
Abstract
Ocular neovascularization is a comprehensive process involved in retinal vascular development and several blinding diseases such as age-related macular degeneration and retinopathy of prematurity, with vascular endothelial growth factor (VEGF) regarded as the master regulator. However, the qualified effect of anti-VEGF therapy reveals that the underlying mechanisms are still not clearly identified. To initialize angiogenesis, endothelial cells undergo a phenotype switching to generate highly migratory and invasive cells. This process shares certain similar characters observed in endothelial–mesenchymal transition (EndMT). Here, we found that SNAI1, an EndMT transcription factor, was expressed by endothelial cells in both physiological and pathological ocular neovascularization. SNAI1 overexpression triggered cell morphological change and enhanced cell motility, while loss of SNAI1 attenuated migration, invasion and sprouting. RNA sequence analysis further revealed that SNAI1 knockdown decreased the expression of genes related to cytoskeleton rearrangement and ECM remodeling. Moreover, intravitreal injection of small interfering RNA of SNAI1 suppressed new vessel formation in developing retina as well as mice model of choroidal neovascularization and oxygen-induced retinopathy. Therefore, we propose that the EndMT transcription factor SNAI1 promotes the early phase of ocular neovascularization and may provide a potential therapeutic target.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ocular angiogenesis is a highly coordinated process involved in retinal vasculature development and several ocular diseases such as age-related macular degeneration (AMD), proliferative diabetic retinopathy (PDR) and retinopathy of prematurity (ROP) [1, 2]. During neovascularization, sprouting endothelial cells (ECs) degrade the basement membrane (BM) with a series of diffusing and membrane-bound proteolytic enzymes, which are mainly involved in two classes: plasminogen activator (PA)/plasmin system and matrix metalloproteinases (MMPs) [3]. Then, ECs begin to proliferate, invade and migrate along components of the extracellular matrix (ECM) and take part in the formation of an immature capillary structure and deposition of a new complex BM [4]. This process involves complex and highly dynamic interactions between ECs and their environment [5]. Though extensive researches have suggested that multiple factors participate into this growth program, with vascular endothelial growth factor (VEGF) signaling pathway emerging as a master regulator [6, 7], the underlying cellular and molecular processes still need to be fully identified.
Endothelial cell plasticity is critical for development and multiple biological processes including angiogenesis [8]. During angiogenesis, activated ECs act more invasively compared to quiescent phase, including formation of lamellipodia and filopodia, degradation of BM, migration into ECM and participation in ECM remodeling. These functional changes share several similarities with endothelial–mesenchymal transition (EndMT), in which process ECs undergo a phenotype switching to generate elongated, spindle-shaped mesenchymal-like cells which are highly migratory and invasive [9,10,11]. These common points implied a possible role for EndMT transcription factors in angiogenesis.
The recognized EndMT transcription factors include the zinc-finger binding transcription factors Snail family and several basic helix-loop-helix (bHLH) factors such as ZEB1, ZEB2 and TWIST [12]. Among them, the Snail family plays the most critical role. In vertebrates, the Snail family contains three members, characterized up to now, which are SNAI1 (Snail), SNAI2 (Slug) and SNAI3 [13]. SNAI1 and SNAI2 were reported to trigger EndMT under certain conditions [14,15,16,17,18]. Apart from its functions in EndMT, evidences have indicated that Snail family had a role in angiogenesis. ECs-specific deletion of SNAI1 was embryonic lethal, while inducible SNAI1 loss of function caused vessel defects [19]. Both SNAI1 and SNAI2 were reported to be increased in angiogenic ECs in vitro, and decreased as lumen stabilizing, which suggested their roles in initializing angiogenesis [20, 21]. Therefore, it would be intriguing to investigate if Snail family members have a role in ocular angiogenesis. Previous expression array data showed that SNAI1 was abundant in retina among the three family members [22]. Thus, in this study, we focused on SNAI1 expression and function during physiological and pathological ocular angiogenesis. We further tested its function under VEGF stimulus and evaluated its therapeutic potential as well.
Materials and Methods
Animals
Pups and 8–10-week-old male C57/BL6 mice were maintained under specific pathogen-free conditions. All animal experiments were approved and followed the guidelines issued by the Animal Experiment Administration Committee of the Fourth Military Medical University and also complied with the ARVO Statement.
Small interfering RNA (siRNA) of mouse SNAI1 (siSNAI1) for in vivo studies was selected among 3 sets (RiboBio) and manufactured as in vivo modified STABLE siSNAI1 by RiboBio. 5 nmol Cy3-labeled negative control (NC) or siSNAI1 was dissolved in 25 μL RNA-free phosphate-buffered saline (PBS) to prepare working solution.
CNV model was induced by laser photocoagulation as described before [23, 24]. Briefly, mice were anesthetized with pupils dilated. Laser photocoagulation was performed on both eyes of mice using a 532 nm laser (Iris Radiation Systems, USA; spot size 75 μm, power 100 mw, duration 0.1 s) through a contact lens. Five laser spots were made uniformly between the major retinal vessels, 1.5–2 disk diameters from the optical disk. Disruption of Bruch’s membrane was confirmed when a cavitation bubble appeared without hemorrhage. Modified NC or siSNAI1 were injected intravitreally at 0.1 nmol in either eye of adult mice on day 1 (d1) after photocoagulation if needed. Mice were killed at d7.
Oxygen-induced retinopathy (OIR) model was induced as described before [25]. The pups and their mothers were placed in a high-oxygen chamber (75 ± 2%) by post-natal day 7 (P7) for 5 days and then returned to room air (normoxic) conditions. Modified NC or siSNAI1 were injected intravitreally at 0.1 nmol in either eye of pups on P12 if needed. Pups were killed at P17.
Cell culture, adenovirus infection and siRNA transfection
Human umbilical vein endothelial cells (HUVECs) were isolated from human umbilical cord biopsies, which were obtained from the Department of Gynecology and Obstetrics of Xijing Hospital, Fourth Military Medical University, by collagenase treatment. Informed consent was obtained from all individual participants included in the study. Cells at passages 2–6 were used and cultured in EC medium (ECM) (ScienCell) supplemented with 5% FBS, EC growth supplements (ECGS), 100 U/mL penicillin and 100 μg/mL streptomycin in a humidified atmosphere with 5% CO2 at 37 °C. For cytokine stimuli, HUVECs were starved in ECM with 0.5% FBS and without ECGS for 24 hours (24 h) and then treated with VEGF-165 (Sino Biological) at 25, 50 or 100 ng/mL for 6 h. Cells were treated with 20 μM cyclohexane (CHX) or 50 μM MG132 if necessary. MRC5 and bEnd.3 cells (ATCC) were cultured in Dulbecco’s modified eagle medium (DMEM) (Gibco) containing 10% FBS.
HUVECs were infected with SNAI1 overexpression adenovirus (Ad-SNAI1) and GFP-labeled ctrl adenovirus (Ad-ctrl) (Vigene) at a multiplicity of infection (MOI) of 50 according to the operation procedures. HUVECs or bEnd.3 cells were transfected with NC or siSNAI1 at a concentration of 50 nM using Lipofectamine 2000 reagent (Invitrogen), according to the manufacturer’s instructions. siRNA for human SNAI1 (RiboBio) was selected from 2 sets.
