
Abstract
Slavonian oak (Quercus robur subsp. slavonica (Gáyer) Mátyás) is currently gaining interest in forestry due to forest restructuring in Germany caused by climate change. Slavonian oaks originating from Croatia have been introduced into Germany mainly in the Münsterland region of North Rhine-Westphalia since the second half of the nineteenth century. They are characterized by their late bud burst, long clear bole, stem straightness and faster height and diameter growth compared to indigenous oaks in Germany. In this study, the genetic differentiation of adult trees and their respective progeny of two Slavonian and two indigenous stands in Hamm-Westtünnen, was evaluated. Genetic diversity and structure were estimated using 23 nuclear simple sequence repeat (SSRs) and 5 maternally inherited chloroplast microsatellite markers (cpSSRs). The mean expected heterozygosity of 0.545 and allelic richness of 6.23 indicate high genetic diversity in the studied populations. The group of progenies (AR = 8.40, Ho = 0.524, He = 0.559, FIS = 0.064) shows similar levels of genetic variation as the adult stands (AR = 8.37, Ho = 0.513, He = 0.554, FIS = 0.075). The genetic differentiation between adult stands and progeny was low (FST = 0.013). Genetic assignment of individuals using STRUCTURE revealed that the studied populations were divided into two clusters. There was no evidence of extensive hybridization or gene flow between Slavonian and native populations, possibly due to the different timing of bud burst of the two taxa.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In addition to forest fragmentation and urbanization, more frequent extreme weather events and the associated spread of pests and diseases, as well as the maladaptation of tree populations that have been translocated to new sites, threaten existing forests (Ivetić et al. 2016). Due to global warming and associated temperature extremes, forest trees will have to adapt to the major climate changes in the future. Thus, translocated populations also offer the possibility to study adaptation processes and to evaluate their suitability in the face of climate change. The adaptability of a plant is determined by its genetic material and by environmental factors at its growth site (Finkeldey and Hattemer 2010). A prerequisite for both this adaptation and for maintaining adaptive capacity for future generations is sufficient genetic variation in populations (Vornam et al. 2004; Gailing et al. 2008). The conservation of genetic variation plays an important role in forestry in the context of restructuring towards more climate-stable forests in order to sustain vital and productive stands. Therefore, the response of forest trees to climate change is determined by the interplay of genetic variation, adaptation, gene flow, and interspecific hybridization (Kremer 2010).
White oaks (section Quercus, family Fagaceae) are dominant and diverse forest tree species widely distributed across the Northern Hemisphere (Nixon 1993; Aldrich and Cavender-Bares 2011; Leroy et al. 2020). With regard to climate change, it is assumed that oaks will play an increasingly important role in forests, as they have a high adaptation potential as well as drought tolerance and could thus respond positively to a deficit in the climatic water balance. Due to their major ecological as well as economic significance, and their propensity for hybridization and gene flow (e.g., Burger 1975; Rushton 1993; Lepais et al. 2009; Kremer and Hipp 2020) oaks have been intensively studied, particularly with regard to their genetic diversity and especially their geographic structure (Dumolin et al. 1995; Curtu et al. 2007; Aldrich and Cavender-Bares 2011).
Pedunculate oak (Quercus robur L.) is one of the dominant oak species in Germany as well as most of Europe (Barreneche et al. 1998; Reif et al. 2016). Within the species Quercus robur, numerous taxonomic subspecies or ecological varieties have evolved over time in geographic sub-areas throughout its range, such as Quercus robur subsp. slavonica (Gáyer) Mátyás with distribution in Slavonia (eastern region of Croatia) (Burger and Gailing 2021). Slavonian oak is in demand as imported timber throughout Europe, mainly because of its very good qualities (Rieger 2018), and is currently gaining interest for forestry due to forest restructuring in Germany caused by climate change (Burger and Gailing 2021). Especially from a yield point of view, it has a great number of remarkable characteristics like high growth performance, straightness, a long clear bole as well as fine branches (Wachter 2001; Gailing et al. 2003). Slavonian oaks have been introduced into the western part of Germany, especially in the region around Münster, in the second half of the nineteenth century with the beginning of extensive seed trade through steam engines (Wachter 2001; Gailing et al. 2007a). Accordingly, the Slavonian oak stands are first generation stands in Germany.
Over the last two decades, the development and accessibility of several molecular markers enabled precise characterization of forest genetic resources as well as the study of hybridization and gene flow among oak species (Crăciunesc et al. 2013; Lind and Gailing 2013). For instance, nuclear microsatellites (nSSRs) can be used to study genetic differentiation and structures as well as gene flow between related species (e.g., Lexer et al. 2005; Lepais et al. 2009; Lind and Gailing 2013; Pérez-Pedraza et al. 2021), and chloroplast DNA markers (cpSSRs) for studying postglacial recolonization patterns (e.g., Petit et al. 2002b) in order to describe past episodes of introgression.
Here, we employed molecular markers (nSSRs and cpSSRs) to examine the genetic variation in adult stands and their natural regeneration and the presence of gene flow between populations of Quercus robur subsp. robur and Quercus robur subsp. slavonica. The present paper will address the following questions: (1) Is there any evidence of gene flow between the two closely related oak taxa, based on genetic assignment analysis comparing adult and offspring generations? (2) Is the haplotype composition of the natural regeneration in accordance with the one of the adult stands? (3) Does the amount of genetic variation vary among varieties and their natural regeneration?
Materials and methods
Plant material and DNA isolation
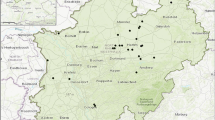
Plant material (leaves) of in total 270 trees from four neighboring stands (SLAV_161 A2, SLAV_161 A1, IND_160 B, IND_159 B) in Hüls (Hamm-Westtünnen, NRW) owned by Freiherr v. Boeselager was collected by shooting branch tips in early July 2019, Fig. 1, Table 1). Samples for SLAV_161 A2, SLAV_161 A1 and IND_160 B were randomly distributed across the plots, while for IND_159 B only the northern part of the plots were sampled (Fig. 1). Due to topographical and local conditions, as well as a very heterogeneous inventory picture, the northern part of 159 B was regarded as an independent stand from the rest of the entire stand. The stands are between 125 and 133 years old (Table 1). In addition, leaves of 369 seedlings from the natural regeneration were sampled in all stands at 7–12 sample points evenly distributed throughout the sampled part of each stand. Gailing et al. (2003) characterized the four stands based on 10 individuals at chloroplast - DNA markers and classified them at the tree level into early, intermediate, and late flushing according to Wachter (2001) (Table 1). The stands IND_159 B and IND_160 B showed an early bud burst and also the indigenous haplotype 1 (only one tree in IND_159 B showed haplotype 2), while the trees in the stand SLAV_161 A1 consistently sprouted later (Gailing et al. 2003). Stand SLAV_161 A1 was characterized by the Slavonian haplotype 2 (8 individuals) and haplotype 5 (two individuals, haplotype present in both Germany and Croatia). Stand SLAV_161 A2 showed a heterogeneous picture with early and late sprouting trees (intermediate) and was characterized by the haplotypes HP2 (4 individuals), HP5 (4 individuals) and HP10 (two individuals) (Table 1). Based on morphological and phenological classification of stands according to Wachter (2001) and genotyping with chloroplast markers by Gailing et al. (2003), two indigenous (IND_159 B, IND_160B) and two Slavonian (SLAV_161 A1, SLAV_161 A2) were identified.

