
Abstract
The aim of presented research concerned synthesis of a novel lignosulfonate-based sorbent and its application in batch adsorption tests related to removal of an active pharmaceutical ingredient (ibuprofen). Obtained hybrid material was thoroughly characterized with respect to morphology, porosity (BET method and BJH algorithm), electrokinetic stability, and characteristic functional groups. As a result of the proposed synthesis route, an active MgO–SiO2/lignosulfonate hybrid was obtained as confirmed by significant amount of functional groups present in its structure and relative good parameters of the porous structure (ABET = 71 m2/g, Vp = 0.2 mL/g and Sp = 11.8 nm). Next stage concerned typical batch adsorption tests with respect to active pharmaceutical ingredient (API)—ibuprofen. It was proved that efficiency of removal of ibuprofen was mostly determined by its structure as well as variable process parameters. Affinity of sorption material towards model organic impurity was established based on equilibrium and kinetics study of adsorption process performed. A key element of the investigations was an interaction and mechanism study resulted from interpretation how the parameters of the adsorption affect the nature of the adsorbent/adsorbate interactions. Experimental data collected, supplemented with regeneration tests, proved significant affinity of ibuprofen to the hybrid sorbent and enabled determination of nature of the adsorbent/adsorbate interactions. Moreover, suggested mechanism of ibuprofen sorption was proposed.
Similar content being viewed by others


1 Introduction
Due to the dynamic development of the pharmaceutical industry, the amount of consumed drugs is increasing and the problem of their removal from the natural environment is growing. Pharmaceuticals most often pollute water systems, where they get along with municipal sewage from households and hospitals. However, the results of water monitoring also prove the presence of pharmaceuticals in treated and drinking water. Their high content in the environment is mainly caused by their resistance to biodegradation and subsequent accumulation in aquatic ecosystems. Drugs consumed by people and animals are not fully metabolised, as a result of which they are excreted, both as original drugs and their metabolites, and then go to the sewerage system (Khetan and Collins 2007; Evgenidou et al. 2015; Madikizela and Chimuka 2016). On the other hand, large amounts of drugs enter the aquatic environment in an unchanged form, when their expiry dates expires and are not properly disposed. An important source of pharmaceuticals are also hospitals wastewaters and waste sewage rising from plants dealing with the production of pharmacologically active substances (Escher et al. 2011; Gadipelly et al. 2014).
Classic wastewater treatment is not effective in neutralizing pharmaceuticals, because it is not aimed at eliminating such specific micro-pollutants. Therefore, new methods are sought, or ways to improve existing ones, used in processes carried out in municipal wastewater treatment plants, to ensure complete degradation of pharmacologically active compounds in water (Kovalova et al. 2012). Among purification techniques exhibiting significant effectiveness, membrane methods, advanced oxidation processes or more popular and universal methods based on adsorption, are mentioned (Ikehata et al. 2006; Jones-Lepp et al. 2010; Zhang et al. 2013). The advantages of adsorption methods include, above all, low cost and simplicity of implementation. In turn, the efficiency of removal of micro-pollutants from aqueous solutions using adsorption process is affected by the method of adsorbent synthesis, its physicochemical attributes, as well as the parameters of the adsorption process, such as contact time, stirring intensity, solution pH, adsorbent dosage (Nam et al. 2014; Reza et al. 2014; Behera et al. 2012; Wang et al. 2015; Bui and Choi 2009). The primary aspect of the adsorption process is to evaluate the equilibrium contact time of the adsorbent with the adsorbate. Another important factor that determines the effectiveness of adsorption of micro-pollutants from aqueous solutions is the pH, appropriately selected for the ionic nature of the adsorbent surface (type of functional groups present on the surface) and the charge of the active part of the sorbed micro-pollutant. The adsorptive abilities of a given material are also determined by the method of its synthesis, as it affect the properties of the obtained adsorbent, in particular the degree of development of its surface area, which can be additionally controlled by appropriate modification of the starting material (Grabicka and Jaroniec 2010; Michot and Villiéras 2006; Morais et al. 2013).
Effective water purification and removal of micro-pollutants from wastewater can be achieved by using adsorption on activated carbons. Activated carbon meets the requirements of a good adsorbent, is characterized by high surface area, significant porosity and mechanical strength (Calisto et al. 2015; Rigobello et al. 2013). Another good adsorption systems are inorganic materials, both natural and synthetic ones, including mono- and multi-component oxide systems (Sheng et al. 2009; Singhon et al. 2012; Ciesielczyk et al. 2017). These include silicates, aluminosilicates, zeolites, kaolins and bentonites (Tahar et al. 2014; Martucci et al. 2012). An interesting group include also natural biosorbents, among which cellulose, chitin, lignin, chitosan, cotton, waste bark, seaweed, algae and many others can be mentioned (Moro et al. 2017; Kyzas et al. 2013; Ribeiro et al. 2011; Kyzas and Deliyanni 2015). The management of waste biomass is currently of significant importance to many scientists worldwide. The advantage of using biomass, such as lignin or lignosulfonates, as compared to activated carbons is that they contain various functional groups on their surface and are waste products from paper industry. They offer good adsorption efficiency, maybe lower in some cases than activated carbons, but their application is much cheaper. Intensive works are being carried out on technological solutions for using wood biomass, the main component of which is lignin. On the other hand, the lignosulfonates can be mentioned. Those are calcium, magnesium and sodium salts of ligninsulfonic acids. Lignosulfonates owe their unique properties to the presence of sulfonic, hydroxyl and phenol groups, they contain more elemental sulfur than other derivatives of technical lignin, and that is the reason that they are expect to act effectively in adsorption of organic pollutants (Aso et al. 2013; Klapiszewski et al. 2018; Ciesielczyk et al. 2017).
Taking into account development tendencies and the fact that new adsorbents and methods of their preparation are being searched more and more often, as well as that the conditions of designed synthesis are modified in various directions, in presented research, a comprehensive experiment was performed in order to synthesize hybrid MgO–SiO2/lignosulfonate and its use in removal of an active pharmaceutical ingredient commonly present in wastewaters (ibuprofen). The scientific novelty of the work relates to the application, for the first time, of this type of hybrid adsorbent in removal of active pharmaceutical ingredient from water solutions. Such a type of material wasn’t previously analyzed in batch adsorption tests and described in available scientific papers.