Immunostaining
Retinal flat mounts were performed on P7, P14 or P17 for retinal vascular development analysis, while choroidal flat mounts on d7 for CNV analysis, as described previously [23, 25, 26]. Retinal and RPE–choroid–sclera complex tissues were post-fixed in 4% paraformaldehyde (PFA) for 6 h at 4 °C and then blocked and permeabilized in PBS containing 1% bull serum albumin (BSA) and 0.5% Triton X-100 overnight. Isolectin B4 (IB4) (1:100, FL-1201 or HL-1102, Vector) and anti-SNAI1 antibody (1:80, MABE167, Millipore or 1:200, ab180714, Abcam) in PBS containing 1% BSA and 0.5% Triton X-100 were incubated overnight at 4 °C, followed by incubation with the following secondary antibodies in PBS overnight at 4 °C: Alexa Fluor 488-conjugated goat anti-mouse IgM mu chain secondary antibody (1:400, ab150121, Abcam) or DyLight 488-conjugated goat anti-rabbit IgG(H + L) secondary antibody (1:400, A-23220, Abbkine). Each step was followed by three washes in PBS for 10 minutes (10 min). The retinal or RPE–choroid–sclera complex tissues were flat-mounted on a glass slide and then covered under a dissecting microscope (Olympus). All flat mounts were examined and photographed under a confocal laser scanning microscopy (FV1000, Olympus).
Frozen sections of eyeballs were made as described before [23]. Sections were dried for 2 h at room temperature and then washed in PBS and blocked with PBS containing 1% BSA and 0.5% Triton X-100 at room temperature for 1 h. Slides were incubated with primary antibodies overnight at 4 °C and secondary antibodies for 2 h at room temperature and then counterstained with Hoechst (94403, Sigma-Aldrich) for 15 min at room temperature. Each step was followed by three washes in PBS for 10 min. Slides were closed with coverslips. Primary antibodies included anti-CD31 antibody (1:400, 102502, BioLegend) and anti-SNAI1 antibody (1:80, MABE167, Millipore or 1:200, ab180714, Abcam). Secondary antibodies were Alexa Fluor 594-conjugated donkey anti-Rat IgG (H + L) secondary antibody (1:400, A-21209, Invitrogen), Alexa Fluor 488-conjugated goat anti-mouse IgM mu chain secondary antibody (1:400, ab150121, Abcam) and DyLight 488-conjugated goat anti-rabbit IgG (H + L) secondary antibody (1:400, A-23220, Abbkine). Nuclei were stained with Hoechst (94403, Sigma-Aldrich). All photographs were taken under a confocal laser scanning microscopy (FV1000, Olympus).
Cells were fixed with 4% PFA for 30 min and blocked with PBS containing 1% BSA and 0.5% Triton X-100 for 30 min at room temperature. F-actin was stained with FITC-conjugated Phalloidine (Yeasen), and nuclei were stained with Hoechst (94403, Sigma-Aldrich). Each step was followed by three washes in PBS for 10 min. Slides were covered and analyzed with a fluorescence microscope (BX51, Olympus).
Quantitative reverse transcription polymerase chain reaction (qRT-PCR)
Total RNA was purified using TRIzol reagent (Invitrogen) according to manufacturer’s instructions. cDNA was synthesized using reverse transcription kit (Takara). qRT-PCR was performed, as described previously [23]. The primer sequences are shown in Table 1. Gene expression levels were reported as relative fold change, with β-actin as an internal control.
Western blotting
Cells were harvested in radio immunoprecipitation assay (RIPA) buffer (Beyotime) containing phenylmethylsulfonyl fluoride (PMSF) (Sigma-Aldrich) and then separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). Immunoblotting was performed with primary antibodies including: anti-SNAI1 antibody (1:1000, 3879, Cell Signaling Technology), anti-MMP2 antibody (1:1000, 10373-2-AP, Proteintech), anti-MMP9 antibody (1:1000, 10375-2-AP, Proteintech), anti-FURIN antibody (1:1000, 18413-1-AP, Proteintech), anti-CTSK antibody (1:1000, 11239-1-AP, Proteintech), anti-CTSS antibody (1:1000, ab134157, Abcam), anti-FN antibody (1:1000, 15613-1-AP, Proteintech), anti LAMB1 antibody (1:1000, 23498-1-AP, Proteintech), anti COL IV antibody (1:1000, 55131-1-AP, Proteintech) and anti-β-ACTIN antibody (1:5000, A5316, Sigma-Aldrich) on polyvinylidene fluoride (PVDF) membranes followed by incubation of horseradish peroxidase (HRP)-linked goat anti-rabbit IgG or horse anti-mouse IgG secondary antibody (1:2000, 7074 or 7076, Cell Signaling Technology). Each step was followed by three washes in PBS containing 0.1% Tween-20 (PBST) for 10 min. Membranes were observed using an enhanced chemoluminescence system (Clinx Science Instruments).
Cell proliferation assay
EdU incorporation was performed as described before [27]. HUVECs were seeded on 48-well plates and incubated with 50 μM EdU (5-ethynyl-2′-deoxyuridine) (RiboBio) in complete ECM for 2 h and then fixed with 4% PFA at room temperature for 30 min, followed by staining with Apollo 567 and Hoechst according to manufacturer’s instructions. Images were captured under a fluorescence microscope (BX51, Olympus).
Cell migration assay
Cell migration ability was tested by wound healing assay as described before [28]. In brief, a scratch was created with a p20 pipette tip, and the medium was then replaced by ECM supplemented with 0.5% FBS and without ECGS. Wound closure was observed under a microscope (CKX41, Olympus) with a CCD camera (DP70, Olympus) and measured at 12 h after the scratch was made.
Cell invasion assay
Cell invasion ability was observed by spheroid collagen type I invasion assay and tested by Transwell invasion assay based on published protocol [29, 30]. For spheroid collagen type I invasion assay, HUVECs at a total number of 5.0 × 104 in 100 μL EGM-2 medium (Lonza) were seeded on 96-well ultra-low attachment surface plates (Corning) for 2 days to generate cell spheres. Collagen type I (C8062, Solarbio) solution with a final concentration of 1 mg/mL was prepared according to the manufacturer’s instructions. The spheres were then embedded in the collagen gel, and 100 μL complete ECM with or without additional VEGF at 50 ng/mL was added. Invasion of spheres was observed the next day under a microscope (CKX41, Olympus) with a CCD camera (DP70, Olympus). For Transwell invasion assay, the upper chamber of Transwell chamber (Corning) was pre-coated with 100 μL Matrigel Basement Membrane Matrix (matrigel) (1:20, BD Biosciences) overnight at 37 °C and then cultivated with HUVECs at a total number of 1.0 × 105 in 200 μl ECM containing 0.5% FBS without ECGS. 500 μL complete ECM with 50 ng/ml VEGF-165 (Sino Biological) was added to the lower chamber. Cells were cultured for 12 h and were fixed by 4% PFA and stained with crystal violet. Number of cells invading to the lower chamber was observed under a microscope (CKX41; Olympus) with a CCD camera (DP70; Olympus).