Location of the four stands in Hüls (Hamm-Westtünnen, NRW) with all sampled trees (red dots) and the sampling points of natural regeneration (blue dots). White lines are the borders of all stands. IND = indigenous stand, SLAV = Slavonian stand. Map created in ArcGIS Online (Esri, California, USA)
The genomic DNA was extracted from in total 749 freshly collected leaves (approx. 1 cm2 piece of leaf) using the DNeasy 96 Plant Kit from Qiagen (Hilden, Germany). Subsequently, the amount of DNA was checked on 1% agarose gels.
Nuclear microsatellites
For the genotyping with nuclear microsatellites one genomic simple sequence repeat (Aldrich et al. 2002), three genomic simple sequence repeats (gSSRs) (Sullivan et al. 2013) and 19 gene-based expressed sequence tag–simple sequence repeats (EST-SSRs) (Durand et al. 2010; Burger et al. 2018; Müller and Gailing 2018) were used (Table 2).
Thereof, 11 primer pairs were originally developed for Quercus rubra, of which 7 were derived from a Quercus rubra EST-library (https://www.hardwoodgenomics.org/Transcriptome-assembly/1963023?tripal_pane=group_description_download) and the other four markers were from Aldrich et al. (2002) and Sullivan et al. (2013). The other 12 markers were originally developed for Quercus robur by Durand et al. (2010). We chose these 23 markers because of the high transferability of EST-SSRs between Quercus species, and the availability of a large number of nSSRs and EST-SSRs developed for Q. robur, Q. petraea, and related species (Steinkellner et al. 1997; Barreneche et al. 1998; Tanaka et al. 1999; Durand et al. 2010) that have been used in previous studies on indigenous and Slavonian oaks (e.g., Burger et al. 2021). The annotation of the sequences was obtained by searching the individual primer sequences in the respective contigs to identify the complete contig sequences for similarity searches against the UniProt Viridiplantae database (The UniProt Consortium 2021) using BLASTx (Basic Local Alignment Search Tool) (Altschul et al. 1990).
We used the same multiplex reactions for PCR as described in Burger et al. (2021) with the exception that 3 additional primers (one in multiplex 2 and two in multiplex 3) were used.
Microsatellite loci from each of the 639 individual tree samples were amplified with six different multiplexes in a 13 µl PCR mix. The PCR mix of multiplex 1 (2P24, 3A05, 3D15) and multiplex 2 (FIR013, FIR028, FIR035, FIR043) consisted of 1.5 μl reaction buffer (containing 0.8 M Tris–HCl and 0.2 M (NH4)2SO4), 1.5 μl MgCl2 (25 mM), 1 μl dNTPs (2.5 mM of each dNTP), 0.2 μl HOTFIREPol Taq polymerase (Solis BioDyne, Estonia) (5 units·μl−1), 5.8 µl H2O, 0.5 µl of each forward primer (5 pmol·μl−1), 0.5 µl of each reverse primer (5 pmol·μl−1) and 1 µl DNA (ca. 0.6 ng·μl−1). The PCR mix of multiplex 3 (PIE040, PIE125, GOT040, OC11, VIT023, VIT107) consisted of 6.5 µl Multiplex Taq PCR Master Mix Kit (QIAGEN, Germantown, Maryland, USA, providing a final concentration of 3 mM MgCl2), 2.8 µl H2O, 0.4 µl of each forward and reverse primer PIE125 (5 pmol·μl−1), 0.7 µl of each forward and reverse primer VIT107 (5 pmol·μl−1), 0.25 µl of each forward and reverse primer VIT023 (5 pmol·μl−1) 0.5 µl of each forward and reverse primer GOT040, PIE040, OC11 (5 pmol·μl−1) and 1 µl DNA (ca. 0.6 units·μl−1). For multiplex 4 (PIE102, FIR104, PIE267), the PCR mix consisted of 1.5 μl reaction buffer (containing 0.8 M Tris–HCl and 0.2 M (NH4)2SO4), 1.5 μl MgCl2 (25 mM), 1 μl dNTPs (2.5 mM of each dNTP), 0.2 μl HOTFIREPol Taq polymerase (Solis BioDyne, Estonia) (5 units·μl−1), 6.2 µl H2O, each 0.5 µl forward and reverse primer PIE102 (5 pmol·μl−1) and FIR104 (5 pmol·μl−1), 0.3 µl of each forward and reverse primer PIE267 (5 pmol·μl−1) and 1 µl DNA (ca. 0.6 ng·μl−1). For PCR amplifications of multiplex 5 (FS_C032, FS_C2660, FS_C2791, FS_C8183) and 6 (Qr0057, Qr1423, FS_C2361) a cost-effective tailed-primer approach was used (Schuelke 2000; Kubisiak et al. 2013) consisting of 1.5 μl reaction buffer (containing 0.8 M Tris–HCl and 0.2 M (NH4)2SO4), 1.5 μl MgCl2 (25 mM), 1 μl dNTPs (2.5 mM of each dNTP), 0.2 μl HOTFIREPol Taq polymerase (Solis BioDyne, Estonia) (5 units·μl−1), 5.5 μl H2O, 0.2 μl M13 (5’-CACGACGTTGTAAAACGAC-3 ́) (Kubisiak et al. 2013) tailed forward primer (5 pmol·μl−1), 0.5 μl PIG-tailed reverse primer (5ʹ-GTTTCTT-3ʹ) (5 pmol·μl−1) (Brownstein et al. 1996; Schuelke 2000; Kubisiak et al. 2013), 1 μl M13 (6-FAM/HEX) primer (5 pmol·μl−1), 5 µl H2O (5.7 µl H2O for multiplex 6) and 1 µl DNA (ca. 0.6 ng·μl−1).
Each PCR reaction was performed in a Biometra Thermal Cycler (MJ Research PTC 200, Analytik Jena, Germany) using the following touchdown program:
An initial denaturation at 95 °C for 15 min, followed by 10 touchdown cycles at 94 °C for 1 min, 1 min at 60 °C (decreasing 1 °C each cycle) and 1 min at 72 °C, followed by 25 cycles at 94 °C for 1 min, annealing at 50 °C for 1 min and elongation at 72 °C for 1 min, and a final extension step at 72 °C for 20 min. Amplification products were separated on an ABI 3130xl Genetic Analyzer (Applied Biosystems, Foster City, USA) using the GeneScan™ Rox-500 and Liz-500 (only for multiplex 3) size markers. For the fragment length analysis, multiplexes 5 and 6 were run together. Scoring of alleles was conducted using GeneMapper® version 4.1 (Applied Biosystems, Foster City, USA).