2 Experimental
2.1 Materials and methods
2.1.1 Synthesis of MgO–SiO2/lignosulfonate hybrid adsorbent
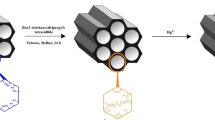
The synthesis of hybrid MgO–SiO2/lignosulfonate adsorbent was realized via in situ sol–gel route, using organic precursors of magnesium and silicon—magnesium ethoxide (C2H5O)2Mg and tetraethoxysilane (C2H5O)4Si (both purchased from Sigma-Aldrich), 25% solution of ammonia as promoter of hydrolysis and methyl alcohol as a solvent (both purchased from Chempur). Additionally, during the synthesis of inorganic matrix, the biopolymer—commercial calcium lignosulfonate was introduced. The synthesis was performed on a quarter-technical scale, using a reactor 2 L in capacity, equipped with a high-speed stirrer. At the first stage, a specific quantity (45 g) of magnesium ethoxide was dissolved in 1500 mL of methyl alcohol. The system was stirred for 15 min, and after that time tetraethoxysilane (TEOS—the organic precursor of silica) and the promoter of hydrolysis (25% ammonia solution) were introduced simultaneously. The quantities of the reactants were selected so as to achieve a TEOS:magnesium ethoxide mass ratio of 1.5:1. The methodology was described in details in previously published papers (Ciesielczyk et al. 2014a, b). The difference is that at the final stage of MgO–SiO2 oxide system synthesis calcium lignosulofante was introduced into the reaction mixture (Klapiszewski et al. 2018). The amount of biopolymer used for this purpose was 20 wt./wt. Next, the mixture was stirred for 60 min with a stirrer speed of 300 rpm, to enable gel formation. The resulting alcogel combined with lignosulfonate was placed in a vacuum evaporator to remove the solvent, and then washed several times with hot distilled water. Finally, the product was dried at 105 °C for 48 h. The dry MgO–SiO2/lignosulfonate adsorbent was classified and thoroughly analyzed. The scheme of the synthesis together with a part of the structure of calcium lignosulfonate is presented in Fig. 1.

Synthesis route used for the preparation of MgO–SiO2/lignosulfonate hybrid adsorbent (Klapiszewski et al. 2018)
2.1.2 Batch adsorption experiments
The key element of the research concerned typical batch adsorption tests performed using synthesized hybrid MgO–SiO2/lignosulfonate adsorbent and model solutions of an active pharmaceutical ingredient, which characteristic is presented in Table 1.
Batch adsorption tests were conducted in 100 mL flasks, to which 50 mL of ibuprofen solution in defined concentration was added. Next, an appropriate amount of hybrid MgO–SiO2/lignosulfonate adsorbent was introduced into the reaction system, which was furthermore stirred using an RO5 magnetic stirrer (Ika Werke GmbH). After defined time of adsorption the whole mixture was filtered. The filtrate was subjected to HPLC evaluation in order to establish the content of unadsorbed pharmaceutical. The idea was to verify the influence of variable parameters of adsorption process onto its effectiveness.
During evaluation of model organic impurity concentration and adsorption time, pharmaceutical solutions in a concentration range of 0.5–1.5 mg/L, a sorbent mass of 0.5 g, a time range of 1–120 min, and initial pH 5–6 of ibuprofen solution were used. The adsorption took place at room temperature. Those results supported with commonly known theoretical models were included in description of equilibrium and kinetic aspects of adsorption of pharmaceutical onto synthesized hybrid adsorbent. To evaluate the influence of the adsorbent dosage, the process was performed within 60 min, using 0.1, 0.3, 0.5, 0.7 and 1 g of the MgO–SiO2/lignosulfonate hybrid material and constant concentration of model organic impurity (1.0 mg/L). Evaluation of the influence of pH was carried out in range of 1–12 using 0.5 g of the hybrid adsorbent and adsorbate solutions at a concentration of 1.0 mg/L. To adjust the pH of the system NaOH and HCl solutions, both at a concentration of 0.1 M, were used.
Determination of ibuprofen was performed with a chromatographic system UltiMate 3000 RSLC (Dionex, Thermo, USA) coupled to an API 4000 QTRAP triple quadrupole mass spectrometer with electrospray ionization (ESI) (from AB Sciex, Foster City, CA, USA) in both positive and negative ionization modes (UHPLC–MS/MS). A Hypersil Gold C18 RP (100 mm × 2.1 mm, 1.9 μm particle size) column from Thermo Scientific, USA was used for chromatographic separation of these compounds. The column temperature was maintained at 35 °C and the injection volume was 5.0 μL. For UHPLC–MS/MS analysis in positive ionization mode, the mobile phase was a gradient prepared from Milli-Q water containing
5 mmol/L ammonium acetate (component A) and MeOH (component B). The gradient program was: 50% B at 0 min, increased to 100% B in 3 min and held for 6.0 min; the flow rate was 0.20 mL/min. For the analysis in negative ionization mode, the mobile phase was a gradient prepared from Milli-Q water containing 5 mmol/L ammonium acetate (component A) and MeOH (component B). The gradient program was: 50% B at 0 min, increased to 67% B in 3 min and held for 2 min, stepped to 100% in 3 min and held for 5.0 min; the flow rate was 0.2 mL/min. A post run time was set at 4.0 min for column equilibration before the next injection. The operating conditions for mass spectrometry for ibuprofen were as follow: curtain gas 10 (20) psi, nebuliser gas and auxiliary gas 55 (50) psi, source temperature 4500 (400 °C), ion spray voltage 5500 (− 4500) V and collision gas set to medium. Quantitative analysis of the compounds was performed in multiple reaction monitoring (MRM) mode. For analytes one transitions of the deprotonated molecular ion (for ibuprofen) and its respective ion product were chosen. These transitions (m/z) with associated declustering potentials (V) and collision energies (V) were: 205 → 161, − 50, − 12 for ibuprofen.
