisher Scientific).
Library preparation, cluster generation, and sequencing of cDNA were performed by ServiceXS (Leiden, The Netherlands) using Illumina sequencing. The reads are deposited in NCBI GEO with accession number GSE93990. Tophat version 2.1.13 (Trapnell et al. 2009) was used to align sequence reads to the Aspni7 version of the A. niger ATCC 1015 genome (Andersen et al. 2011), which was obtained from Mycocosm (Grigoriev et al. 2014). Functional annotation of the predicted genes was described previously (Teertstra et al. 2017). Cuffdiff (version 2.2.1), which is part of Cufflinks (Trapnell et al. 2010), was used to identify reads mapping to predicted genes and to identify differentially expressed genes. The bias correction method was used while running Cuffdiff (Roberts et al. 2011). The expression level of each predicted gene was normalized to fragments per kilobase of exon model per million fragments (FPKM). In addition to Cuffdiff’s requirements for differential expression the following requirements were applied: a ≥ 2-fold change and a minimal expression level of 4 FPKM in at least one of the samples. The quality of these results was analyzed using CummeRbund (Goff et al. 2013).
Q-PCR
Expression of fum10 (proteinID 1117227), fum8 (ProteinID 1117230), and fum1 (ProteinID 1162446) in A. niger wild type, Δfum21, and 4 complemented strains was assessed by Q-PCR using beta-tubulin (ProteinID 208263) and 18S rRNA (GenBank sequence ID KC545869.1) as reference genes. Total RNA was isolated as described above from biological duplicates of CYA- and YES-grown sandwiched colonies, after which it was purified using NucleoSpin® RNA (Macherey-Nagel, Düren, Germany) and reverse transcribed using the QuantiTect Reverse Transcription Kit (Qiagen, Venlo, The Netherlands). The cDNA (1 ng) was used for SYBR Green Q-PCR using 200 nM of the primer pairs 15/16 for fum10, (efficiency 98.7%), 17/18 for fum8 (efficiency 104.4%), 19/20 for fum1 (efficiency 109.7%), 21/22 for beta-tubulin (efficiency 102.1%), and 23/24 for 18S (efficiency 95.8%) (Supplemental Table 1). No-template controls (NTCs) were included as a negative control and total reaction volumes were 10 µl. Samples were run on a ViiA™ 7 Real-Time PCR System (Applied Biosystems, Wilmington DE, USA) and analyzed using the ΔΔCt method.
SDS-PAGE
Proteins in culture medium were precipitated overnight at − 20 °C after adding 4 volumes of acetone. They were pelleted twice for 30 min at 4 °C and 21,000×g with an intermediate washing step using − 20 °C acetone. After drying the pellets, sample buffer (125 mM Tris pH 6.8, 4% sodium dodecyl sulfate (SDS), 17.4% glycerol, 5% β-mercaptoethanol, 200 µg/ml bromophenol blue) was added resulting in a 20-fold volume reduction when compared to the culture medium. Samples were incubated at 100 °C for 10 min and proteins were separated in 12.5% SDS-PAGE gels using Pierce™ Prestained Protein Molecular Weight Marker (Thermo Fisher Scientific) as reference. Gels were fixed with 50% methanol and 10% acetic acid for 10 min, stained overnight with 0.1% Coomassie Brilliant Blue R-250, and destained with 25% methanol, 10% acetic acid. Gels were imaged using the Universal Hood III with Image Lab software (Bio-Rad Laboratories BV, Veenendaal, The Netherlands).
Localization of protein secretion
Protein secretion was monitored as described (Krijgsheld et al. 2013b) by labelling sandwiched colonies that had been grown on MM-XA for 6 days and transferred for 24 h to fresh MM-XA containing 14C-amino acids (NEC-850E amino acid mixture, L-[14C(U)]-, Perkin Elmer, Waltham MA, USA).
Protease activity
A. niger was grown as a sandwiched colony (see above) on MM containing 33% skimmed milk and 1.5% agar. After 5 days of growth, the sandwiched culture was removed and presence of halos monitored.