Tube formation assay
Assessment of tube formation ability of HUVECs was performed as described previously [31]. HUVECs (1 × 105 cells per well) were loaded onto 200 μL matrigel (1:1, BD Biosciences) which was pre-coated in 48-well plates and incubated at 37 °C for 4 h. The number of loops and total length of cell cords in the enclosed lumen structures were determined.
Fibrin beads assay
A fibrin beads sprouting assay was conducted, as described previously, using a fibrin beads assay kit (Amersham-Pharmacia Biotech) according to the supplier’s instructions [31, 32]. Briefly, HUVECs were transfected and changed medium into EGM-2 medium (Lonza) the day before beading. Cytodex 3 microcarrier beads (Amersham) were incubated with HUVECs (400 cells per bead) at 37 °C for 4 h and then transferred to 12-well plates and left overnight. The beads were embedded in fibrinogen (Sigma-Aldrich) containing 0.625 U/mL thrombin (Sigma-Aldrich) at a density of 100 beads/mL in a 48-well plate the next day, and MRC5 cells (2 × 105 cells per well) in 0.5 mL EGM-2 medium were added. The medium was changed every other day. Sprouting was quantified by measuring the number and length of sprouts.
Aortic ring sprouting assay
Aortic ring assay was performed following previous protocol [33]. In brief, aortas were removed from adult mice, cleaned and transfected with 200 nM siSNAI1 or NC using Lipofectamine 2000 reagent (Invitrogen) overnight. The aortas were then embedded in 80 μL matrigel in a 48-well plate containing 500 μL opti-MEM Reduced Serum Media (Gibco) supplemented with 2.5% FBS and 30 ng/mL VEGF-164 (Sino Biological). Endothelial sprouts were allowed to grow over 4 days with medium changing every other day. The number and length of sprouts were measured.
RNA sequence analysis
HUVECs were transfected with siSNAI1 or NC as mentioned above and cultured in complete ECM for 24 h. Cells were then starved in ECM with 0.5% FBS and without ECGS for 24 h following VEGF-165 stimulus at 50 ng/mL for 6 h. RNA was extracted, sequenced and analyzed by RiboBio. In brief, total RNA was isolated from cells using TRIzol reagent (Invitrogen) according to the manufacturer’s protocol. RNA purity was assessed using the ND-1000 Nanodrop. Each RNA sample had an A260:A280 ratio above 1.8 and A260:A230 ratio above 2.0. RNA integrity was evaluated using the Agilent 2200 TapeStation (Agilent Technologies), and each sample had the RINe above 7.0. mRNAs were isolated from total RNA and fragmented to approximately 200 bp and subsequently subjected to first strand and second strand cDNA synthesis following by adaptor ligation and enrichment with a low-cycle according to instructions of TruSeq® RNA LT/HT Sample Prep Kit (Illumina). The purified library products were evaluated using the Agilent 2200 TapeStation and Qubit®2.0 (Life Technologies) and then diluted to 10 pmol/L for cluster generation in situ on the HiSeq 3000 pair-end flow cell followed by sequencing (2 × 150 bp) on HiSeq 3000. Prior to sequencing, the raw data were filtered to produce high-quality clean data. All the subsequent analyses were performed using the clean data.
The reference genome and gene model annotation files were downloaded directly from the NCBI library. The clean reads were aligned to the reference genome using TopHat (Broad Institute). During read alignment, a maximum of two mismatches and a gap length of 2 bps were allowed. The functional annotations of genetic variants were generated using ANNOVAR [34]. Read alignments were processed by Cufflinks software package [35] to determine the differential expression of genes. Gene expression values were quantified as reads per kilobase of transcript per million mapped reads (RPKM). Original data were deposited in the Gene Expression Omnibus (GEO) database (accession # GSE111386).
Retinal vascular development, CNV and OIR lesion assessment
The assessment of retinal vessels were based on published protocol [36]. Digitized whole retina images were reconstructed using Photoshop CS6 software (Adobe Systems). Average distance from optic disk to the edge of vasculature was assessed as the overall situation of vascular development. For vasculature density, capillary plexus area situated between an artery and a vein was selected and branching points were defined as junctions of capillary segments. Tip cells were counted per endothelial vessel length of 1000 μm in the angiogenic front. For each time point, six pups were treated in two independent experiments and four to six fields per retina were analyzed.
CNV lesion was analyzed as described formerly [23, 24]. Briefly, horizontal optical sections. (2 μm step) were obtained under a confocal microscopy from flat-mount RPE–choroid–sclera complex. The deepest focal plane in which the surrounding choroidal vascular network connecting to the lesion could be identified was judged to be the floor of the lesion. Any IB4 positive vessels in the laser-treated area and superficial to this reference plane was judged as CNV. Images of each section were digitally stored, followed three-dimensional (3D) reconstruction by IMARIS software (Bitplane). The summation of whole fluorescent area in each horizontal section was used as an index for the volume of CNV. Six mice were treated in two independent experiments, and five CNV spots per eye were analyzed.
OIR lesion was evaluated according to the proposed protocol [37]. The neovascular (NV) and total areas were manually outlined and quantified by a masked observer, and the ratio of NV to total retinal area was then calculated. Three pups were analyzed.
Statistics
Statistical analysis was performed with the Image-Pro Plus 6.0 and GraphPad Prism 5 software. All quantitative data were presented as mean ± SEM. Statistical significance was calculated using unpaired or paired Student’s t test. P < 0.05 was considered as significant.
Results
SNAI1 was expressed in sprouting vessels in both physiological and pathological ocular neovascularization
We first investigated whether SNAI1 expressed on sprouting vessels in developing retinal vasculature, CNV and OIR mice models. In the retina of neonatal mice, which displays a complete physiological vasculature development, SNAI1 mRNA showed a significant increase at P7 and remained increasing during neovascularization process till P14 (Fig. 1a). Whole flat-mount and fluorescent staining analysis at P7 and P14 showed SNAI1 expression in active vessels (IB4 staining) in both superficial and deep plexus, especially in deep-penetrating vessels from vein (Fig. 1b, d), which was further confirmed by 3D reconstruction (Fig. 1c) and double staining with SNAI1 and CD31 on retinal sections (Fig. 1e). In CNV and OIR models, which classically represent pathological choroidal and retinal neovascularization, respectively, an increase in SNAI1 mRNA was also detected by qRT-PCR (Fig. 1f). Whole flat-mount analysis of both choroidal tissues at CNV d7 and retinal tissues at OIR P17 showed SNAI1 immunoreactivity in areas of new vessels (Fig. 1g). To find out the source of SNAI1, we further carried out immunofluorescent double staining in eye ball sections of CNV and OIR and found that SNAI1 was mainly expressed by endothelial cells (Fig. 1h). These results demonstrated that SNAI1 was expressed in the sprouting vessels in both physiological and pathological ocular neovascularization.