Chloroplast microsatellites (cpSSRs)
The chloroplast microsatellites udt1, ucd4 and udt4 specifically developed for oaks (Deguilloux et al. 2003) (Table S. 1), were analyzed and amplified in all samples. Ucd4 and udt4 were used to differentiate between the main haplotypes (Table S. 2). In addition, udt1 was used to distinguish the southwestern European chloroplast haplotypes HP10-11 and HP12 from each other. Finally, the universal chloroplast markers ccmp2 and ccmp10 (Weising and Gardner 1999) were used for additional validation of 28 samples with haplotype 17 (Table S. 2).
The haplotypes HP5 and HP7-26 showed the same length at all used cpSSRs and could therefore not be distinguished (Gailing et al. 2007a, b) (Table S. 2). Consequently, we consider them as one haplotype HP5/7–26.
Each PCR reaction of the udt and ucd cpSSRs was also performed in a Biometra Thermal Cycler (MJ Research PTC 200, Analytik Jena, Germany) using the following touchdown program:
An initial denaturation at 95 °C for 15 min, followed by 8 touchdown cycles at 94 °C for 1 min, 1 min at 53 °C (decreasing 1 °C each cycle) and 1 min at 72 °C, followed by 33 cycles at 94 °C for 1 min, annealing at 45 °C for 1 min and elongation at 72 °C for 1 min, and a final extension step at 72 °C for 20 min. The PCR profile for the universal cpSSRs (ccmps) consisted of an initial denaturation at 95 °C for 15 min, followed by 35 cycles at 94 °C for 1 min, a 1 min annealing step at 53 °C, an elongation step at 72 °C for 1 min and a final extension step at 72 °C for 10 min.
The forward primers used for PCR reactions are labelled with the fluorescence dyes 6-FAM or HEX (Table 4). The amplification products were diluted (1:200) and separated on an ABI 3130xl Genetic Analyzer (Applied Biosystems, Foster City, USA) in a multiplex analysis. Fragment sizes were determined with the GeneScan™ Rox-500 size marker using the software GeneMapper® version 4.1 (Applied Biosystems, Foster City, USA).
Statistical analysis of genetic diversity and differentiation based on nSSRs
The genetic variation indices observed heterozygosity (Ho) and expected heterozygosity (He), as well as the fixation index FST and Hedrick’s standardized GST (G’ST(Hed)) for individual markers and across all markers were calculated in GenAlEx version 6.51b2 (Peakall and Smouse 2006; Smouse et al. 2017). With the software Fstat version 2.9.4 (Goudet 2003) allelic richness (Ar) (here: based on a minimum sample size of 48 individuals) as well as the inbreeding coefficients (FIS) (Weir and Cockerham 1984) were determined. Differences in levels of AR, Ho, He and FIS between adults and progeny were analyzed using the ‘comparison among groups of samples test’ in Fstat computing 1000 permutations. In addition, significant deviations from zero of FIS were determined after standard Bonferroni correction (α = 0.05, p < 0.00022) implemented in the software Fstat (Goudet 2003). Linkage disequilibrium (LD) was calculated for each pair of loci in the 8 populations using Genepop version 4.7.2 (Rousset 2008) based on the following settings for the Markov chain: dememorization 10,000, batches 100 and iterations per batch 5000.
A dendrogram with UPGMA based on Nei’s standard genetic distance DS (Nei 1972) was calculated using the software program Populations version 1.2.32 (Langella 1999), with bootstrap values based on 1000 permutations across loci. Applying the software TreeView version 1.6.6 (Page 1996), the dendrogram was visualized.
To detect signatures of recent genetic bottlenecks and to investigate whether the introduced seeds established from seeds representing a limited number of seed parents, the software BOTTLENECK version 1.2.02 (Piry et al. 1999) was used. Therefore, we performed a Wilcoxon signed-rank test (one-tailed) for heterozygosity excess for ‘the two phase-mutation model’ (TPM) and ‘the stepwise mutation model’ (SMM) and a mode-shift analysis to test for a distortion in the allele frequency distribution.
Genotyping errors due to non-amplified alleles (null alleles) which can lead to overestimates of the inbreeding coefficient (FIS) were identified using the Windows®-based software MicroChecker version 2.2.3 (Van Oosterhout et al. 2004). Furthermore, Arlequin version 3.5.2.2 (Excoffier and Lischer 2010) was run with 50,000 simulations of 100 demes per group with the infinite island model based on FST in order to detect outliers which deviate significantly from the variation and differentiation expected under neutrality among all populations and between the groups of indigenous and Slavonian oaks.
An analysis of molecular variance (AMOVA) was used to analyze population differentiation and performed with GenAlEx using 9999 permutations.
Analysis of population structure
Population structure was inferred using model-based cluster analysis implemented in the software STRUCTURE version 2.3.4 (Pritchard et al. 2000) to identify possible subpopulations based on the microsatellite dataset. For this purpose, the admixture model was chosen because of its consideration of the ancestry of admixed individuals, and correlated allele frequencies were selected because of their potential to improve clustering in closely related populations, while using a burn-in period of 50,000, Markov chain Monte Carlo (MCMC) replicates of 100,000, and the LOCPRIOR model. However, we used additionally the default setting in STRUCTURE ‘admixed model without LOCPRIOR’ to identify population structure solely based on genetic information. In addition, to determine the "best K" from the logarithmic results and the ΔK method (Evanno et al. 2005), the online software STRUCTURE HARVESTER v. 0.6.94 (Earl and vonHoldt 2012) was used. The CLUMPAK software (Cluster Markov Packager Across K) was applied for post-processing and graphical representation of the results of the model-based population STRUCTURE analysis (Kopelman et al. 2015).
Besides, clustering of populations was analyzed using a principal coordinate analysis (PCoA) with the pairwise, individual-by-individual (N x N) genetic distance implemented in GeneAlEx in order to find and plot the major patterns within this dataset (Peakall and Smouse 2006, 2012).
Results
Chloroplast microsatellites (cpSSRs) and the frequency of cpDNA haplotypes
A total of 5 haplotypes could be distinguished in the studied populations corresponding to the haplotypes HP1, HP10-11, HP2, HP5/7–26, and HP17 (Petit et al. 2002b). Haplotype 1 is the most frequent haplotype in western Germany and dominant in Central Europe, but completely absent in the Balkan region (Bordács et al. 2002; König et al. 2002; Petit et al. 2002a, b). Its glacial refugium was most likely located in southern Italy (Petit et al. 2002a). The haplotypes HP10-11 show a center of distribution in southwestern and western Europe and had their glacial refugia on the Iberian Peninsula (Petit et al. 2002b). Moreover, these haplotype (HP10-11) are also apparently missing in the Balkan region (Petit et al. 2002a). In addition to these presumably indigenous haplotypes, HP2, HP5/7–26 and HP17 have a center of distribution in the Balkan region with presumed southern Italian origin (Bordács et al. 2002; Petit et al., 2002a, b). Although HP2, HP5/7–26, and HP17 are very frequent in the Balkan region (Petit et al. 2002a), none of them are restricted to this region, but only haplotype 5/7–26 occurs naturally in oak populations in both Germany and the Balkan region.