Secondary metabolite profiling
CYA and YES plates were inoculated in duplicate with 104 conidia. After 7 days of growth at 25 °C, 56-mm wide mycelial plugs were taken along the diameter of the colony and freeze-dried. Secondary metabolites were extracted in duplicate from the plugs with ethylacetate/dichloromethane/methanol (3:2:1, vol/vol/vol) with 1% formic acid using ultrasonic treatment for 50 min (Smedsgaard 1997). The extracts were transferred to a 2 ml dram vial and taken up in 300 µl methanol after removing the organic solvents by evaporation. Pyranonigrin A, pyranonigrin X (i.e. pyranonigrin B, C, D, E, or S), kotanin, BMS 192548, aurasperone B, tensidol B, and funalenone were analyzed using ultra-high performance liquid chromatography (UHPLC) (Houbraken et al. 2012). These compounds were identified using diode array detection (UV spectra from 190-600 nm) with purified compounds as standard. The relative quantity of the metabolites was estimated based on absorption at 210 nm (milli absorption units, mAU). Fumonisin B2, B4, and B6 were quantified by UHPLC-High Resolution Mass Spectrometry (UHPLC-HRMS) using an Agilent Infinity 1290 UHPLC system (Agilent Technologies, Santa Clara, CA, USA) equipped with a diode array detector. Separation was obtained on an Agilent Poroshell 120 phenyl-hexyl column (2.1 × 250 mm, 2.7 μm) with a linear gradient consisting of water (A) and acetonitrile (B) both buffered with 20 mM formic acid, starting at 10% B, increased to 100% B in 15 min, and held for 2 min at this composition, returned to 10% B in 0.1 min and held for 3 min at this composition (0.35 ml min−1, 60 °C). An injection volume of 1 μl was used. MS detection was performed on an Agilent 6540 QTOF MS equipped with Agilent Dual Jet Stream electrospray ion source with a drying gas temperature of 250 °C, gas flow of 8 l min−1, sheath gas temperature of 300 °C and flow of 12 l min−1. Capillary and nozzle voltage were set at 4000 and 500 V, respectively. Mass spectra were recorded at 10, 20, and 40 eV as centroid data for m/z 85–1700 in MS mode and m/z 30–1700 in MS/MS mode, with an acquisition rate of 10 spectra/s. Lock mass solution in 70:30 methanol: water was infused in the second sprayer using an extra LC pump at a flow of 15 μl min−1 using a 1:100 splitter. The solution contained 1 μM tributylamine (Sigma-Aldrich) and 10 μM Hexakis (2,2,3,3-tetrafluoropropoxy)phosphazene (Apollo Scientific Ltd., Cheshire, UK) as lock masses. The [M + H] + ions (m/z 186.2216 and 922.0098, respectively) of both compounds was used (Kildgaard et al. 2014; Klitgaard et al. 2014).
Results
Functional characterization of fum21
Thirty eight predicted transcription factor genes are differentially expressed in xylose-grown colonies of ΔflbA when compared to wild type (Krijgsheld and Wösten 2013). Of these genes, An01g06900 (Cerqueira et al. 2014) is the most down-regulated predicted transcription factor gene with a 22, 42, and 31 fold-change in the central, middle, and the outermost concentric zone of the colony, respectively (Krijgsheld and Wösten 2013). This gene showed a bi-directional hit with the fumonisin regulator fum21 of Fusarium (Proctor et al. 2013) showing 29% identity at amino acid level and sharing the GAL4 DNA binding domain and the Middle Homology Region (MHR) domain (Supplemental Fig. 2). The fact that An01g06900 (i.e. fum21) is located in the predicted fumonisin gene cluster of A. niger (Supplemental Fig. 2) supports a role of this gene in fumonisin production.
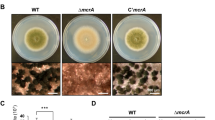
Gene fum21 was deleted in A. niger strain MA234.1, resulting in strain Δfum21 (Supplemental Fig. 1). Growth and conidiation were not affected in the deletion strain. Biomass of 7-day-old wild-type and Δfum21 sandwiched colonies was 17.20 ± 3.4 mg and 17.43 ± 2.0 mg (± SD, n = 7, p > 0.05), while colony diameter was 4.6 ± 0.63 cm and 4.9 ± 0.29 cm (± SD, n = 6, p > 0.05). Conidiation in the two strains took place in the sub-peripheral zone and the center of the colonies (Fig. 1a, b). These sporulation zones did not secrete proteins (Fig. 1c, d). SDS-PAGE protein profiles obtained in liquid cultures were not different between the strains (data not shown). Degradation of skimmed milk (data not shown) and the number of spores that were produced also did not differ (Fig. 1e). These data show that conidiation and sporulation-inhibited secretion are still functional in Δfum21.