2.1.3 Equilibrium and kinetic study
Taking into the consideration the physicochemical properties of selected adsorbent and in order to evaluate the kinetic parameters the pseudo-first (established by Lagergren) (Lagergren 1898) and pseudo-second order (described by Ho) (Ho and McKay 1999) were used. Pseudo-first and pseudo-second order models describe the dependence of the amount of absorbed substance in a function of time. However, they have different starting assumptions. The Lagergren model based on the description of the adsorption rate depending on the sorption capacity, and Ho gave the assumption that the adsorption process can be treated as a second order reaction in relation to the difference between the amount of adsorbed substance at specified time and the amount of adsorbed substance at equilibrium. The linear equation of Lagergen (1) and Ho (2) are presented below:
where: qe and qt are the adsorptive capacities at equilibrium and after specified time (mg/g), t means time (min) and k1/2 is the pseudo-first or pseudo-second order rate constant (g/mg min). Since the Eq. (2) can be presented using different approach, four different types of pseudo-second order kinetic model can be distinguish (Table 2).
In addition, for the pseudo-second-order model the rate of adsorption (h) was determined using formula (3):
where h means the rate of adsorption (mg/mL min), qe denotes the quantity of drug adsorbed at equilibrium (mg/g), k2 is the rate constant for the process for the pseudo-second-order model (g/mg min).
For the kinetics of the adsorption process the standard deviation was calculated using the formula (4):
where qexp are the experimental data, n means the number of data points, and qcal are the data caculated by the model. The higher the value of the correlation coefficient (r2), and the smaller the standard deviation (SD), the better the model fits the experimental data.
In order to determine adsorption isotherms and indirect sorption capacity of synthesized material, tests were carried out for adsorbate solutions in the concentration range of 25–400 mg/L within 1 h. The equilibrium concentration (qe) was calculated using the Eq. (5):
where qe is the quantity of pharmaceutical adsorbed at equilibrium (mg/g), C0 is the initial concentration of pharmaceutical (mg/L), Ce is the equilibrium concentration of pharmaceutical (mg/L), V is the volume of model adsorbate solution (L), and m is the mass of adsorbent (g).
To investigate the adsorption isotherms the Langmuir and Freundlich models were used. The Langmuir isotherm assumes monolayer adsorption on energetically homogenous sites (like functional groups, defect in crystalline lattice etc.) (Langmuir 1918) and it can be described using the Eq. (6):
where qmax means the maximum amount of adsorbed pharmaceutical based on the sorbent mass (mg/g), KL is sorption energy (L/mg) and Ce is the adsorbate concentration (mg/L). However, the Eq. 5 can be linearized in four different way, therefore the Langmuir isotherm model can be divided into four different type (Table 3).
In contrast, the Freundlich isotherm model based on the adsorption onto heterogenous surface (Freundlich 1906) and can be described using the formula (7):
where qe is the amount of adsorbed drug (mg/g), Ce is the equilibrium concentration of the adsorbate (mg/L), KF and n are the Freundlich constants associated with the efficiency and intensity of adsorption.
Chi square test was used to measure the goodness of fit. The Chi square test can be defined by Eq. (8):
where m is the number of experimental data.
The percentage removal of ibuprofen (W) was calculated using Eq. (9):
where C0 and Ck are the concentrations of pharmaceutical in the solution before and after adsorption respectively (mg/L).
2.1.4 Desorption process
In order to evaluate the stability of the bonds between the API and hybrid adsorbent surface, and the potential regeneration of the material-desorption tests were performed. Selected samples of materials with adsorbed API were placed in a flask 250 mL in capacity, to which 100 mL of distilled water was introduced. The whole mixture was stirred using an RO5 magnetic stirrer (Ika Werke GmbH) for 15 min at room temperature, and after that time it was filtered. The filtrate underwent analysis performed with chromatographic system UltiMate 3000 RSLC (described earlier) to establish the quantity of desorbed API.
The efficiency of desorption (Y) was calculated using Eq. (10):
where Ca is the amount (concentration) of the adsorbed API (according to the efficiency of adsorption) and Cd is the concentration of the adsorbate that was removed from the surface of the tested sorbent during the desorption tests (mg/L).
2.1.5 Physicochemical evaluations
The initial MgO–SiO2 oxide material, calcium lignosulfonate and synthesized hybrid material were subjected to physicochemical evaluation. First the morphology (scanning electron microscopy—SEM) of the tested materials was evaluated. The observations enabled evaluation of the degree of dispersion, the structure of particles and their tendency towards aggregation or agglomeration. SEM images were recorded from an EVO40 scanning electron microscope (Zeiss). Before testing the samples were coated with Au for a time of 20 s using a Balzers PV205P coater. The electrokinetic properties of mentioned materials were also established. First, the electrophoretic mobility was measured at a constant ionic strength of 0.001 M NaCl, and then the zeta potential value was calculated based on the Henry equation (Zetasizer Nano ZS equipped with autotitrator—Malvern Instruments Ltd.). The ABET surface area (BET method) as well as the pore size distribution (BJH method) were obtained based on measurement data from low-temperature (− 196 °C) adsorption of nitrogen. The isotherms of nitrogen adsorption/desorption were recorded using an ASAP 2020 apparatus (Micromeritics Instrument Co.). Before the measurements, samples were degassed under low pressure for 4 h at 120 °C. Additionally, the used adsorbents after adsorption tests with API were analyzed with respect to FTIR spectroscopy in order to investigate the ability of the API to the adsorbent surface and to suggest a mechanism of their interactions. The FTIR spectra were obtained using a Vertex 70 spectrometer (Bruker). The samples were analyzed in the form of tablets, made by pressing a mixture of anhydrous KBr (ca. 0.25 g) and 1 mg of the tested substance in a special steel ring, under a pressure of 10 MPa. Analysis was performed over a wavenumber range of 4000–400 cm−1 (resolution of: 0.5 cm−1; number of scans: 64).