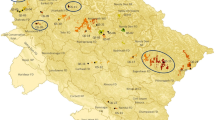
The haplotype composition of each of the four stands (two Slavonian and two indigenous) and their natural regeneration is summarized in Table 3 and visualized in Fig. 2.

Haplotype composition of each of the four analyzed stands and their natural regeneration (NR). HP1: Italy-Scandinavia line (lineage C), HP2: Croatia-Sicily line (lineage C), HP5: Italy-Eastern Balkan-Germany line (lineage A), HP7-26: Croatia-Catalonia line (lineage A), HP10: Western Europe-Portugal line (lineage B), HP11: Western Europe line (lineage B), HP17: Italy-Balkan line (lineage E) (as described in Petit et al. (2002b)). SLAV = Slavonian stand, IND = indigenous stand, NR = natural regeneration. Map generated with ArcGIS online
Stand SLAV_161 A2 consists to 72% of Slavonian haplotypes with HP5/7–26 as the most frequent one (n = 50) followed by HP2 (n = 28), HP17 (n = 11) and a low proportion of haplotypes HP10-11 (n = 14) and HP1 (n = 5). All sampled trees of stand SLAV_161 A1 show only Slavonian haplotype 2 except for one tree with HP5/7–26. One of the indigenous stands, IND_160 B, consists to 98% of HP1 (n = 49) and only one individual shows haplotype 2. The other indigenous stand IND_159 B includes only 75% native oaks with haplotype 1 (n = 46) and 25% with Slavonian haplotype HP2 (n = 4), and HP17 (n = 2) as well as HP5/7–26 (n = 9).
The natural regeneration of SLAV_NR_161 A2, SLAV_NR_161 A1, IND_160 B and IND_159 B shows comparable genetic structures to their adult stands.
Variation within and among stands at nuclear microsatellite markers
Inbreeding coefficients across all markers were not significantly different from zero in any population. An overview of the genetic diversity indices (N, AR, Ho, He, FIS, GST(Hed), FST) at all 23 markers in the 8 populations can be found in Online-Resource 1. The mean observed heterozygosity (Ho) ranged from 0.499 in SLAV_161 A1 to 0.538 in IND_NR_159 B, and the expected heterozygosity (He) ranged from 0.532 in IND_NR_160 B to 0.556 in IND_159 B and SLAV_NR_161 A2. A summary of the genetic parameters across all loci is given in Table 4.
The mean values for effective number of alleles, allelic richness and inbreeding coefficients (not significant after Bonferroni correction (5%)) were lower in the natural regeneration (AR: 6.02, He: 0.546, FIS: 0.045) compared to adult stands (AR: 6.43, He: 0.545, FIS: 0.068). However, the observed and expected heterozygosity was slightly lower in the group of adult stands (Ho: 0.513, He: 0.545) than in the natural regeneration (Ho: 0.524, He: 0.546).
The mean observed heterozygosity per locus ranged from 0.092 at locus 2P24–0.818 at locus GOT040, and the mean expected heterozygosity per locus ranged from 0.088 at locus 2P24 – 0.879 at locus OC11 (Table S. 3).
The pooled sample of naturally established seedlings across all stands showed no significantly higher (exact test, 1000 permutations, p = 0.045) allelic richness (AR) in adult trees (6.43) than in the seedling cohort (6.02; Table 4). Higher levels of AR in adults compared with seedlings were detected for each population separately. In addition to the mean AR across populations, observed heterozygosity (Ho), expected heterozygosity (He) and inbreeding coefficient (FIS) were also not different between the adult and progeny cohort (exact test, 1000 permutations, p > 0.05) (Table 2).
The genetic differentiation (FST) between the 8 populations (adult and natural regeneration) was relatively low at most loci ranging from 0.009 for FS_C2791 – 0.174 for Qr0332. Overall, the mean differentiation among all populations was 0.022 (G’ST(Hed) = 0.044) (Online-Resource 1). The mean pairwise FST between adult stands is 0.010 (G’ST(Hed) = 0.022), between progeny 0.014 (G’ST(Hed) = 0.046) and between adult stands and progeny 0.013 (G’ST(Hed) = 0.038) (Table S. 4). The mean pairwise FST between indigenous oaks is 0.010 (G’ST(Hed) = 0.027), between Slavonian oaks 0.009 (G’ST(Hed) = 0.022) and between indigenous and Slavonian oaks 0.014 (G’ST(Hed) = 0.042) (Table S. 4). In addition, Table S. 5 shows also the pairwise FST values between adult and progeny separately for Slavonian and indigenous stands. The low differentiation between adult and offspring generations as well as between indigenous and Slavonian oaks and results of genetic assignment analyses reveal similar genetic structures in both generations and no evidence for extensive gene flow between Slavonian and indigenous stands.
Further, the outlier test with Arlequin showed that there are no markers with signature of selection among populations and between clearly indigenous (IND_160_B) and Slavonian (SLAV_161 A1) stands (Fig. S. 1).
The percentage of all pairs of loci in LD was 0.79% in the natural regeneration SLAV_NR_161 A2, as well as 0.40% in the natural regenerations SLAV_NR_161 A1, IND_NR_159 B and IND_NR_160 B (mean over all populations: 0.20%). All adult populations had no loci in LD.
Null alleles were detected in each of the 10 populations at least at one of the markers OC11, 3D15, FIR028, FIR035, FIR043, GOT040, PIE040 (highest null allele frequencies in IND_NR_159 B: 0.1716, in SLAV_NR_161 A1: 0.1848 and IND_159 B: 0.1716), PIE125, VIT023, VIT107, FS_C2660 and FS_C2791 (highest null allele frequency in SLAV_161 A1: 0.1615) (Table S. 6). However, in all populations null alleles only occurred with a frequency between 0.51% and 2.51% over all loci (Table S. 6). Furthermore, null alleles across all populations per locus ranged from 0.006 at locus PIE125 and GOT040 to 0.092 at locus PIE040 (Table S. 3). The comparison of null alleles between marker types showed that two of the four gSSRs (OC11, 3D15) showed null alleles with a low frequency (< 0.040), null allele frequencies of EST-SSRs developed for Q. robur and Q. petraea ranged from 0.006 at locus PIE125 to 0.092 at locus PIE040 and null allele frequencies of EST-SSRs developed for Q. rubra ranged from 0.013 at locus Qr0057 and FS_C2660 to 0.016 at locus FS_C2791 (Table S. 3). Across all loci, EST-SSRs developed for Q. robur and Q. petraea have the highest null allele frequency (0.021), followed by gSSRs (0.012) and EST-SSRs developed for Q. rubra (0.006).