Spatial distribution of sporulation (a, b), protein secretion (c, d), and the number of spores that were produced (e) in 8-day-old xylose-grown colonies of the wild-type strain MA234.1 (a, c) and Δfum21 (b, d). Secretion was monitored by immobilizing 14C-labeled secreted proteins on a PVDF membrane that had been placed underneath the colony. Localization and quantification of sporulation was monitored 48 h after removal of the upper membrane of sandwiched colonies
Strain Δfum21 and its progenitor were grown for 7 days on CYA. Fumonisin B2, B4, and B6 could be extracted from mycelial plugs from different zones of wild-type colonies. In contrast, Δfum21 did not produce these metabolites (Fig. 2a). Similar results were obtained when colonies were grown on YES medium (data not shown). Production of pyranonigrin A was also reduced by 25% in the deletion strain but production of other secondary metabolites was not affected when Δfum21 and the wild type were grown on CYA (Fig. 2b) and YES (data not shown) medium.

Amount of fumonisin B2, B4, and B6 (FB2, FB4, and FB6, respectively) (a) and other secondary metabolites (b) in arbitrary units (a.u.), produced by the wild-type strain MA234.1 (open bars) and Δfum21 (gray shaded bars) in CYA medium. Asterisk indicates significant differences (p ≤ 0.05) between the two strains indicated by the horizontal line below the asterisk
Gene expression analysis
Gene expression in Δfum21 and the wild-type progenitor strain was assessed by RNA sequence analysis after growth in TM for 16 h followed by transfer to MM-X for 4 h. This analysis revealed that only 74 genes were differentially expressed in Δfum21 relative to the wild type. Of these genes, 63 were down-regulated and 11 were up-regulated in the deletion strain (Table 1; Supplemental Table 2). Of the up-regulated genes, 7 have a predicted signal sequence for secretion including genes encoding lipases, a protease, a cellobiohydrolase and an anti-fungal protein. Of the down-regulated genes, 12 genes encode a protein without annotation and 24 genes are predicted to be involved in secondary metabolism. Of the latter genes, 20 are located in the secondary metabolism clusters 1 (3 out of 21 genes down-regulated), 12 (4 out of 13 genes down-regulated), 70 (3 out of 6 genes down-regulated), and 15 (10 out of 12 genes down-regulated). The secondary metabolites produced by the proteins encoded by clusters 1 and 12 are unknown but clusters 70 and 15 are predicted to be involved in TAN-1612 and fumonisin production, respectively (Li et al. 2011; Khaldi and Wolfe 2011). The down-regulated genes in the fumonisin cluster included fum10, fum8, and fum1 (Table 2), which are predicted to be involved in the early catalytic steps of fumonisin synthesis in Fusarium (Proctor et al. 2003). Q-PCR analysis showed that their expression was absent in Δfum21 of A. niger and restored in two complemented strains (data not shown).
Expression of the 74 differentially expressed genes in Δfum21 was assessed in the central, intermediate, and outer zones of wild-type and ΔflbA colonies using data of Krijgsheld and Wösten (2013). Six out of 11 up-regulated genes in Δfum21 were down-regulated in ΔflbA (Supplemental Table 3). Conversely, 10 out of the 63 down-regulated genes in Δfum21 were up-regulated in ΔflbA. In addition, 13 of the down-regulated genes in Δfum21 were also down-regulated in ΔflbA among which all genes of the fumonisin cluster (Supplemental Table 4). The genes in the fumonisin cluster were highly expressed at the periphery of xylose-grown colonies of A. niger, 6 of which were statistically significantly more highly expressed (i.e. exhibited > 2-fold higher expression levels) in this zone when compared to the more inner zones. Gene fum21 was not differentially expressed in zones of the wild-type colonies.