SNAI1 was expressed in sprouting vessels in physiological and pathological ocular neovascularization. a qRT-PCR analyses showed SNAI1 mRNA expression level during retinal development at P3, P5, P7, P11 and P14. (n = 6 for each group). b Whole retinal tissues were flat-mounted at P7 and stained with IB4 (red) and anti-SNAI1 (green). The immunoreactivity of SNAI1 on superficial and deep retinal vascular plexus was observed under a confocal microscope. c 3D reconstruction figure of Fig. 1b showed SNAI1-positive descending ECs. d Flat-mounted retinal tissue at P14 was stained with IB4 (red) and anti-SNAI1 (green). The immunoreactivity of SNAI1 was detected on veins and deep venous vessels. e Eye balls of P14 mice were sectioned and stained with anti-CD31 (red), anti-SNAI1 (green) and Hoechst (blue). Cross-sectional confocal images showed SNAI1 expression in the descending vessels. F qRT-PCR analyses showed SNAI1 mRNA expression level during CNV after laser coagulation at d0, d7 and d14 (top), and during OIR at P12, P14 and P17 (bottom). (n = 6 for each group). g Whole choroidal tissues at CNV d7 (left) and retinal tissues at OIR P17 (right) were flat-mounted and stained with IB4 (red) and anti-SNAI1 (green) and were observed under a confocal microscope. SNAI1 was detected in pathological growing vessels. h Eye balls of mice at CNV d7 (left) and OIR P17 (right) were stained with anti-CD31 (red), anti-SNAI1 (green) and Hoechst (blue). Cross-sectional confocal images showed SNAI1 co-expression with CD31 (arrows) on endothelial cells. *p < 0.05, ***p < 0.001 Bar = 100 μm
SNAI1 overexpression improved cell motility and invasion in vitro
We then investigated SNAI1 function using adenovirus-infected HUVECs. SNAI1 mRNA was significantly increased in Ad-SNAI1 group compared with Ad-ctrl, which was further confirmed by Western blotting (Fig. 2a). Moreover, HUVECs treated by Ad-SNAI1 become elongated and spindle compared to original cobblestone-like shape in control group (Fig. 2b). SNAI1 gain of function in HUVECs promoted migration and invasion (Fig. 2c, d) but didn’t affect proliferation and tube formation (Fig. S1A, S1B).

SNAI1 overexpression improved cell motility and invasion in vitro. a qRT-PCR and Western blotting analyses showed SNAI1 mRNA expression level was significantly increased after Ad-SNAI1 infection. b Representative photographs showed cell morphological change after Ad-SNAI1 infection. c HUVECs infected with Ad-ctrl or Ad-SNAI1 for 48 h were wounded with a p20 pipette tip. Photographs were taken immediately and after 12 h, and average migration distance was calculated by the difference of gap widths of the same area (n = 4 for wound assay and 2 fields were calculated per sample). d HUVECs infected with Ad-ctrl or Ad-SNAI1 for 48 h were digested and reseeded on Transwell chamber pre-coated by matrigel. Cells were allowed to invade through the membrane for 12 h and then the percentage of invasive cell area was calculated. (n = 3 for each group and 3 fields were calculated per sample). ***p < 0.001 Bar = 100 μm
VEGF increased SNAI1 expression and generated invasive cells
VEGF is recognized to be highly expressed in angiogenic retina and plays the most important role in ocular angiogenesis. Therefore, we chose VEGF stimulus in vitro to study SNAI1 expression under angiogenic condition. SNAI1 mRNA increased after treated by VEGF-165 (Fig. 3a). Besides, VEGF-165 at 50 ng/mL was approved to be the best concentration to improve SNAI1 mRNA and was used in the following experiments (Fig. 3a). SNAI1 protein is highly unstable and tightly regulated by the ubiquitin–proteasome pathway [17], we thereby treated HUVECs with CHX to block protein synthesis or MG132 to inhibit proteasome-related protein degradation, respectively. Western blotting showed that SNAI1 protein increased after VEGF stimulus, with or without MG132 treatment (Fig. 3b). Moreover, CHX treatment remarkably weakened SNAI1 up-regulation (Fig. 3b). These results showed that VEGF stimulated SNAI1 synthesis in either transcriptional or translational level. Moreover, VEGF stimulus successfully generated elongated and invasive cells with enhanced lamellipodia formation (Fig. 3c).

VEGF increased SNAI1 expression and generated invasive cells. a qRT-PCR analysis showed SNAI1 mRNA expression level of HUVECs after treated with VEGF-165 at different concentration. (n = 3 for each group). b HUVECs were treated with DMSO only, DMSO plus VEGF-165 at 50 ng/mL, MG132 plus VEGF-165 and CHX plus VEGF-165 (left). SNAI1 protein level of HUVECs was detected by Western blotting. SNAI1 protein level of MG132-treated HUVECs with/without VEGF stimulus was also shown (right). c HUVECs were treated with or without VEGF-165 at 50 ng/mL. Bright field photographs showed that VEGF treatment generated spindle-like cells (top). Besides, VEGF-treated HUVEC spheres possessed the ability to invade through collagen type I (middle). Phalloidine (green) and Hoechst (blue) staining showed that VEGF improved lamellipodia formation at the leading edge (bottom). *p < 0.05 Bar = 100 μm
Loss of SNAI1 inhibited angiogenesis due to repressive cell motility and invasion in vitro
SNAI1 mRNA was significantly reduced by 61.21% after transfection, and the inhibition effect was further confirmed by Western blotting (Fig. 4a and 4b). We then examined the effect of SNAI1 knockdown on cell proliferation, migration, invasion, tube formation and finally sprouting ability. SNAI1 knockdown significantly inhibited cell migration and invasion (Fig. 4c, d), while had little effect on proliferation and slightly meliorated tube formation (Fig. S1C, S1D). Moreover, loss of SNAI1 impaired VEGF-induced cell morphological change and lamellipodia formation (Fig. 4e). Finally, we assessed the effect of SNAI1 knockdown on endothelial cell sprouting by aortic sprouting assay and fibrin bead assay, which showed that SNAI1 knockdown attenuated sprouting both in murine aorta and HUVECs (Fig. 5a, b). Taken together, our data confirmed that SNAI1 loss of function inhibited angiogenic sprouting in vitro, which could be attributed to repressive cell motility and invasion.