The results of the program BOTTLENECK mode-shift analysis showed that the allele frequencies of all populations followed a normal L-shaped distribution as expected for mutation-drift equilibrium (Table S. 7). This may indicate that the introduced seeds did not originate from only a limited number of seed parents. The AMOVA overall populations showed that 98% of the molecular variance was within populations (91% within and 7% between Individuals) and only 2% among populations.
Population structure
Using principal coordinate analysis (PCoA), the basic patterns among the populations are presented in Fig. 3. Here, the PCoA results do not show a clear differentiation between the indigenous and the Slavonian populations. Even though there is a separation along axis 1, the natural regeneration of IND_159 B groups with Slavonian stands (Fig. 3, see also Fig. 4). In comparison, PCoA based on chloroplast microsatellite markers shows that each natural regeneration is grouped with their adult stand (Fig. 5).

Principal coordinate analysis (PCoA) based on nuclear microsatellite markers (nSSRs) for all populations. Green and SLAV = Slavonian stand, yellow and IND = indigenous stand, circle = adult stands, square = natural regeneration

Principal coordinate analysis (PCoA) based on chloroplast microsatellite markers (cpSSRs) for all populations. Green and SLAV = Slavonian stand, yellow and IND = indigenous stand, circle = adult stands, NR and square = natural regeneration

STRUCTURE diagram with admixed model and LOCPRIOR for K = 2 displayed using CLUMPAK (Kopelman et al. 2015). Populations are separated by black vertical lines. Blue and SLAV = Slavonian oak, orange = common oak for all populations, NR = natural regeneration, SLAV = Slavonian stand, IND = indigenous stand
Using the Evanno method (Evanno et al. 2005) and STRUCTURE HARVESTER (Earl and vonHoldt 2012) an optimal and significant K = 2 was determined (Fig. S. 2, Fig. S. 3 and Table S. 8). The diagram calculated by STRUCTURE using the admixed model and LOCPRIOR with the population assumption of K = 2 is presented in Fig. 4. In addition, Table 5 shows the proportion of membership of each sampled population in the two clusters, but there is no specific cluster for each origin, since both clusters occur in both origins. A proportion of ancestry > 0.5 or < 0.5 in cluster 1 is characteristic for indigenous and Slavonian stands, respectively. In addition, Fig. 6 combines the STRUCTURE results with the haplotypes and shows that the Slavonian haplotypes (HP2, HP5/7–26, HP17) are differentiated from non-Slavonian HP1, HP10-11 at nSSRs (Fig. 6). Furthermore, a table in the Online-Resource 2 lists a direct comparison of cpSSR haplotype vs. STRUCTURE-based proportion at tree level for each population. However, we found no putative F1 hybrids. Because of the low differentiation between Slavonian and indigenous oaks at nSSRs, the identification of F1 hybrids is challenging. While adult stand and natural regeneration of stand IND_160 B are clearly assigned to the “indigenous” cluster, seedlings and adult trees of IND_159B show a variable assignment which is also reflected in the PCoA analysis and cpSSR haplotype composition (see above, Fig. 3).

Differentiation of haplotypes based on nSSRs (admixed model with LOCPRIOR) displayed using CLUMPAK (Kopelman et al. 2015). Populations separated by black vertical lines. More orange = indigenous haplotype. blue = Slavonian haplotype
To show the genetic relationship between the 8 studied populations (4 adult stands and their natural regeneration), an UPGMA dendrogram was also prepared in the appendix, based on the unbiased genetic distance matrix of Nei (1972) (Fig. S. 4). In accordance with PCoA (Fig. 3), it shows that the Slavonian and native stands differ only slightly at nuclear markers.
Discussion
Genetic diversity of adults and natural regeneration
To our knowledge, nuclear microsatellites are among the most precise tools for genotype determination and are important genetic markers widely used in population genetic analysis in oaks. According to Spence et al. (2021), microsatellites are an accessible method to gain an understanding of genetic diversity and structure.
A previous study (Burger et al. 2021) conducted on a larger number of populations enabled the assessment of genetic differentiation between native and Slavonian adult stands using the same nuclear microsatellite marker set (except for 3 additional markers used in the present study). Accordingly, our dataset also shows high and similar genetic diversity at the microsatellite loci examined (Table 4 and Online Resource 1) across all populations with expected heterozygosity (He) of 0.545 and allelic richness (AR) of 6.23. Such a relatively high genetic variation is common for outcrossing and wind pollinated woody species such as oaks (e.g., Shi et al. 2017: He = 0.707, AR = 7.79; Spence et al. 2021: He = 0.73, AR = 5.29). Furthermore, the overall pattern of genetic variation we observed at nuclear microsatellite loci is typical of long-lived and outcrossing species such as trees with relatively high genetic diversity (AR = 6.23, He = 0.545), a low level of inbreeding within (FIS = 0.059) and a low differentiation among populations (FST = 0.022) (Hamrick et al. 1992). According to Kremer and Petit (1993) such a low species differentiation is typically for European white oaks.
No clear differences between the genetic variation indices of the natural regeneration and the adult trees were found in the stands of Hamm-Westtünnen in North-Rhine Westphalia (Table 4). Other studies, however, showed higher genetic diversity estimates, but also no clear differences between adult and progeny of pedunculate oak stands. For instance, based on 10 genomic microsatellite loci, Vranckx et al. (2014) reported for the adult trees a mean number of AR of 11.5, a mean Ho of 0.798, a mean He of 0.821, and for the progeny a mean AR of 10.7, a mean Ho of 0.804, and a mean He of 0.817. Based on 19 nuclear microsatellite markers (12 genomic and 7 EST-SSRs), also Sandurska et al. (2017) reported slightly higher values (adult: AR = 17.823, Ho = 0.784, He = 0.821; seedlings: AR = 17.128, Ho = 0.759, He = 0.803) as compared to the present study. However, in assessing diversity indices from different studies, it should be mentioned that the values are not directly comparable, as different microsatellite marker sets have been used by different researchers. For instance, higher genetic variation could be due to the generally higher variability at genomic SSRs compared with EST-SSRs (Rungis et al. 2004).
The estimates of FIS were positive and show significant deviations from HWE at some loci (Online-Resource 1). Positive FIS values indicate inbreeding only if they are significant at most loci, which was not the case here (only at 2 loci per population, Online-Resource 1). Otherwise, high FIS values at only certain loci may indicate selection for these loci, selection for genes associated with these loci, or the occurrence of null alleles (Katičić et al. 2018). However, due to the very low null allele frequency (0.51%–2.71%) on average across loci, little impact on population genetic structures and differentiation (FST) can be assumed based on Oddou-Muratorio et al. (2009) and Chapuis und Estoup (2007).