Discussion
Most wild-type A. niger strains produce fumonisin in liquid and solid cultures (Frisvad et al. 2007, 2011). Fumonisins are potent mycotoxins that exhibit neurotoxicity, hepatotoxicity, and nephrotoxicity in various animal models. They have also been linked to tumor formation in humans (esophageal cancer) and animals (Stockmann-Juvala and Savolainen 2008). The fumonisin biosynthesis cluster of A. niger has been proposed to originate from a horizontal gene transfer event (Khaldi and Wolfe 2011). The Fusarium cluster consists of up to 17 genes depending on the species (Proctor et al. 2003, 2008; Brown et al. 2007; Wiemann et al. 2013). Of these genes, expression of at least fum1 and fum8 is controlled by the transcriptional regulator Fum21 (Brown et al. 2007). A bidirectional BLAST showed that gene An01g06900 of A. niger is the orthologue of fum21 of Fusarium. Inactivation of fum21 in A. niger did not impact vegetative growth, mycelium morphology, conidiation, and (spatial) secretion of proteins. In contrast, Δfum21 of A. niger did not produce fumonisin, while expression of 10 out of 12 genes of the fumonisin gene cluster was reduced. These data show that Fum21 controls fumonisin production as was previously shown in Fusarium (Brown et al. 2007).
The function of Fum21 of A. niger and Fusarium verticillioides is remarkably similar despite the evolutionary distance between these species that belong to the Sordariomycetes and the Eurotiomycetes, respectively. Both proteins activate fumonisin production, while not affecting other processes such as growth and sporulation (Brown et al. 2007). In addition, both fum21 homologs are regulated by genes involved in asexual development; i.e. the VeA homolog FvVE1 in F. verticillioides (Myung et al. 2009) and flbA in A. niger. It should be noted that deletion of fum21 in A. niger completely abolished fumonisin production, while some fumonisin can still be detected in the fum21 deletion strain of F. verticillioides. In the latter case the transcription factor genes pac1 and zfr1 also impact biosynthesis of this secondary metabolite (Shim and Woloshuk 2001; Flaherty et al. 2003; Flaherty and Woloshuk 2004; Bluhm and Woloshuk 2006). A. niger has orthologues of both genes. Future studies should confirm a role of these genes in fumonisin production. They might for instance regulate fum15 and fum19 that were shown not to be regulated by fum21. Results also showed that Fum21 of A. niger impacts production of the secondary metabolite pyranonigrin A. Indeed, absence of Fum21 of A. niger affected expression of genes of three other secondary metabolite clusters. Whether Fum21 of F. verticillioides also affects expression of other secondary metabolite clusters is not known.
As mentioned above, fum21 is down-regulated in ∆flbA (Krijgsheld and Wösten 2013). This implies that FlbA is not only involved in conidiation, spatial secretion of proteins, composition of the secretome, cell wall architecture, and lysis of hyphae (Krijgsheld et al. 2013b) but also in controlling secondary metabolism. Indeed, all genes of the fumonisin cluster are down-regulated in ∆flbA (Krijgsheld and Wösten 2013). The fact that Fum21 does not control expression of 2 out of 12 genes of the cluster implies that FlbA impacts expression of another transcription factor involved in fumonisin production. Of interest, the pac1 homologue of A. niger (known as pacC) is up-regulated in ∆flbA (Krijgsheld and Wösten 2013). Possibly, this transcription factor is a repressor of genes in this mycotoxin cluster.
LaeA was initially identified as a regulator of secondary metabolism in A. nidulans (Bok and Keller 2004). It controls expression of several gene clusters, including clusters involved in production of sterigmatocystin, penicillin, and lovastatin. LaeA is also involved in secondary metabolism in A. niger. It does not impact fumonisin production but it represses production of the compounds BMS-192548 and aspernigrin A, while activating production of asperrubrol, atromentin, and JBIR86 (Niu et al. 2015). Experimental data showed that production of aurasperone B, funalenone, kotanin, and tensidol are neither regulated by Fum21 (this work) nor by LaeA (Niu et al. 2015). This implies that other regulatory genes are involved in the production of these secondary metabolites. The fact that expression of laeA is not affected by FlbA in A. niger implies that its regulation is different from that of fum21. Together, these results indicate that fum21, laeA, and other transcriptional regulators are involved in secondary metabolite production in A. niger. These genes are potential targets to improve A. niger as a cell factory by minimizing production of mycotoxins.