3 Results and discussion
3.1 Characterization of the obtained MgO–SiO2/lignosulfonate hybrid adsorbent
3.1.1 Morphology study
The morphology of the precursors and synthesized MgO–SiO2/lignosulfonate hybrid adsorbent was made based on the SEM images (Fig. 2).

SEM images of the: a synthesized MgO–SiO2 oxide system, b calcium lignosulfonate and c obtained MgO–SiO2/lignosulfonate hybrid adsorbent
The SEM images of the MgO–SiO2 oxide system (Fig. 2a) shows that particles are substantially homogeneous and tend to form rather spherical aggregates with average size around 1 µm. The particles of the calcium lignosulfonate reached size of around 5 µm and are characterized by irregular shape (Fig. 2b). The SEM image of the synthesized MgO–SiO2/lignosulfonate hybrid adsorbent indicates that the particles of this material are polydisperse and exhibits varied size, ranging from less than 1 µm up to even few micrometers (Fig. 2c). Moreover these particles have a tendency to form agglomerates with irregular shapes that is related to the morphological structure of the used precursors. Obtained results confirmed effectivity of the proposed synthesis route and stay in agreement with the previously published data, which reported that addition of biopolymer cause the agglomerization of the particles of the hybrid material (Klapiszewski et al. 2013).
3.1.2 Characterization of the surface functional groups
In order to confirm effective synthesis of the hybrid sorbent and to determine functional groups presented onto the surface of the precursors and resulting hybrid material, FTIR spectroscopy was used. In the FTIR spectrum of the MgO–SiO2 system (Fig. 3a) signals with maxima at 3440 and around 1630 cm−1 assigned for stretching vibrations of the hydroxyl groups and bending vibrations of the physically adsorbed water, respectively, can be seen. In addition, several peaks characteristic for the oxide structure can be observed. These are signals at: 1080 and 950 cm−1 (stretching vibrations of Si–O–Si bonds), 700 cm−1 (deformational vibrations of O–Mg–O bonds) and around 520 cm−1 (stretching vibrations of O–Si–O bonds). However, the most important is signal with maximum at 650 cm−1 assigned to the stretching vibrations of Si–O–Mg bonds, which presence indirectly confirm effective synthesis of the MgO–SiO2 oxide system (Klapiszewski et al. 2015a). The FTIR spectrum of the obtained MgO–SiO2/lignosulfonate hybrid adsorbent shows similarity with the spectrum of the pure oxide system (Fig. 3b). However, the presence of an additional signals, with maximum at 2950 cm−1 and in the wavenumber range of 1500–1420 cm−1, assigned for stretching vibrations of ≡C–H bonds in –CH2 and –CH3 groups and for stretching vibrations of CAr=CAr bonds in the aromatic rings, respectively, is observed. These signals are characteristic for the structure of calcium lignosulfonate and their presence indirectly confirms effective synthesis of the inorganic–organic hybrid sorbent and combination of inorganic matrix together with biomaterial, the same as reported in our previous study (Klapiszewski et al. 2015b).

FTIR spectra of the: a synthesized MgO–SiO2 oxide system and b obtained MgO–SiO2/lignosulfonate hybrid adsorbent
3.1.3 Porous structure parameters and electrokinetic stability
Figure 4a shows adsorption/desorption isotherm of the obtained MgO–SiO2/lignosulfonate hybrid adsorbent. The isotherm is classified as type IV with hysteresis loop type H3, which suggests that this material may be classified as mesoporous with disordered pores. MgO–SiO2/lignosulfonate hybrid is characterized with the surface area of 71 m2/g, pore volume of 0.2 mL/g and pore diameter of 11.8 nm. By contrast, as reported earlier, small amount of the adsorbed nitrogen indicates low porosity of calcium lignosulfonate (ABET = 0.92 m2/g, Vp = 0.0008 mL/g and Sp = 29.6 nm) (Klapiszewski et al. 2018). Thus, it could be concluded that combination of the biopolymer with magnesium silicate significantly increase its surface area and make the material suitable for a sorption experiments towards hazardous pollutants.

a Adsorption/desorption isotherms of the obtained MgO–SiO2/lignosulfonate hybrid and b electrokinetic curves of the initial reagents and obtained MgO–SiO2/lignosulfonate hybrid
Analysis of the electrokinetic stability of the used precursors and obtained hybrid material (Fig. 4b) allows to indicate the pH values at which synthesized hybrid form a stable dispersion. This fact is very important concerning practical application of the above-mentioned material as a sorbent of impurities from wastewaters. MgO–SiO2 oxide system has a negative zeta potential over the whole analyzed pH range. The highest value of zeta potential (–6 mV) was noticed at pH 2 due to protonation of the surface hydroxyl groups of MgO–SiO2 with H+ ions (Ciesielczyk et al. 2014a, b). Whereas the lowest (–37 mV) was noticed at pH 10.5. The pH range at which the zeta potential takes the lowest values, at this study pH > 10.5, may be considered as the region at which the MgO–SiO2 oxide system exhibits good stability in water systems. Although the second precursor, calcium lignosulfonate, exhibited similar electrokinetic curve to the previously described, its electrokinetic stability is better, as over whole analyzed pH range (1.5–9) it takes values ranging from − 17 mV to − 34 mV. Calcium lignosulfonate showed the highest dispersion stability at pH above 5 as at this pH it exhibits zeta potential values lower than − 30 mV. The values of the zeta potential obtained for the synthesized hybrid material lies between values of zeta potential of the used precursors and reaches values from − 9 mV at pH 1.8 to − 24 mV at pH 8.3 indicating relatively good electrokinetics stability of the obtained MgO–SiO2/lignosulfonate adsorbent over whole analyzed pH range. These results are in agreement with previously published study and provide indirect confirmation of the effective combination of the both precursors and obtaining of the hybrid material (Ciesielczyk et al. 2014a, b). In addition, it should be emphasized that isoelectric point was not reached for any of the analyzed materials.