Loss of SNAI1 repressed cell motility and invasion in vitro. a qRT-PCR analysis showed SNAI1 mRNA was reduced by 61.21% after transfected with siSNAI1. (n = 3 for each group). b After transfected with siSNAI1 or NC for 24 h, HUVECs were starved for another 24 h and then treated with/without VEGF-165 for 6 h. SNAI1 mRNA and protein expression levels were detected by qRT-PCR and Western blotting. (n = 5 for qRT-PCR and n = 3 for Western blotting). c HUVECs transfected with siSNAI1 or NC for 48 h were wounded with a p20 pipette tip. Photographs were taken immediately and after 12 h, and average migration distance was calculated by the difference of gap widths of the same area. (n = 6 for wound assay and 4 fields were calculated per sample). d HUVECs transfected with siSNAI1 or NC for 48 h were digested and reseeded on Transwell chamber pre-coated by matrigel. Cells were allowed to invade through the membrane for 12 h, and then the percentage of invasive cell area was calculated. (n = 3 for each group and 3 fields were calculated per sample). e Phalloidine (green) and Hoechst (blue) staining showed relatively smooth leading edge combined with impaired lamellipodia formation in siSNAI1-treated HUVECs. The number of lamellipodia (arrows) per field was calculated. (n = 4 for each group and 4–6 fields were calculated per sample). *p < 0.05 **p < 0.01 ***p < 0.001 Bar = 100 μm

SNAI1 knockdown attenuated ECs sprouting. a Aortic rings isolated from adult mice were transfected with siSNAI1 or NC overnight and cultured for 4 days. Images were captured, and sprouting numbers and total sprouting length were compared between the two groups. (n = 4 for each group). b HUVECs were transfected and tested for sprouting with Fibrin Beads assay. Sprouting numbers per bead and total sprouting length from one bead were calculated to quantify endothelial sprouting. (n = 3 for each group and 10 beads were calculated per sample). *p < 0.05, ***p < 0.001 Bar = 100 μm
RNA sequence analysis showed that SNAI1 knockdown decreased genes involved in cytoskeleton arrangement, ECM degrading and remodeling
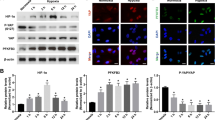
To further investigate how SNAI1 function in angiogenesis, we performed RNA sequence analysis between NC and siSNAI1-treated HUVECs under VEGF stimulus. GSEA results revealed a less enrichment of “Rac1 pathway” and “Rho pathway” in siSNAI1-transfected cells, which was in accordance with impaired cell motility (Fig. 6a). We next focused on genes related to ECM degradation and cell invasion and found a significant enrichment in control compared to siSNAI1-transfected cells (Fig. 6b). GSEA also revealed a decrease of genes related to “ECM component” and “ECM structural constituent” after SNAI1 knockdown (Fig. 6c). The typically changed genes with relatively high RPKM were picked up and shown in heat maps (Fig. 6b, c). Among them, MMP2, MMP9, FURIN, CTSK, MMP10 and CTSS were known ECM degradation-related genes, while FN, LAMA4, LAMB1, LAMC1 and COL IV were ECM components secreted by ECs. By qRT-PCR and Western blotting analysis, most of those genes showed significant decrease at either mRNA (Fig. 6d, e) or protein level in siSNAI1-treated HUVECs (Fig. 6f). Our data suggested that SNAI1 up-regulation upon VEGF stimulus mainly affected genes involved in cytoskeleton arrangement, ECM degrading and remodeling, which may account for its role in cell migration and invasion.

RNA sequence analysis showed that SNAI1 knockdown decreased genes involved in cytoskeleton arrangement, ECM degrading and remodeling. a GSEA results revealed “Rac1 pathway” and “Rho pathway” enriched in NC compared to siSNAI1-transfected cells. b GSEA results revealed genes related to “ECM disassembly” and “invasiveness” enriched in NC compared to siSNAI1-transfected cells. Heat map displayed the expression of typical regulated genes involved in ECM degradation. c GSEA results revealed genes related to “ECM component” and “ECM structural constituent” enriched in NC compared to siSNAI1-transfected cells. Heat map displayed the expression of typical regulated genes involved in ECM component. d qRT-PCR analysis showed that expression of typical regulated genes involved in ECM degradation, including MMP2, MMP9, FURIN, CTSK, MMP10 and CTSS were significantly decreased after SNAI1 knockdown. (n = 4 for each group.) e qRT-PCR analysis showed that expression of typical regulated genes involved in ECM component, including FN, LAMB1, LAMC1, COL4A1 and COL4A2 were significantly decreased after SNAI1 knockdown. However, we did not detect down-regulation of LAMA4 mRNA. (n = 4 for each group). f Western blotting analysis showed that expression of typical regulated genes involved in ECM degradation and component, including MMP2, MMP9, FURIN, CTSK, CTSS, FN, LAMB1 and collagen IV were significantly decreased after SNAI1 knockdown. (typical photographs of 3 pairs of sample were showed). *P < 0.05, **P < 0.01, ***P < 0.001 Bar = 100 μm
SNAI1 knockdown by siSNAI1 intravitreal injection suppressed ocular neovascularization in vivo
At last, we assessed the effect of SNAI1 on ocular neovascularization by intravitreal injection in vivo. Tested by qRT-PCR in vitro, SNAI1 mRNA was significantly reduced by 71.47% after transfected with the second sequence of siSNAI1, which was further chemically modified and used in vivo (Fig. S2A). Pups at P3 were intravitreally injected with siSNAI1 or Cy3-labeled NC and killed at P7 (Fig. 7a). Activity of Cy3 fluorescent dye was observed on Cy3-labeled NC-injected retina, and expression of SNAI1 was significantly decreased in siSNAI1-injected retinas, showing siSNAI1 worked successfully in vivo (Fig. 7b). Whole-mount staining with IB4 was performed to elicit the retinal vasculature, and siSNAI1-injected retinas showed nearly normal vascular development and tip cell formation in the early stage, except for slightly reduced vascular density (Fig. 7c). We then changed the injection time point to P7, based on our data that SNAI1 expression increased greatly at P7 and collected retinas at P14 to observe the effect of SNAI1 knockdown on deep vascular plexus formation (Fig. 7a). SNAI1 was significantly reduced in siSNAI1-injected group (Fig. 7b), combined with impaired formation of deep plexus (Fig. 7d), indicating that SNAI1 plays an important role during deep retinal vasculature formation. Moreover, we injected siSNAI1 or NC intravitreally 1 day after laser coagulation in adult CNV model mice and collected choroidal tissues at d7 (Fig. 8a). qRT-PCR analysis showed reduced SNAI1 mRNA expression in choroidal tissues (Fig. 8b). Compared with the control group, mice treated with siSNAI1 showed a significant decrease in CNV area (Fig. 8c). The same inhibitory effect was also observed in OIR model (Fig. 8d), as siSNAI1 was injected at P12 and retinal tissues were analyzed at P17(Fig. 8a). Consequently, SNAI1 knockdown could suppress physiological and pathological ocular neovascularization in vivo, therefore indicating its potential as a novel therapeutic target.