Positive and higher FIS in the Slavonian adult and offspring cohorts as compared to the indigenous stands could be due to the Wahlund effect and non-random mating (e.g., inbreeding, or preferential mating within groups of different geographic origin, differences in flowering time). However, no clear effects of genetic drift and inbreeding on the genetic diversity of the progeny could be observed, so non-random mating is rather unlikely.
Reproductive isolation of late flushing Slavonian oaks from indigenous stands has likely resulted in the observed pattern of genetic differentiation between Slavonian and indigenous seedling cohorts. Here, it is important to note that the flushing date was assessed a proxy for flowering time, two traits which are highly correlated in oaks (e.g., Chesnoiu et al. 2009). Low coefficients of pairwise fixation levels show also low difference between the two generations (FST = 0.013) of the studied stands, thus providing no evidence for fixation or genetic drift. In addition, the overall effect of genetic drift on allelic richness (AR) was also rather limited, and even when all populations were pooled, allelic richness was not significantly different between adult and seedling cohorts (Table 4).
Likewise, the analysis of molecular variance for the two generations shows greater variability within populations (98%) than among populations (2%), which is commonly observed in pedunculate oaks (Gömöry et al. 2001; Neophytou 2015; Burger et al. 2021; Kesić et al. 2021). Furthermore, a comparison between Slavonian (AR = 6.26, Ho = 0.504, He = 0.542, FIS = 0.077) and indigenous (AR = 6.13, Ho = 0.523, He = 0.548, FIS = 0.055) old-growth stands and their natural regeneration (SLAV_NR: AR = 6.10, Ho = 0.521, He = 0.550, FIS = 0.055; IND_NR: AR = 5.54, Ho = 0.528, He = 0.541, FIS = 0.030) also revealed similar levels of genetic variation. In addition, similar diversity values of the introduced plantations as compared to native stands, suggest that Slavonian stands were established with seed material that was sampled in Croatia from a representative number of trees per population.
The low genetic differentiation between generations and similar levels of expected heterozygosity and allelic richness in the seedlings of Slavonian stands indicated that natural regeneration is efficient in these stands, possibly due to the low density of herbivores/deer. According to Hamrick (2004), sustaining high genetic diversity increases the likelihood of having well-adapted genotypes to ensure the long-term viability of forest tree populations.
Haplotype composition of adult stands and their natural regeneration
Since chloroplast markers (cpSSRs) in angiosperms are usually maternally inherited (Dumolin et al. 1995) and natural regeneration share the same chloroplast information as the maternal tree, they show high uniformity within stands but marked differentiation among them, which is especially true for barochoric tree species such as oaks (Petit et al. 2005). Thus, chloroplast markers are particularly suitable in combination with biparentally inherited microsatellite markers to classify and detect admixture of natural regeneration of Slavonian and indigenous oaks. The five cpSSRs used in this study have been shown to be suitable and useful in previous studies to identify different haplotypes (Gailing et al. 2007a, b, 2009).
The populations analyzed in this study were first examined for haplotype composition in 2003 by Gailing et al. (2003) on 10 representative individuals per population. Thereby, haplotypes 1–4 found in Gailing et al. (2003), correspond to haplotypes 2, 5, 10 and 1 described in Petit et al. (2002a). We found similar haplotype composition for the SLAV_161 A1, SLAV_161 A2, IND_159 B, and IND_160 B stands as described in Gailing et al. (2003), except that HP1 and HP17 were additionally found in individual trees in SLAV_161 A2 and HP2, HP5/7–26, and HP17 in IND_159 B (Table 3, Fig. 2). Since haplotype 5/7–26 also occurs naturally in Germany, it is not possible to infer Slavonian reproductive material from cpSSR markers alone. Therefore, the combination of both marker types (maternally inherited cpSSRs and biparental inherited nSSRs) was useful to detect this admixture and to assign haplotypes, which occur naturally in both Germany and Slavonia, to their geographic origin.
The natural regeneration shows a similar to equal haplotype composition compared to the respective adult stands (Table 3). This can be also seen in the PCoA based on cpSSRs (Fig. 5), which assigns each natural regeneration to its respective old-growth stand with low genetic distances. Only the relative frequencies of haplotypes differ slightly between adults and seedlings (Fig. 2), but the most frequent haplotype remains the same in both generations (Table 3).
Genetic assignment of populations
We used the Bayesian model-based clustering method implemented in the program STRUCTURE (version 2.3.4, Pritchard et al. 2000) to assign individuals to K populations based on 23 nuclear microsatellites. The STRUCTURE analysis (Fig. 4, Table 5) revealed that the studied populations were grouped into two clusters, Slavonian (a higher proportion of blue) and indigenous oaks (a higher proportion of orange). However, it is difficult to identify two distinct groups (Slavonian and Native) based on the PCoA (Fig. 3), since the natural regeneration of each adult stand is shifted substantially against their maternal stands in one direction. On the one hand, a possible reason for the shift in natural regeneration IND_NR_159 B and IND_NR_160 B along axis 1 could be gene flow between native stands (e.g., the surrounding native stands that were not sampled, cf. Figure 1). On the other hand, the shifts in the natural regeneration of the Slavonian stands could suggest a (non-random) gene flow (pollen influx) from another Slavonian stand (Table S. 4, 5, Fig. S.4). However, a PCoA with all Slavonian and native populations in North Rhine-Westphalia from Burger et al. (2021) and the populations investigated in this study also revealed two groups in which the Slavonian populations (SLAV) could be assigned to the Slavonian cluster of other Slavonian populations in North Rhine-Westphalia (Fig. S. 5). Therefore, the nuclear marker set is well suited to distinguish at the population level (but not at the tree level) between Slavonian and native oak in both old-growth stands and their progeny and to identify mixed stands (see stand IND_159 B). In addition to these nuclear marker results, the inclusion of cpDNA haplotypes shows that Slavonian and indigenous oak stands are not always pure stands (Fig. 2). Thus, the Slavonian stand SLAV_161 A2 also showed 18% of the non-Slavonian haplotypes 1 and 10–11, and IND_159 B showed 25% Slavonian old-growth trees (HP5/7–26, 5, 17) (Fig. 2). We demonstrated that chloroplast markers are suitable to identify individual trees (tree level) with non-Slavonian haplotypes in Slavonian old-growth stands and vice versa.
Comparison of STRUCTURE results with cpSSR haplotypes for haplotype 2 showed that within the parent population 85 trees had HP2 and of these 73 were also assigned to the Slavonian cluster (ancestry coefficient > 0.6) based on nSSRs (Online-Resource 2, Fig. 6). Thus, 86% of the parent trees with haplotype 2 were also assigned to the Slavonian variety based on nSSRs.
Furthermore, low genetic differentiation between Slavonian and indigenous oaks did not allow to unambiguously distinguish between varieties at the tree level (Online-Resource 2). The different timing of bud burst (Table 1), as described for these stands in Gailing et al. (2003) suggests that flowering times between taxa show little to no overlap (due to synchronization of flowering with flushing, Chesnoiu et al. 2009), thus restricting gene flow between Slavonian and indigenous stands. In Germany, Slavonian oaks show up to three weeks later bud burst compared to indigenous oak stands (Wachter 2001; Gailing et al. 2003).