3.2 Adsorption study of the ibuprofen
3.2.1 Effect of contact time
The investigation of the effect of contact time on sorption efficiency was performed (Fig. 5a). The results of these experiments, which were conducted for initial ibuprofen concentration of 500, 1000 and 1500 μg/L, indicate a sharp increase of ibuprofen adsorption on the obtained MgO–SiO2/lignosulphonate hybrid adsorbent. After 3 min the efficiency of ibuprofen adsorption exceeded 50% for all pharmaceutical solutions. What is more, the efficiency of removal of ibuprofen increases with increasing concentration of the pharmaceutical. It may be summarized that the initial concentration of model solution is a driving force capable of overcoming the mass transfer resistance between the liquid and solid phases, and also contributes to an increase in the number of collisions of adsorbent with adsorbate molecules. The obtained results correspond with the measurements of amount of adsorbed pharmaceutical (Fig. 5b), where the highest mass of adsorbed ibuprofen was reached also after 30 min of the test for each of measured solutions.

Effect of contact time on: a sorption efficiency and b amount of adsorbed pharmaceutical per unit mass of the adsorbent
3.2.2 Effect of pH and adsorbent dosage
The pH of tested solution is one of the key parameter which affect the adsorption process. The experiments were conducted for initial concentration of adsorbate, which was 1000 μg/L. The results (Fig. 6a) show that the highest efficiency of adsorption of pharmaceutical (75%) was obtained at the highly acidic solution (pH 2), which was consistent with literature reports that the pH value determines the degree of ionisation of ibuprofen (Nam et al. 2014; Reza et al. 2014). The lower pH value, the more undissociated molecules for which the adsorption is much more effective than for dissociated molecules. The reduction of removal efficiency of ibuprofen with an increase in alkalinity may therefore be a consequence of the mutual repulsion of ibuprofen anions and negatively charged adsorbent molecules. At the pH range between 3 and 8, the removal efficiency of ibuprofen was 60%, but above pH 8 an increase to the value of 70% can be seen. It is caused by the additional activation of functional groups of the lignosulphonate at basic pH, and next it may cause them to have greater affinity for the adsorbed pharmaceutical.

Effect of: a pH of the solution and b adsorbent dosage on the sorption efficiency of ibuprofen by MgO–SiO2/lignosulfonate hybrid adsorbent
The adsorbent dosage is another parameter which significantly may affect the adsorption process of pharmaceuticals. The results of measurements (Fig. 6b) show that the higher adsorbent dosage, the more efficient the adsorption process. This is due to an increase of the contact surface at the adsorbent-adsorbate interface and the increase of the number of active sites, i.e. functional groups capable of adsorbing a pharmaceutically active substance.
3.2.3 Kinetic study
Kinetics of ibuprofen adsorption by MgO–SiO2/lignosulfonate hybrid adsorbent was determined using the pseudo-first order and pseudo-second order model (Lagergren 1898; Ho and McKay 1999). The kinetics parameters were determined graphically from the linearized forms of kinetic equations. Obtaining a linear graph enabled the adaptation of experimental data of ibuprofen adsorption to a given order of reaction (Table 4, Fig. 7). On the basis of kinetic parameters as well as the curves obtained, it can be concluded that the experimental data describes the pseudo-second kinetic equation, type I, which is confirmed by the high value of the determined correlation coefficient (R2 = 0.99). It should be noted that the calculated values of the qe.cal parameter describing the amount of adsorbed pharmaceutical in equilibrium correlate with the experimental values of the qe.exp parameter. Lower values of correlation coefficients R2 obtained in the case of pseudo-first and pseudo-second models for the other types (II, III and IV) indicate significantly worse correlation between experimental and theoretical data. The obtained data also indicate an increase in the adsorption constant rate h together with an increase in the concentration of the pharmaceutical, which indicates that the driving force of the process is the increasing concentration of the adsorbate. This fact is in an agreement with earlier presented statements.

Adaptation of experimental data of ibuprofen adsorption to theoretical models of sorption kinetics: a pseudo-first order, b pseudo-second order type I, c pseudo-second order type II, d pseudo-second order type III and e pseudo-second order type IV
To determine the adsorption isotherms using the Langmuir and Freundlich models the measurements were made at equilibrium conditions for initial pharmaceutical concentrations: 1000; 2000; 3000; 4000 μg/L (Langmuir 1918; Freundlich 1906). This enabled determination of the sorption capacity of the prepared adsorbent towards used pharmaceutical. The parameters of the Langmuir adsorption isotherms are summarized in Table 5. The adjustment curves of experimental data for Langmuir isotherms is shown in Fig. 8. The Langmuir type III and IV equations best match the description of experimental isotherms, for which the highest correlation coefficient values were obtained, R2 equal to 0.996 and 0.995, respectively. The lower values of R2 in the case of type I and II indicate a worse match of experimental results to these forms of the Langmuir linear isotherm. Freundlich’s isotherm model showed a correlation at the level of R2 = 0.657, which proves this with a lower matching of this model to the description of experimental data than the Langmuir isotherm model. Based on the correlation coefficient between experimental data and theoretical (model), it was found that the sorption process of ibuprofen on the MgO–SiO2/lignosulfonate hybrid adsorbent better describes the Langmuir model (type III and IV), which indicates a single-layer coverage of the adsorbent with adsorbate molecules. This may correlate with the adsorption yields obtained at a level of 60–70%. The implementation of the adsorption process is effective until the adsorbent is coated with the adsorbate layer, the adsorbent is not able to bind further molecules of the pharmaceutical. Observed efficiency increase at pH > 8 may also indicate the activation of certain groups in the molecule of adsorbed ibuprofen, which to a certain extent may become active centers for further adsorption. The maximum sorption capacity of the hybrid adsorbent (calculated from the Langmuir isotherm model) reaches value of 2.318 mg/g which is acceptable one taking into account relative low concentration of model ibuprofen solutions used in the batch adsorption tests.