SNAI1 knockdown by siSNAI1 intravitreal injection impaired retinal deep vascular development. a Illustration of siSNAI1 or NC injection strategy in mice. Pups received intravitreal injection of siSNAI1 or NC at P3 or P7 after birth and were killed at P7 or P14, respectively. b Immunofluorescence showed Cy3-labeled NC staining on the whole-mount retina of P7. qRT-PCR analysis demonstrated SNAI1 mRNA knockdown at retina in siSNAI1-injected eyes. (n = 5 for each group in d7 and n = 4 in d14). c Whole retinal tissues were flat-mounted and stained with IB4 (green) at P7. Confocal images were taken in the superficial plexus. White squares presented the areas for quantification. Bar charts showed quantification of distance from optic nerve to the vascular margin, number of branches per field and number of tip cells per vascular edge of 1000 μm. Development of superficial vessels was almost normal in siSNAI1-injected retina except for a slightly decrease in vascular density. (n = 6 for each group). d Whole retinal tissues were flat-mounted and stained with IB4 (green) at P14. Confocal images were taken in the superficial plexus and then taken in the deep plexus below the superficial plexus. White squares presented the areas for quantification. Bar charts showed quantification of distance from optic nerve to the vascular margin (both superficial and deep vasculature) and branches per field in the deep vascular plexus at P14. The deep plexus formation was disturbed in siSNAI1-injected retina. (n = 6 for each group.) *P < 0.05, **P < 0.01 ***P < 0.001 ns = not significant A = artery V = vein Bar = 100 μm

SNAI1 knockdown by siSNAI1 intravitreal injection suppressed CNV and OIR. a Illustration of siSNAI1 or NC injection strategy in mice. Mice received intravitreal injection of siSNAI1 or NC at d1 after laser coagulation and were killed at d7 for CNV analysis (left), or received injection at P12 after oxygen uptake and were killed at P17 for OIR analysis (right), respectively. b qRT-PCR analysis demonstrated SNAI1 mRNA knockdown at choroidal tissues in siSNAI1-injected eyes. (n = 4 for each group). c Whole choroidal tissues were flat mounted and stained with IB4 (green) at d7 and examined under a laser scanning confocal microscope, followed by reconstruction of the CNV lesions. The CNV volumes were compared between siSNAI1 and NC-injected mice. (n = 6 for each group and 5 laser burns per sample). d Whole retinal tissues were flat mounted and stained with IB4 (green) at P17. Confocal images were taken. The percentage of NV area were calculated and compared between siSNAI1 and NC-injected pups. (n=3 for each group) *P < 0.05, ***P < 0.001 O.N = optic nerve Bar = 100 μm
Discussion
Findings from this study showed that (a) endothelial cells could express SNAI1 in both physiological and pathological ocular neovascularization; (b) SNAI1 expression, partly by VEGF stimulus, improved ECs motility and invasion; and (c) intravitreal injection of siSNAI1 suppressed new vessel formation in developing retina, CNV and OIR model. Taken together, our data suggested that SNAI1, an EndMT transcription factor, promoted the early phase of ocular neovascularization and might provide a potential therapeutic target.
Ocular neovascularization is orchestrated by complicated cellular interactions and vascular growth factors. Above all, VEGF, due to its role in endothelial proliferation, survival and migration, has proved to be the master regulator [38]. Elevated intraocular levels of VEGF have been reported within the setting of iris neovascularization, retinal vein occlusion (RVO), PDR, ROP and CNV, which led to the clinical development of a number of approved anti-VEGF therapies [38]. Activation of ECs acts as the initial stage of angiogenesis. Responding to angiogenic stimuli, ECs undergo a compound morphogenetic process including polarization, lamellipodia and filopodia formation, cell–cell and cell–matrix contact rebuilding and ECM degradation and therefore migrate through the BM to form new vessels. The series of activities generate highly invasive phenotype, which have resemblance to EndMT process. Accordingly, it has been proposed that angiogenesis may involve a “partial EndMT”, as ECs display a fundamental transition in both morphology and functions but remain most endothelial characteristics [11]. However, much more are needed to elucidate the correlation between angiogenesis and EndMT.
As a comparison of angiogenesis and EndMT reveals several similarities, it seems reasonable to assume that factors inducing EndMT may also play a role in ocular angiogenesis. First of all, we observed that SNAI1 was expressed in ECs in the early phase of physiological and pathological ocular angiogenesis. In retinal development model, SNAI1 mRNA level was highly up-regulated at P7 and remained high level till P14. The expression of SNAI1 in veins, venules and descending ECs was further revealed by immunofluorescence, which was consistent with previous report [21]. In pathological angiogenesis, newly growing ECs also expressed SNAI1 in CNV and OIR model. We then found that both SNAI1 mRNA and protein level increased responding to VEGF stimulus. As SNAI1 is highly unstable and degraded by proteasome via phosphorylation by GSK-3β [17], we further tested whether this up-regulation was resulted from increased synthesis or decreased degradation by adding proteasome inhibitor MG132 or protein synthesis inhibitor CHX. Our data showed that VEGF stimulus promoted SNAI1 protein synthesis rather than restrained it degradation.
It was reported that SNAI1 could induce EndMT under certain conditions [14, 15, 17, 39,40,41,42]. Besides, SNAI1 also had a role in embryonic vessel formation and in vitro angiogenesis [19, 20]. Recently, Park et al. have reported that ECM-induced SNAI1 expression contributed to vertical vessel sprouting via the regulation of VEGFR3 [21]. However, more details about how SNAI1 functions in ocular neovascularization need to be elucidated. Here, by using SNAI1 overexpression adenovirus and SNAI1 siRNA, we found that SNAI1 expression in ECs mainly affected cell motility, migration and invasion. These activities are coincident with properties involving in EndMT. SNAI1 knockdown also partly blocked ECs response to VEGF stimulus. To our surprise, in lumen formation assay, total length of siSNAI1-treated HUVECs slightly increased compared with control. Previous data showed that SNAI1 expression rose up temporarily during ECs network formation and disappeared when it was completed [21]. As the complicated mechanisms involved in ECs lumen formation in vitro, this result indicated, though need more support, that down-regulation of SNAI1 may stabilize vessel structure.
The downstream targets of Snail family in ECs are barely studied. Several studies pointed out that delta like canonical Notch ligand 4 (Dll4), MMP15, membrane type 1-matrix metalloproteinase (MT1-MMP) and VEGFR3 may mediated part of SNAI1 or SNAI2 function in ECs [14, 21, 42, 43]. Here, we performed RNA sequence and put forward that genes involving in cytoskeleton arrangement, ECM degradation and remodeling might account for the phenotype changes in SNAI1 activation. Firstly, ECs require remodeling of the F-actin cytoskeleton in order to migrate into perivascular areas. Previous studies have shown that Rho family GTPase could be activated by VEGF to generate protrusions in leading endothelial tip cells that guide newly formed sprouts [44, 45]. The best studied Rho family GTPase are Rho, Rac and Cdc42 [46]. Here, we detected a decreased enrichment of genes involved in Rho and Rac1 pathway, which indicated that SNAI1 down-regulation impaired cell motility. Secondly, during angiogenic sprouting, the basal lamina is degraded and replaced by new matrix molecules like laminin, fibronectin, and collagen type IV [47]. The former process required a series of diffusing and membrane-bound proteolytic enzymes, which mainly fell into two classes: PA and MMPs [3]. Here, the decrease in genes related to ECM disassembly including MMP2, MMP9, FURIN, CTSK, MMP10 and CTSS accounted for the impaired migration, invasion and sprouting after SNAI1 knockdown. Besides, we noticed that genes involved in ECM component, including FN, LAMB1, LAMC1, COL4A1 and COL4A2 were also decreased in siSNAI1-treated HUVECs, which indicated that SNAI1 also played a role in following ECM synthesis. Therefore, our data inferred that SNAI1 up-regulation in activated ECs mainly promotes genes related to cytoskeleton arrangement and ECM remodeling. These changes on gene expression enabled ECs to form prominence, invade through the ECM and deposit new BM and might result in the observed SNAI1-induced pro-angiogenic characters.