Practical applicability
For sustainable forest management, it is important to provide forest enterprises with reproductive material that complies with both economic objectives, such as increment and wood quality, and ecological objectives, such as genetic diversity and local adaptation (Konrad et al. 2012). In this context, Slavonian oak is currently gaining interest as a seed source for forestry not only because of its very good economic characteristics (wood quality, fast height growth, long clear stem), but also with regard to climate change-induced forest conversion due to its broad site amplitude, possibly better adaption and late bud burst and associated lower susceptibility to the oak leaf roller (Schirmer 2017; Rieger 2018; Burger and Gailing 2021). Currently, an alternative form of sustainable forest management is practiced, based on certification systems such as certification of the origin of reproductive material, which provides the forest owner with detailed information about the origin of the planting material. The microsatellite marker set (cpSSRs and nSSRs) used in this study can reliably be used for identification and certification of Slavonian and native forest reproductive material. For this purpose, a previous study (Burger et al. 2021) already indicated that this nuclear microsatellite marker set is suitable for identifying mixed stands, detecting the origin of reproductive material, and distinguishing between Slavonian and native oak stands.
Furthermore, the marker set is also suitable for establishment and test of admixture in seed orchards. Thus, Katičić et al. (2018) based on other nSSRs and the same cpSSRs were able to investigate clonal seed orchards in Croatia for their genetic diversity and differentiation as well as spatial genetic structure and transfer of forest reproductive material between clonal seed orchards.
Meanwhile, there are also some late flushing Slavonian stands in Germany for seed production and breeding, such as the selected provenances (“Sonderherkünfte”) "Münsterländer Späteiche – Slawonische Stieleiche", "Königsforst" as well as "Kottenforst" from North Rhine-Westphalia and the "Späteiche Burg Eltz" from Rhineland-Palatinate (DKV 2022).
In a further study, however, it would be necessary to investigate the extent to which gene flow occurs between intermediate and early flushing stands. Especially seed stands must be chosen to avoid outside pollen influx. The advantage in this case could be that plantations or seed orchard with the particularly late-flushing haplotype 2 may not require geographic isolation from conspecific indigenous stands, as they are reproductively isolated from early-flushing neighboring stands due to different flowering times.
Conclusion and future perspectives
Our study shows that the genetic information of old-growth stands is almost completely passed on to the next generation. Minor changes in the genetic structure from adult to offspring generation suggest the maintenance of the same level of genetic variation in the progeny. Since genetic diversity has a strong impact on the adaptability of populations to changing environmental conditions and survival, the main objective for effective conservation of forest genetic resources is to achieve the highest possible level of genetic variability (Andonovski and Velkovski 2019). In addition, the genetic variability of tree populations ensures the stability and sustainability of forest ecosystems. Moreover, limited gene flow was revealed between the native and Slavonian oaks, likely due to differences in bud burst and flowering time between the two oak taxa, especially between the very late-flushing (predominantly haplotype 2) and early-flushing (predominantly haplotype 1) oaks.
In conclusion, seed production stands and seed orchards of late flushing Slavonian oak would require little geographic isolation from early flushing indigenous oak stands.
References
Aldrich PR, Cavender-Bares J (2011) Quercus. In: Kole C (Ed.), Wild Crop Relatives: Genomic and Breeding Resources: Forest Trees. Springer Berlin Heidelberg 89–129 https://doi.org/10.1007/978-3-642-21250-5_6
Aldrich PR, Michler CH, Sun W, Romero-Severson J (2002) Microsatellite markers for northern red oak (Fagaceae: Quercus rubra). Mol Ecol Notes 2:472–474. https://doi.org/10.1046/j.1471-8286.2002.00282.x
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410. https://doi.org/10.1016/S0022-2836(05)80360-2
Andonovski V, Velkovski N (2019) Conservation and management of forest genetic resources in Macedonia. In: Šijačić-Nikolić M, Milovanović J, Nonić M (eds) Forests of southeast Europe under a changing climate. Advances in Global Change Research 65. Springer, Cham. https://doi.org/10.1007/978-3-319-95267-3_4
Barreneche T, Bodenes C, Lexer C, Trontin J-F, Fluch S, Streiff R, Plomion C, Roussel G, Steinkellner H, Burg K, Favre J-M, Glössl J, Kremer A (1998) A genetic linkage map of Quercus robur L. (pedunculate oak) based on RAPD, SCAR, microsatellite, minisatellite, isozyme and 5S rDNA markers. Theor Appl Genet 97:1090–1103. https://doi.org/10.1007/s001220050996
Bordács S, Flaviu P, Slade D, Csaikl U, Lesur I, Borovics A, Kézdy P, König A, Gömöry D, Brewer S, Burg K, Petit R (2002) Chloroplast DNA variation of white oaks in the Northern Balkans and in the Carpathian Basin. For Ecol Manage 156:197–209. https://doi.org/10.1016/S0378-1127(01)00643-0
Brownstein MJ, Carpten JD, Smith JR (1996) Modulation of non-templated nucleotide addition by Taq DNA polymerase: primer modifications that facilitate genotyping. Biotechniques 20:1004–1010. https://doi.org/10.2144/96206st01
Burger WC (1975) The species concept in Quercus. Taxon 24:45–50. https://doi.org/10.2307/1218998
Burger K, Gailing O (2021) Die slawonische Stieleiche (Quercus robur subsp. slavonica (Gáyer) Mátyás) als Alternative für den Klimawandel: Experimentelle und genomische Ressourcen. Allg Forst u J-Ztg 192:26–37
Burger K, Müller M, Gailing O (2018) Characterization of EST-SSRs for European beech (Fagus sylvatica L.) and their transferability to Fagus orientalis Lipsky, Castanea dentata Bork., and Quercus rubra L. Silvae Genet 67:127–132. https://doi.org/10.2478/sg-2018-0019
Burger K, Müller M, Rogge M, Gailing O (2021) Genetic differentiation of indigenous (Quercus robur L.) and late flushing oak stands (Q. robur L. subsp. slavonica (Gáyer) Mátyás) in western Germany (North Rhine-Westphalia). Eur J for Res 140:1179–1194. https://doi.org/10.1007/s10342-021-01395-8