References
Andersen MR, Salazar MP, Schaap PJ, van de Vondervoort PJ, Culley D, Thykaer J, Frisvad JC, Nielsen KF, Albang R, Albermann K, Berka RM, Braus GH, Braus-Stromeyer SA, Corrochano LM, Dai Z, van Dijck PW, Hofmann G, Lasure LL, Magnuson JK, Menke H, Meijer M, Meijer SL, Nielsen JB, Nielsen ML, van Ooyen AJ, Pel HJ, Poulsen L, Samson RA, Stam H, Tsang A, van den Brink JM, Atkins A, Aerts A, Shapiro H, Pangilinan J, Salamov A, Lou Y, Lindquist E, Lucas S, Grimwood J, Grigoriev IV, Kubicek CP, Martinez D, van Peij NN, Roubos JA, Nielsen J, Baker SE (2011) Comparative genomics of citric-acid-producing Aspergillus niger ATCC 1015 versus enzyme-producing CBS 513.88. Genome Res 21:885–897
Arenthorst M, Jing N, Ram AFJ (2015) Efficient generation of Aspergillus niger knock out strains by combining NHEJ mutants and a split Marker Approach. In: van den Berg MA, Maruthachalam K (eds) Genetic transformation systems in fungi, vol 1. Springer, Basel, pp 263–272
Bluhm BH, Woloshuk CP (2006) Fck1, a C-type cyclin-dependent kinase, interacts with Fcc1 to regulate development and secondary metabolism in Fusarium verticillioides. Fungal Genet Biol 43:146–154
Bok JW, Keller NP (2004) LaeA, a regulator of secondary metabolism in Aspergillus spp. Eukaryot Cell 3:527–535
Brown DW, Butchko RAE, Busman M, Proctor RH (2007) The Fusarium verticillioides FUM gene cluster encodes a Zn(II)2Cys6 protein that affects FUM gene expression and fumonisin production. Eukaryot Cell 6:1210–1218
Carvalho ND, Arentshorst M, Jin Kwon M, Meyer V, Ram AFJ (2010) Expanding the ku70 toolbox for filamentous fungi: establishment of complementation vectors and recipient strains for advanced gene analyses. Appl Microbiol Biotechnol 87:1463–1473
Cerqueira GC, Arnaud MB, Inglis DO, Skrzypek MS, Binkley G, Simison M, Miyasato SR, Binkley J, Orvis J, Shah P, Wymore F, Sherlock G, Wortman JR (2014) The Aspergillus Genome Database: multispecies curation and incorporation of RNA-Seq data to improve structural gene annotations. Nucl Acids Res 42:D705–D710
de Bekker C, Wiebenga A, Aguilar G, Wösten HAB (2009) An enzyme cocktail for efficient protoplast formation in Aspergillus niger. J Microbiol Methods 76:305–306
de Vries RP, Burgers K, van de Vondervoort PJ, Frisvad JC, Samsom RA, Visser J (2004) A new black Aspergillus species, A. vadensis, is a promising host for homologous and heterologous protein production. Appl Environ Microbiol 70:3954–3959
Flaherty JE, Woloshuk CP (2004) Regulation of fumonisin biosynthesis in Fusarium verticillioides by a zinc binuclear cluster-type gene ZFR1. Appl Environ Microbiol 70:2653–2659
Flaherty JE, Pirttila AM, Bluhm BH, Woloshuk CP (2003) PAC1, a pH-regulatory gene from Fusarium verticillioides. Appl Environ Microbiol 69:5222–5227
Frisvad JC, Samson RA (2004) Polyphasic taxonomy of Penicillium subgenus Penicillium. A guide to identification of the food and air-borne terverticillate Penicillia and their mycotoxins. Stud Mycol 49:1–173
Frisvad JC, Smedsgaard J, Samson RA, Larsen TO, Thrane U (2007) Fumonisin B2 production by Aspergillus niger. J Agric Food Chem 55:9727–9732
Frisvad JC, Larsen TO, Thrane U, Meijer M, Varga J, Samson RA, Nielsen KF (2011) Fumonisin and ochratoxin production in industrial Aspergillus niger strains. PLoS ONE 6:e23496
Goff L, Trapnell C, Kelley D (2013) CummeRbund: analysis, exploration, manipulation, and visualization of Cufflinks high-throughput sequencing data. R package version 2.16.0