Adaptation of experimental data concerning adsorption of analysed pharmaceutical onto oxide adsorbent, to the Langmuir isotherms models (types 1–4)
Figure 9 presents the efficiency of desorption of ibuprofen from the adsorbent surface. Results proved high affinity of the tested pharmaceutical to the adsorbent as confirmed by desorption efficiency in the range of 2.6–4.8%. Despite the percentage uptake of the ibuprofen, which do not exceed 80%, the interactions of adsorbate and the adsorbent are rather strong. The higher the concentration of model organic impurity, higher percentage desorption of API was noted (the highest one—4.8% for model solution in concentration of 1500 µg/L). The same situation was observed in case of samples rising from pH analysis, where solution of API in the concentration of 1000 µg/L was used.

Results of desorption tests of selected adsorbent with adsorbed ibuprofen
Therefore, suggested mechanism of interaction between synthesized hybrid adsorbent and ibuprofen was proposed (Fig. 10). It was confirmed that adsorbent/adsorbate interaction results mainly from the chemical bonds formation between positively charged functional groups of the hybrid MgO–SiO2/calcium lignosulfonate adsorbent and undissociated molecules of the ibuprofen (e.g. see desorption test results). However, the weak physical and electrostatic interactions are not negligible.

The comparison of obtained results to the published so far information concerning adsorption of ibuprofen is presented in Table 6. Many researchers studied the removal of ibuprofen using different type of adsorbents. As it can be seen the time needed to reach the equilibrium of adsorption is in most cases determined by the adsorbent nature. Mestre et al. (2007) and Guedidi et al. (2017) reported that equilibrium time of adsorption of ibuprofen onto activated carbons is 4 and 10 h, respectively. On the other hand, 15 and 30 min was enough to reach equilibrium, when using silica SBA-15 (Bui and Choi 2009), mature sausage fruit modified with ionic liquid (Lawal and Moodley, 2017) or magnetic nanomaterial (Kollarahithlu and Balakrishnan 2018). Surprisingly, 17 h were needed to established the equilibrium of adsorption of ibuprofen onto bentonite (Salihi and Mahramanlioglu 2014). The analyzed literature reports state that such adsorption process is quite complex and in most cases is described by Langmuir or Freundlich isotherm models. Additionally, there are some arguable elements concerning initial adsorbate concentration used for batch adsorption experiments. Bany-Aiesh et al. (2015) studied the adsorption of ibuprofen onto β-cyclodextrin-chitosan polymer using its solution in the concentration of 20 ppm. Finally, authors have received the sorption capacity of 4.83 mg/g. Most of the researchers described the adsorption of ibuprofen from the solutions in concentration of 10–100 mg/L. Differences were also noted in the case of optimal pH of adsorption process. This parameter is very important when analyzing adsorption process because its affect both the nature of surface functional groups of the adsorbents as well as the form in which ibuprofen exist in water solution. Analysis of the experimental data suggest that adsorption of ibuprofen can be realized in a wide range of pH, what is determined mostly by the type of the sorption material used. More acidic environment is the most suitable when adsorbing ibuprofen onto activated carbons (Maestre et al. 2007; Guedidi et al. 2017) as neutral or basic are better one to adsorb it onto natural clays (Martín et al. 2019; Rafati et al. 2018; Khazri et al. 2017). Comparison of maximum sorption capacities of different type sorbents confirmed that the most important is appropriate selection of the material. Significant capacities were noted in case of activated carbons (Maestre et al. 2007; Guedidi et al. 2017), materials of natural origin (Lawal and Moodley 2017) or synthetic oxide based materials (Kollarahithlu and Balakrishnan 2018). It is worth to mention that experimental data collected in the manuscript are in agreement with analyzed literature reports, and confirm the complexity of adsorption of active pharmaceutical ingredients. Among many different adsorbents reported, synthesized hybrid MgO–SiO2/calcium lignosulfonate system exhibits relative good sorption capacity towards ibuprofen but it should be mentioned that obtained qm value results from high dilution of adsorbate solution.
4 Conclusions
The proposed in situ sol–gel method enabled effective synthesis of a functional hybrid material, which is a combination of an MgO–SiO2 oxide system and an active biopolimer (calcium lignosulfonate), as confirmed with physicochemical analysis results. The porous structure parameters proved a relatively high surface area (71 m2/g), pore diameter and their volume, which made it possible to classify hybrid adsorbent to the group of mesoporous materials. It is worth to emphasize that the advantage of the synthesized material are not only the parameters of the porous structure, but also the presence of multifunctional calcium lignosulphonate determining its surface activity.
The hybrid system obtained proved to be effective in the adsorption of ibuprofen—the efficiency of its removal from the model aqueous solution exceeded 70%. Adsorption tests allowed to confirm the relationship between the effectiveness of the removal of the pharmaceutical and the conditions of the adsorption process. The adsorption process itself was relatively rapid, and high efficiency was achieved in the first 3–5 min of the process. The increase in the mass of the adsorbent used contributed to the increasing efficiency of drug removal. The pH of the solution also implied the adsorption efficiency. The best results of drug removal were obtained in the acidic reaction environment (pH 2). Those results also confirmed the participation of functional groups in the adsorption mechanism. The theoretical description of the process confirmed that the obtained experimental data best describes the pseudo-second kinetic model and the Langmuir isotherm model (type III and IV), which indicates single-layer adsorption. Based on the assessment of the influence of individual parameters of the adsorption process and analyzing its kinetic and equilibrium aspects, the mechanism of interactions at the adsorbent-adsorbate interface was determined. It was suggested that the binding of the pharmaceutical may results from condensation of ‒OH groups (present in the ibuprofen structure and on the adsorbent surface) and on the principle hydrogen bonds (weak electrostatic and Van der Waals interactions).