The development of ocular new vessels is companied by ECs sprouting and invasion. In developing retina, deep retinal vascular plexus emerge from venous vessels and penetrate the retina perpendicular to the primary plexus from P7 [48]. In CNV, choroidal ECs break through the Bruch’s membrane and grow into the avascular subretinal space [1]. In OIR, after transferred from hyperoxic to normoxic conditions, the retinal vessels are dilated and tufts of pathological neovasculature grows on the surface of the retina [1]. Studies once revealed that systemically inhibition of SNAI1 limited retinal deep vascular formation without impacting superficial vessels [21]. Therefore, we speculated that SNAI1 could provide a promising therapeutic target for the following reasons: (a) based on our results, activated ECs expressed SNAI1, and inhibition of SNAI1 suppressed angiogenesis in vitro; (b) VEGF was the master regulator in ocular neovascularization and SNAI1 suppression limited VEGF response; and (c) SNAI1 is quickly degraded and retain silence in mature vasculature, and thus, its inhibition should not cause undesirable side effects [17, 20]. To test this hypothesis, we performed siSNAI1 intravitreal injection in both pups and adult mice. We found that local SNAI1 inhibition could significantly reduce retinal deep sprouting as well as OIR and CNV formation. In summary, our data suggested a crucial role of SNAI1 in ocular angiogenesis initialization, which might partly attribute to its promotion on genes related to cell motility and ECM remodeling under VEGF stimulus, and thus, SNAI1 could be regarded as a novel target for ocular neovascularization diseases.
References
Campochiaro PA (2015) Molecular pathogenesis of retinal and choroidal vascular diseases. Prog Retinal Eye Res 49:67–81. https://doi.org/10.1016/j.preteyeres.2015.06.002
Dou GR, Wang L, Wang YS, Han H (2012) Notch signaling in ocular vasculature development and diseases. Mol Med 18:47–55. https://doi.org/10.2119/molmed.2011.00256
Pepper MS (2001) Role of the matrix metalloproteinase and plasminogen activator-plasmin systems in angiogenesis. Arterioscler Thromb Vasc Biol 21(7):1104–1117
Neve A, Cantatore FP, Maruotti N, Corrado A, Ribatti D (2014) Extracellular matrix modulates angiogenesis in physiological and pathological conditions. Biomed Res Int 2014:756078. https://doi.org/10.1155/2014/756078
Eilken HM, Adams RH (2010) Dynamics of endothelial cell behavior in sprouting angiogenesis. Curr Opin Cell Biol 22(5):617–625. https://doi.org/10.1016/j.ceb.2010.08.010
Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N (1989) Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 246(4935):1306–1309
Campochiaro PA (2013) Ocular neovascularization. J Mol Med 91(3):311–321. https://doi.org/10.1007/s00109-013-0993-5
Medici D, Shore EM, Lounev VY, Kaplan FS, Kalluri R, Olsen BR (2010) Conversion of vascular endothelial cells into multipotent stem-like cells. Nat Med 16(12):1400–1406. https://doi.org/10.1038/nm.2252
Medici D, Kalluri R (2012) Endothelial–mesenchymal transition and its contribution to the emergence of stem cell phenotype. Semin Cancer Biol 22(5–6):379–384. https://doi.org/10.1016/j.semcancer.2012.04.004
Lamouille S, Xu J, Derynck R (2014) Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 15(3):178–196. https://doi.org/10.1038/nrm3758
Welch-Reardon KM, Wu N, Hughes CC (2015) A role for partial endothelial–mesenchymal transitions in angiogenesis? Arterioscler Thromb Vasc Biol 35(2):303–308. https://doi.org/10.1161/ATVBAHA.114.303220
Gonzalez DM, Medici D (2014) Signaling mechanisms of the epithelial–mesenchymal transition. Sci Signal 7(344):re8. https://doi.org/10.1126/scisignal.2005189
Nieto MA (2002) The snail superfamily of zinc-finger transcription factors. Nat Rev Mol Cell Biol 3(3):155–166. https://doi.org/10.1038/nrm757
Lee SW, Won JY, Kim WJ, Lee J, Kim KH, Youn SW, Kim JY, Lee EJ, Kim YJ, Kim KW, Kim HS (2013) Snail as a potential target molecule in cardiac fibrosis: paracrine action of endothelial cells on fibroblasts through snail and CTGF axis. Mol Ther J Am Soc Gene Ther 21(9):1767–1777. https://doi.org/10.1038/mt.2013.146
Villarejo A, Cortes-Cabrera A, Molina-Ortiz P, Portillo F, Cano A (2014) Differential role of Snail1 and Snail2 zinc fingers in E-cadherin repression and epithelial to mesenchymal transition. J Biol Chem 289(2):930–941. https://doi.org/10.1074/jbc.M113.528026
Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA (2000) The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol 2(2):76–83. https://doi.org/10.1038/35000025
Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, Hung MC (2004) Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol 6(10):931–940. https://doi.org/10.1038/ncb1173
Cobaleda C, Perez-Caro M, Vicente-Duenas C, Sanchez-Garcia I (2007) Function of the zinc-finger transcription factor SNAI2 in cancer and development. Annu Rev Genet 41:41–61. https://doi.org/10.1146/annurev.genet.41.110306.130146
Wu ZQ, Rowe RG, Lim KC, Lin Y, Willis A, Tang Y, Li XY, Nor JE, Maillard I, Weiss SJ (2014) A Snail1/Notch1 signalling axis controls embryonic vascular development. Nat Commun 5:3998. https://doi.org/10.1038/ncomms4998
Welch-Reardon KM, Ehsan SM, Wang K, Wu N, Newman AC, Romero-Lopez M, Fong AH, George SC, Edwards RA, Hughes CC (2014) Angiogenic sprouting is regulated by endothelial cell expression of Slug. J Cell Sci 127(Pt 9):2017–2028. https://doi.org/10.1242/jcs.143420
Park JA, Kim DY, Kim YM, Lee IK, Kwon YG (2015) Endothelial snail regulates capillary branching morphogenesis via vascular endothelial growth factor receptor 3 expression. PLoS Genet 11(7):e1005324. https://doi.org/10.1371/journal.pgen.1005324
She X, Rohl CA, Castle JC, Kulkarni AV, Johnson JM, Chen R (2009) Definition, conservation and epigenetics of housekeeping and tissue-enriched genes. BMC Genom 10:269. https://doi.org/10.1186/1471-2164-10-269
Dou GR, Li N, Chang TF, Zhang P, Gao X, Yan XC, Liang L, Han H, Wang YS (2016) Myeloid-specific blockade of notch signaling attenuates choroidal neovascularization through compromised macrophage infiltration and polarization in mice. Sci Rep 6:28617. https://doi.org/10.1038/srep28617
Lambert V, Lecomte J, Hansen S, Blacher S, Gonzalez ML, Struman I, Sounni NE, Rozet E, de Tullio P, Foidart JM, Rakic JM, Noel A (2013) Laser-induced choroidal neovascularization model to study age-related macular degeneration in mice. Nat Protoc 8(11):2197–2211. https://doi.org/10.1038/nprot.2013.135