Grigoriev IV, Nikitin R, Haridas S, Kuo A, Ohm R, Otillar R, Riley R, Salamov A, Zhao X, Korzeniewski F, Smirnova T, Nordberg H, Dubchak I, Shabalov I (2014) MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Res 42:D699–D704
Houbraken J, Spierenburg H, Frisvad JC (2012) Rasamsonia, a new genus for thermotolerant and thermophilic Talaromyces and Geosmithia species. Antonie van Leeuwenhoek J Gen Microbiol 101:403–421
Khaldi N, Wolfe KH (2011) Evolutionary origins of the fumonisin secondary metabolite gene cluster in Fusarium verticillioides and Aspergillus niger. Int J Evolut Biol. doi:10.4061/2011/423821
Kildgaard S, Mansson M, Dosen I, Klitgaard A, Frisvad JC, Larsen TO, Nielsen KF (2014) Accurate dereplication of bioactive secondary metabolites from marine-derived fungi by UHPLC-DAD-QTOFMS and MS/HRMS library. Marine Drugs 12:3681–3705
Klitgaard A, Iversen A, Andersen MR, Larsen TO, Frisvad JC, Nielsen KF (2014) Aggressive dereplication using UHPLC-DAD-QTOF – screening extracts for up to 3000 fungal secondary metabolites. Anal Bioanal Chem 406:1933–1943
Krijgsheld P, Wösten HAB (2013) Transcriptome analysis of zones of colonies of the ΔflbA strain of Aspergillus niger. Fungal Genomics Biol 3:1000109
Krijgsheld P, Bleichrodt R, van Veluw GJ, Wang F, Müller WH, Dijksterhuis J, Wösten HAB (2013a) Development in Aspergillus. Stud Mycol 74:1–29
Krijgsheld P, Nitsche BM, Post H, Levin AM, Müller WH, Heck AJR, Ram AFJ, Altelaar AFM, Wösten HAB (2013b) Deletion of flbA results in increased secretome complexity and reduced secretion heterogeneity in colonies of Aspergillus niger. J Prot Res 12:1808–1819
Kusters-van Someren MA, Harmsen JA, Kester HC, Visser J (1991) Structure of the Aspergillus niger pelA gene and its expression in Aspergillus niger and Aspergillus nidulans. Curr Genet 20:293–299
Li Y, Chooi YH, Sheng Y, Valentine JS, Tang Y (2011) Comparative characterization of fungal anthracenone and naphthacenedione biosynthetic pathways reveals an α-hydroxylation-dependent Claisen-like cyclization catalyzed by a dimanganese thioesterase. J Am Chem Soc 133:15773–15785
Meyer V, Arentshorst M, El-Ghezal A, Drews AC, Kooistra R, van den Hondel CA, Ram AF (2007) Highly efficient gene targeting in the Aspergillus niger kusA mutant. J Biotechnol 128:770–775
Meyer V, Wu B, Ram AFJ (2011) Aspergillus as a multi-purpose cell factory: current status and perspectives. Biotechnol Lett 33:469–476
Myung K, Li S, Butchko RA, Busman M, Proctor RH, Abbas HK, Calvo AM (2009) FvVE1 regulates biosynthesis of the mycotoxins fumonisins and fusarins in Fusarium verticillioides. J Agric Food Chem 57:5089–5094
Niu J, Arentshorst M, Nair PD, Dai Z, Baker SE, Frisvad JC, Nielsen KF, Punt PJ, Ram AFJ (2015) Identification of a classical mutant in the industrial host Aspergillus niger by systems genetics: LaeA is required for citric acid production and regulates the formation of some secondary metabolites. G3 6(1):193–204
Park J, Hulsman M, Arentshorst M, Breeman M, Alazi E, Lagendijk EL, Rocha MC, Malavazi I, Nitsche BM, van den Hondel CA, Meyer V, Ram AFJ (2016) Transcriptomic and molecular genetic analysis of the cell wall salvage response of Aspergillus niger to the absence of galactofuranose synthesis. Cell Microbiol 18:1268–1284
Proctor RH, Brown DW, Plattner RD, Desjardins AE (2003) Co-expression of 15 contiguous genes delineates a fumonisin biosynthetic gene cluster in Gibberella moniliformis. Fungal Genet Biol 38:237–249