References
Aso, T., Koda, K., Kubo, S., Yamada, T., Nakajima, I., Uraki, Y.: Preparation of novel lignin-based cement dispersants from isolated lignins. J. Wood Chem. Technol. 33, 286–298 (2013)
Bany-Aiesh, H., Banat, R., Al-Sou, K.: Kinetics and adsorption isotherm of ibuprofen onto grafted β-CD/chitosan polymer. Am. J. Appl. Sci. 12, 917–930 (2015)
Behera, S.K., Oh, S.Y., Park, H.S.: Sorptive removal of ibuprofen from water using selected soil minerals and activated carbon. Int. J. Environ. Sci. Technol. 9, 85–94 (2012)
Bui, T.X., Choi, H.: Adsorptive removal of selected pharmaceuticals by mesoporous silica SBA-15. J. Hazard. Mater. 168, 602–608 (2009)
Calisto, V., Ferreira, C.I.A., Oliveira, J.A.B.P., Otero, M., Esteves, V.I.: Adsorptive removal of pharmaceuticals from water by commercial and waste-based carbons. J. Environ. Manag. 152, 83–90 (2015)
Ciesielczyk, F., Bartczak, P., Klapiszewski, Ł., Jesionowski, T.: Treatment of model and galvanic waste solutions of copper(II) ions using a lignin/inorganic oxide hybrid as an effective sorbent. J. Hazard. Mater. 328, 150–159 (2017)
Ciesielczyk, F., Klapiszewski, Ł., Szwarc-Rzepka, K., Jesionowski, T.: A novel method of combination of Kraft lignin with synthetic mineral support. Adv. Powder Technol. 25, 695–703 (2014a)
Ciesielczyk, F., Przybysz, M., Zdarta, J., Piasecki, A., Paukszta, D., Jesionowski, T.: The sol–gel approach as a method of synthesis of xMgO–ySiO2 powder with defined physicochemical properties including crystalline structure. J. Sol-Gel Sci. Technol. 71, 501–513 (2014b)
Escher, B.I., Baumgartner, R., Koller, M., Treyer, K., Lienert, J., McArdell, C.S.: Environmental toxicology and risk assessment of pharmaceuticals from hospital wastewater. Water Res. 45, 75–92 (2011)
Evgenidou, E.N., Konstantinou, I.K., Lambropoulou, D.A.: Occurrence and removal of transformation products of PPCPs and illicit drugs in wastewaters: a review. Sci. Total Environ. 505, 905–926 (2015)
Freundlich, H.M.F.: Over the adsorption in solution. J. Phys. Chem. 57, 385–470 (1906)
Gadipelly, C., Pérez-González, A., Yadav, G.D., Ortiz, I., Ibáñez, R., Rathod, V.K., Marathe, K.V.: Pharmaceutical industry wastewater: review of the technologies for water treatment and reuse. Ind. Eng. Chem. Res. 53, 11571–11592 (2014)
Grabicka, B.E., Jaroniec, M.: Adsorption properties of ordered mesoporous silicas synthesized in the presence of block copolymer Pluronic F127 under microwave irradiation. Adsorption 16, 385–396 (2010)
Guedidi, H., Reinert, L., Soneda, Y., Bellakhl, N., Duclaux, L.: Adsorption of ibuprofen from aqueous solution on chemically surface-modified activated carbon cloths. Arab. J. Chem. 10, S3584–S3594 (2017)
Ho, Y.S., McKay, G.: Pseudo-second order model for sorption processes. Proc. Biochem. 34, 451–465 (1999)
Ikehata, K., Naghashkar, N.J., El-Din, M.G.: Degradation of aqueous pharmaceuticals by ozonation and advanced oxidation processes: a review. J. Int. Ozone Assoc. 28, 353–414 (2006)
Jones-Lepp, T.L., Sanchez, C.A., Moy, T., Kazemi, R.: Method development and application to determine potential plant uptake of antibiotics and other drugs in irrigated crop production systems. J. Agric. Food Chem. 58, 11568–11573 (2010)
Khazri, H., Ghorbel-Abid, I., Kalfat, R., Trabelsi-Ayadi, M.: Removal of ibuprofen, naproxen and carbamazepine in aqueous solution onto natural clay: equilibrium, kinetics, and thermodynamic study. Appl. Water Sci. 7, 3031–3040 (2017)
Khetan, S.K., Collins, T.J.: Human pharmaceuticals in the aquatic environment: a challenge to green chemistry. Chem. Rev. 107, 2319–2364 (2007)
Klapiszewski, Ł., Bartczak, P., Wysokowski, M., Jankowska, K., Kabat, K., Jesionowski, T.: Silica conjugated with kraft lignin and its use as a novel ‘green’ sorbent for hazardous metal ions removal. Chem. Eng. J. 260, 684–693 (2015a)
Klapiszewski, Ł., Nowacka, M., Milczarek, G., Jesionowski, T.: Physicochemical and electrokinetic properties of silica/lignin biocomposites. Carbohydr. Polym. 94, 345–355 (2013)
Klapiszewski, Ł., Rzemieniecki, T., Krawczyk, M., Malina, D., Norman, M., Zdarta, J., Majchrzak, I., Dobrowolska, A., Czaczyk, K., Jesionowski, T.: Kraft lignin/silica-AgNPs as a functional material with antibacterial activity. Colloid Surf. B 134, 220–228 (2015b)
Klapiszewski, Ł., Zietek, J., Ciesielczyk, F., Siwinska-Stefanska, K., Jesionowski, T.: Magnesium silicate conjugated with calcium lignosulfonate: in situ synthesis and comprehensive physicochemical evaluations. Physicochem. Probl. Miner. Process. 54(3), 793–802 (2018)
Kollarahithlu, S.C., Balakrishnan, R.M.: Adsorption of ibuprofen using cysteine-modified silane-coated magnetic nanomaterial. Environ. Sci. Pollut. Res. (2018). https://doi.org/10.1007/s11356-018-3272-8
Kovalova, L., Siegrist, H., Singer, H., Wittmer, A., McArdell, C.S.: Hospital wastewater treatment by membrane bioreactor: performance and efficiency for organic micropollutant elimination. Environ. Sci. Technol. 46, 1536–1545 (2012)
Kyzas, G.Z., Deliyanni, E.A.: Modified activated carbons from potato peels as green environmental-friendly adsorbents for the treatment of pharmaceutical effluents. Chem. Eng. Res. Design 97, 135–144 (2015)