Xu W, Yin J, Sun L, Hu Z, Dou G, Zhang Z, Wang H, Guo C, Wang Y (2017) Impact of minocycline on vascularization and visual function in an immature mouse model of ischemic retinopathy. Sci Rep 7(1):7535. https://doi.org/10.1038/s41598-017-07978-z
Dou GR, Wang YC, Hu XB, Hou LH, Wang CM, Xu JF, Wang YS, Liang YM, Yao LB, Yang AG, Han H (2008) RBP-J, the transcription factor downstream of Notch receptors, is essential for the maintenance of vascular homeostasis in adult mice. FASEB J Off Publ Fed Am Soc Exp Biol 22(5):1606–1617. https://doi.org/10.1096/fj.07-9998com
Yan X, Yang Z, Chen Y, Li N, Wang L, Dou G, Liu Y, Duan J, Feng L, Deng S, Han H, Zhang P (2015) Endothelial cells-targeted soluble human Delta-like 4 suppresses both physiological and pathological ocular angiogenesis. Sci China Life Sci 58(5):425–431. https://doi.org/10.1007/s11427-015-4834-3
Jonkman JE, Cathcart JA, Xu F, Bartolini ME, Amon JE, Stevens KM, Colarusso P (2014) An introduction to the wound healing assay using live-cell microscopy. Cell Adhes Migr 8(5):440–451. https://doi.org/10.4161/cam.36224
Hall DM, Brooks SA (2014) In vitro invasion assay using matrigel: a reconstituted basement membrane preparation. Methods Mol Biol 1070:1–11. https://doi.org/10.1007/978-1-4614-8244-4_1
De Wever O, Hendrix A, De Boeck A, Eertmans F, Westbroek W, Braems G, Bracke ME (2014) Single cell and spheroid collagen type I invasion assay. Methods Mol Biol 1070:13–35. https://doi.org/10.1007/978-1-4614-8244-4_2
Yan XC, Cao J, Liang L, Wang L, Gao F, Yang ZY, Duan JL, Chang TF, Deng SM, Liu Y, Dou GR, Zhang J, Zheng QJ, Zhang P, Han H (2016) miR-342-5p Is a Notch downstream molecule and regulates multiple angiogenic pathways including notch, vascular endothelial growth factor and transforming growth factor beta signaling. J Am Heart Assoc. https://doi.org/10.1161/JAHA.115.003042
Zhao XC, Dou GR, Wang L, Liang L, Tian DM, Cao XL, Qin HY, Wang CM, Zhang P, Han H (2013) Inhibition of tumor angiogenesis and tumor growth by the DSL domain of human Delta-like 1 targeted to vascular endothelial cells. Neoplasia 15(7):815–825
Baker M, Robinson SD, Lechertier T, Barber PR, Tavora B, D’Amico G, Jones DT, Vojnovic B, Hodivala-Dilke K (2011) Use of the mouse aortic ring assay to study angiogenesis. Nat Protoc 7(1):89–104. https://doi.org/10.1038/nprot.2011.435
Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38(16):e164. https://doi.org/10.1093/nar/gkq603
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7(3):562–578. https://doi.org/10.1038/nprot.2012.016
Pitulescu ME, Schmidt I, Benedito R, Adams RH (2010) Inducible gene targeting in the neonatal vasculature and analysis of retinal angiogenesis in mice. Nat Protoc 5(9):1518–1534. https://doi.org/10.1038/nprot.2010.113
Connor KM, Krah NM, Dennison RJ, Aderman CM, Chen J, Guerin KI, Sapieha P, Stahl A, Willett KL, Smith LE (2009) Quantification of oxygen-induced retinopathy in the mouse: a model of vessel loss, vessel regrowth and pathological angiogenesis. Nat Protoc 4(11):1565–1573. https://doi.org/10.1038/nprot.2009.187
Rubio RG, Adamis AP (2016) Ocular angiogenesis: vascular endothelial growth factor and other factors. Dev Ophthalmol 55:28–37. https://doi.org/10.1159/000431129
Grande MT, Sanchez-Laorden B, Lopez-Blau C, De Frutos CA, Boutet A, Arevalo M, Rowe RG, Weiss SJ, Lopez-Novoa JM, Nieto MA (2015) Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat Med 21(9):989–997. https://doi.org/10.1038/nm.3901
Gras B, Jacqueroud L, Wierinckx A, Lamblot C, Fauvet F, Lachuer J, Puisieux A, Ansieau S (2014) Snail family members unequally trigger EMT and thereby differ in their ability to promote the neoplastic transformation of mammary epithelial cells. PLoS ONE 9(3):e92254. https://doi.org/10.1371/journal.pone.0092254
Li H, Wang H, Wang F, Gu Q, Xu X (2011) Snail involves in the transforming growth factor beta1-mediated epithelial-mesenchymal transition of retinal pigment epithelial cells. PLoS ONE 6(8):e23322. https://doi.org/10.1371/journal.pone.0023322
Xu X, Tan X, Tampe B, Sanchez E, Zeisberg M, Zeisberg EM (2015) Snail is a direct target of hypoxia-inducible factor 1alpha (HIF1alpha) in hypoxia-induced endothelial to mesenchymal transition of human coronary endothelial cells. J Biol Chem 290(27):16653–16664. https://doi.org/10.1074/jbc.M115.636944
Tao G, Levay AK, Gridley T, Lincoln J (2011) Mmp15 is a direct target of Snai1 during endothelial to mesenchymal transformation and endocardial cushion development. Dev Biol 359(2):209–221. https://doi.org/10.1016/j.ydbio.2011.08.022
van Nieuw Amerongen GP, Koolwijk P, Versteilen A, van Hinsbergh VW (2003) Involvement of RhoA/Rho kinase signaling in VEGF-induced endothelial cell migration and angiogenesis in vitro. Arterioscler Thromb Vasc Biol 23(2):211–217
Soga N, Namba N, McAllister S, Cornelius L, Teitelbaum SL, Dowdy SF, Kawamura J, Hruska KA (2001) Rho family GTPases regulate VEGF-stimulated endothelial cell motility. Exp Cell Res 269(1):73–87. https://doi.org/10.1006/excr.2001.5295
van Buul JD, Geerts D, Huveneers S (2014) Rho GAPs and GEFs: controling switches in endothelial cell adhesion. Cell Adhes Migr 8(2):108–124
Bahramsoltani M, Slosarek I, De Spiegelaere W, Plendl J (2014) Angiogenesis and collagen type IV expression in different endothelial cell culture systems. Anat Histol Embryol 43(2):103–115. https://doi.org/10.1111/ahe.12052
Fruttiger M (2007) Development of the retinal vasculature. Angiogenesis 10(2):77–88. https://doi.org/10.1007/s10456-007-9065-1
Acknowledgement
We thank Ms. Qing-Yi Wang for her kind help on correcting the galley proof.
Funding
This study was funded by National Natural Science Foundation of China (Nos. 81770936, 81670863), Youth Science and Technology Nova Program of Shaanxi Province (No. 2016KJXX-19) and Foundation for the Author of National Excellent Doctoral Dissertation of the People’s Republic of China (No. 201481).
Author information
Authors and Affiliations
Contributions
JXS, GRD, YSW and HH designed experiments. JXS and TFC performed most of the studies and analyzed data. MHL, LJS and YL carried out CNV model. WQX and JBS carried out OIR model. XCY, ZYY and YL helped with in vitro experiments. LL and HH provided reagents and advised on experimental design. JXS wrote manuscript. GRD and YSW revised manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sun, JX., Chang, TF., Li, MH. et al. SNAI1, an endothelial–mesenchymal transition transcription factor, promotes the early phase of ocular neovascularization. Angiogenesis 21, 635–652 (2018). https://doi.org/10.1007/s10456-018-9614-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10456-018-9614-9