Proctor RH, Busman M, Seo JA, Lee YW, Plattner RD (2008) A fumonisin biosynthetic gene cluster in Fusarium oxysporum strain O-1890 and the genetic basis for B versus C fumonisin production. Fungal Genet Biol 45:1016–1026
Proctor RH, Van Hove F, Susca A, Stea G, Busman M, van der Lee T, Waalwijk C, Moretti A, Ward TJ (2013) Birth, death and horizontal transfer of the fumonisin biosynthetic gene cluster during the evolutionary diversification of Fusarium. Mol Microbiol 90:290–306
Punt PJ, Oliver RP, Dingemanse MA, Pouwels PH, Van den Hondel CA (1987) Transformation of Aspergillus based on the hygromycin B resistance marker from Escherichia coli. Gene 56:117–124
Roberts A, Trapnell C, Donaghey J, Rinn JL, Pachter L (2011) Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol 12:R22
Samson RA, Visagie CM, Houbraken J, Hong S-B, Hubka V, Klaassen CHW, Perrone G, Seifert KA, Susca A, Tanney JB, Varga J, Kocsubé S, Szigeti G, Yaguchi T, Frisvad JC (2014) Phylogeny, identification and nomenclature of the genus Aspergillus. Stud Mycol 78:141–173
Shim WB, Woloshuk CP (2001) Regulation of fumonisin B1 biosynthesis and conidiation in Fusarium verticillioides by a cyclin-like (C-type) gene, FCC1. Appl Environ Microbiol 67:1607–1612
Smedsgaard J (1997) Micro-scale extraction procedure for standardized screening of fungal metabolite production in cultures. J Chromatogr A 760:264–270
Stockmann-Juvala H, Savolainen K (2008) A review of the toxic effects and mechanisms of action of fumonisin B1. Hum Exp Toxicol 27:799–809
Teertstra WR, Tegelaar M, Dijksterhuis J, Golovina EA, Ohm RA, Wösten HAB (2017) Maturation of conidia on conidiophores of Aspergillus niger. Fungal Genet Biol 98:61–70
Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25:1105–1111
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28:511–515
Wiemann P, Sieber CM, von Bargen KW, Studt L, Niehaus EM, Espino JJ, Huß K, Michielse CB, Albermann S, Wagner D, Bergner SV, Connolly LR, Fischer A, Reuter G, Kleigrewe K, Bald T, Wingfield BD, Ophir R, Freeman S, Hippler M, Smith KM, Brown DW, Proctor RH, Münsterkötter M, Freitag M, Humpf HU, Güldener U, Tudzynski B (2013) Deciphering the cryptic genome: genome-wide analyses of the rice pathogen Fusarium fujikuroi reveal complex regulation of secondary metabolism and novel metabolites. PLoS Pathog 6:e1003475
Wieser J, Lee BN, Fondon JW III, Adams TH (1994) Genetic requirements for initiating asexual development in Aspergillus nidulans. Curr Genet 27:62–69
Wösten HAB, Moukha SM, Sietsma JH, Wessels JGH (1991) Localization of growth and secretion of proteins in Aspergillus niger. J Gen Microbiol 137:2017–2023
Wösten HAB, van Veluw GJ, de Bekker C, Krijgsheld P (2013) Heterogeneity in the mycelium: implications for the use of fungi as cell factories. Biotechnol Lett 35:1155–1164
Yu J-H, Wieser J, Adams TH (1996) The Aspergillus FlbA RGS domain protein antagonizes G protein signaling to block proliferation and allow development. EMBO J 15:5184–5190
Funding
This study was funded by BE-BASIC under Flagship 10.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Aerts, D., Hauer, E.E., Ohm, R.A. et al. The FlbA-regulated predicted transcription factor Fum21 of Aspergillus niger is involved in fumonisin production. Antonie van Leeuwenhoek 111, 311–322 (2018). https://doi.org/10.1007/s10482-017-0952-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10482-017-0952-1