Kyzas, G.Z., Kostoglou, M., Lazaridis, N.K., Lambropoulou, D.A., Bikiaris, D.N.: Environmental friendly technology for the removal of pharmaceutical contaminants from wastewaters using modified chitosan adsorbents. Chem. Eng. J. 222, 248–258 (2013)
Lagergren, S.: About the theory of so-called adsorption of soluble substances. Vetenskapsakad. Handl. 24, 1–39 (1898)
Langmuir, I.: The adsorption of gases on plane surfaces of glass, mica and platinum. J. Am. Chem. Soc. 40, 1361–1403 (1918)
Lawal, I.A., Moodley, B.: Sorption mechanism of pharmaceuticals from aqueous medium on ionic liquid modified biomass. J. Chem. Technol. Biotechnol. 92, 808–818 (2017)
Liao, R., Li, M., Li, W., Lin, X., Liu, D., Wang, L.: Efficient absorption of ibuprofen in aqueous solution using eco-friendly C3N4/soot composite. J. Mater. Sci. 53, 5929–5941 (2018)
Madikizela, L.M., Chimuka, L.: Synthesis, adsorption and selectivity studies of a polymer imprinted with naproxen, ibuprofen and diclofenac. J. Environ. Chem. Eng. 4, 4029–4037 (2016)
Martín, J., del Mar Orta, M., Medina-Carrasco, S., Santos, J.L., Aparicio, I., Alonso, E.: Evaluation of a modified mica and montmorillonite for the adsorption of ibuprofen from aqueous media. Appl. Clay. Sci. 171, 29–37 (2019)
Martucci, A., Pasti, L., Marchetti, N., Cavazzini, A., Dondi, F., Alberti, A.: Adsorption of pharmaceuticals from aqueous solutions on synthetic zeolites. Micropor. Mesopor. Mater. 148, 174–183 (2012)
Mestre, A.S., Pires, J., Nogueira, J.M.F., Carvalho, A.P.: Activated carbons for the adsorption of ibuprofen. Carbon 45, 1979–1988 (2007)
Michot, L.J., Villiéras, F.: Surface area and porosity. Dev. Clay Sci. 1, 965–978 (2006)
Morais, E.C., Correa, G.G., Santos, R.B., Fisch, A.G.: Selective silica-based sorbent materials synthesized by molecular imprinting for adsorption of pharmaceuticals in aqueous matrices. J. Sep. Sci. 36, 636–643 (2013)
Moro, T.R., Henrique, F.R., Malucelli, L.C., Ribas de Oliveira, C.M., Filho, M.A., de Vasconcelos, E.C.: Adsorption of pharmaceuticals in water through lignocellulosic fibers synergism. Chemosphere 171, 57–65 (2017)
Nam, S.W., Choi, D.J., Kim, S.K., Her, N., Zoh, K.D.: Adsorption characteristics of selected hydrophilic and hydrophobic micropollutants in water using activated carbon. J. Hazard. Mater. 270, 144–152 (2014)
Rafati, L., Ehrampoush, M.H., Rafati, A.A., Mokhtari, M., Mahvi, A.H.: Removal of ibuprofen from aqueous solution by functionalized strong nano-clay composite adsorbent: kinetic and equilibrium isotherm studies. Int. J. Environ. Sci. Technol. 15, 513–524 (2018)
Reza, R.A., Ahmaruzzaman, M., Sil, A.K., Gupta, V.K.: Comparative adsorption behavior of ibuprofen and clofibric acid onto microwave assisted activated bamboo waste. J. Am. Chem. Soc. 53, 9331–9339 (2014)
Ribeiro, A.V.F.N., Belisário, M., Galazzi, R.M., Balthazar, D.C., de Godoi Pereira, M., Ribeiro, J.N.: Evaluation of two bioadsorbents for removing paracetamol from aqueous media. Electronic J. Biotechnol. 14, 1–10 (2011)
Rigobello, E.S., Dantas, A., Di Bernardo, L., Vieira, E.M.: Removal of diclofenac by conventional drinking water treatment processes and granular activated carbon filtration. Chemosphere 92, 184–191 (2013)
Salihi, E.Ç., Mahramanlıoğlu, M.: Equilibrium and kinetic adsorption of drugs on bentonite: presence of surface active agents effect. Appl. Clay Sci. 101, 381–389 (2014)
Sheng, G., Wang, S., Hu, J., Lu, Y., Li, J., Dong, Y., Wang, X.: Adsorption of Pb(II) on diatomite as affected via aqueous solution chemistry and temperature. Colloids Surf. A Physicochem. Eng. Aspects 339, 159–166 (2009)
Singhon, R., Husson, J., Knorr, M., Lakard, B., Euvrard, M.: Adsorption of Ni(II) ions on colloidal hybrid organic–inorganic silica composites. Colloids Surf. B Biointerfaces 93, 1–7 (2012)
Tahar, A., Choubert, J.M., Miège, C., Esperanza, M., Le Menach, K., Budzinski, H., Wisniewski, C., Coquery, M.: Removal of xenobiotics from effluent discharge by adsorptionon zeolite and expanded clay: an alternative to activated carbon? Environ. Sci. Pollut. Res. 21, 5660–5668 (2014)
Wang, J., Li, H., Shuang, C., Li, A., Wang, C., Huang, Y.: Effect of pore structure on adsorption behavior of ibuprofen by magnetic anion exchange resins. Micropor. Mesopor. Mater. 210, 94–100 (2015)
Zhang, J., Chang, V.W.C., Giannis, A., Wang, J.-Y.: Removal of cytostatic drugs from aquatic environment: a review. Sci. Total Environ. 445–446, 281–298 (2013)
Acknowledgements
This work was supported by the Polish Ministry of Science and Higher Education (Grant No. 03/32/SBAD/0906).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article belongs to S.I. ISSHAC10, but it reach the press at the time the special issue was published.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ciesielczyk, F., Żółtowska-Aksamitowska, S., Jankowska, K. et al. The role of novel lignosulfonate-based sorbent in a sorption mechanism of active pharmaceutical ingredient: batch adsorption tests and interaction study. Adsorption 25, 865–880 (2019). https://doi.org/10.1007/s10450-019-00099-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10450-019-00099-1