Chapuis MP, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24:621–631. https://doi.org/10.1093/molbev/msl191
Chesnoiu EN, Șofletea N, Curtu AL, Toader A, Radu R, Enescu M (2009) Bud burst and flowering phenology in a mixed oak forest from Eastern Romania. Annal For Res 52(1):199–206
Crăciunesc I, Şofletea N, Curtu A (2013) Identification of hybrids between oak species. In: Proceedings of the Biennial International Symposium. Brașov, Romania: 51–56
Curtu AL, Gailing O, Leinemann L, Finkeldey R (2007) Genetic variation and differentiation within a natural community of five oak species (Quercus spp.). Plant Biol 9:116–126. https://doi.org/10.1055/s-2006-924542
Deguilloux M-F, Dumolin-Lapègue S, Gielly L, Grivet D, Petit RJ (2003) A set of primers for the amplification of chloroplast microsatellites in Quercus. Mol Ecol Notes 3:24–27. https://doi.org/10.1046/j.1471-8286.2003.00339.x
DKV (2022) Stieleiche (Quercus robur) – DKV Gütegemeinschaft für forstliches Vermehrungsgut eV Available from: https://dkv-net.de/stieleiche-quercus-robur/ (February 2, 2022)
Dumolin S, Demesure B, Petit RJ (1995) Inheritance of chloroplast and mitochondrial genomes in pedunculate oak investigated with an efficient PCR method. Theor Appl Genet 91:1253–1256. https://doi.org/10.1007/BF00220937
Durand J, Bodénès C, Chancerel E, Frigerio J-M, Vendramin G, Sebastiani F, Buonamici A, Gailing O, Koelewijn H-P, Villani F, Mattioni C, Cherubini M, Goicoechea PG, Herrán A, Ikaran Z, Cabané C, Ueno S, Alberto F, Dumoulin P-Y, Guichoux E, de Daruvar A, Kremer A, Plomion C (2010) A fast and cost-effective approach to develop and map EST-SSR markers: oak as a case study. BMC Genom 11:570. https://doi.org/10.1186/1471-2164-11-570
Earl DA, vonHoldt BM (2012) Structure harvester: a website and program for visualizing structure output and implementing the evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Finkeldey R, Hattemer HH (2010) Genetische Variation in Wäldern – Wo stehen wir? Forstarchiv 81:123–129. https://doi.org/10.2376/0300-4112-81-123
Gailing O, Wachter H, Leinemann L, Hosius B, Finkeldey R, Schmitt H-P, Heyder J (2003) Charakterisierung verschiedener Ursprungsgebiete der Späteiche (Quercus robur L.) mit Hilfe von DNS-Markern aus dem Chloroplastengenom. Allg Forst- u J-Ztg 174:227–231
Gailing O, Wachter H, Schmitt H-P, Curtu A, Finkeldey R (2007a) Characterization of different provenances of slavonian pedunculate oaks (Quercus robur L.) in Münsterland (Germany) with chloroplast DNA markers: PCR-RFLPs and chloroplast microsatellites. Allg Forst- u J-Ztg 178:85–90
Gailing O, Wachter H, Heyder J, Schmitt H-P, Finkeldey R (2007b) Chloroplast DNA analysis in oak stands (Quercus robur L.) in North Rhine-Westphalia with presumably slavonian origin: Is there an association between geographic origin and bud phenology? J Appl Bot Food 81:165–171
Gailing O, Langenfeld-Heyser R, Polle A, Finkeldey R (2008) Quantitative trait loci affecting stomatal density and growth in a Quercus robur progeny: implications for the adaptation to changing environments. Global Change Biol 14:1934–1946. https://doi.org/10.1111/j.1365-2486.2008.01621.x
Gailing O, Wachter H, Heyder J, Rogge M, Finkeldey R (2009) Chloroplast DNA analyses of very old, presumably autochthonous Quercus robur L. stands in North Rhine-Westphalia. Allg Forst- u J-Ztg 180:221–227
Gömöry D, Yakovlev I, Zhelev P, Jedináková J, Paule L (2001) Genetic differentiation of oak populations within the Quercus robur/Quercus petraea complex in Central and Eastern Europe. J Hered 86:557–563. https://doi.org/10.1046/j.1365-2540.2001.00874.x
Goudet J (2003) FSTAT (version 2.9.4), a program (for Windows 95 and above) to estimate and test population genetics parameters: 1–54
Hamrick JL (2004) Response of forest trees to global environmental changes. For Ecol Manage 197:323–335. https://doi.org/10.1016/j.foreco.2004.05.023
Hamrick JL, Godt MJW, Sherman-Broyles SL (1992) Factors influencing levels of genetic diversity in woody plant species. In: Adams WT, Strauss SH, Copes DL, Griffin AR (Eds), Population genetics of forest trees: Proceedings of the international Symposium on population genetics of Forest Trees Corvallis, Oregon, U.S.A., July 31–August 2 1990 Forestry Sciences. Springer Netherlands, Dordrecht 95–124. https://doi.org/10.1007/978-94-011-2815-5_7
Ivetić V, Devetaković J, Nonić M, Stanković D, Šijačić-Nikolić M (2016) Genetic diversity and forest reproductive material – from seed source selection to planting. iForest – Biogeosciences and Forestry 9: 801–812. https://doi.org/10.3832/ifor1577-009
Katičić Bogdan I, Kajba D, Šatović Z, Schüler S, Bogdan S (2018) Genetic diversity of pedunculate oak (Quercus robur L.) in clonal seed orchards in Croatia, assessed by nuclear and chloroplast microsatellites. South-east Eur for SEEFOR 9(1):29–46
Kesić L, Cseke K, Orlović S, Stojanović DB, Kostić S, Benke A, Borovics A, Stojnić S, Avramidou EV (2021) Genetic diversity and differentiation of pedunculate oak (Quercus robur L.) Populations at the southern margin of its distribution range—implications for conservation. Diversity 13:371. https://doi.org/10.3390/d13080371
König AO, Ziegenhagen B, van Dam BC, Csaikl UM, Coart E, Degen B, Burg K, de Vries SMG, Petit RJ (2002) Chloroplast DNA variation of oaks in western Central Europe and genetic consequences of human influences. For Ecol Manage 156:147–166. https://doi.org/10.1016/S0378-1127(01)00640-5
Konrad H, Sieberer K, Golesch G, Litschauer R (2012) Qualitätssaatgut aus Samenplantagen. BFW-Praxisinformation 29: 22–23.
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15:1179–1191. https://doi.org/10.1111/1755-0998.12387
Kremer A (2010) Evolutionary responses of European oaks to climate change. Irish Forestry 67:53–65
Kremer A, Hipp AL (2020) Oaks: an evolutionary success story. New Phytol 226:987–1011. https://doi.org/10.1111/nph.16274
Kremer A, Petit RJ (1993) Gene diversity in natural populations of oak species. Ann Sci for 50(Suppl. 1):186s–202s. https://doi.org/10.1051/forest:19930717
Kubisiak TL, Nelson CD, Staton ME, Zhebentyayeva T, Smith C, Olukolu BA, Fang GC, Hebard FV, Anagnostakis S, Wheeler N, Sisco PH, Abbott AG, Sederoff RR (2013) A transcriptome-based genetic map of Chinese chestnut (Castanea mollissima) and identification of regions of segmental homology with peach (Prunus persica). Tree Genet Genomes 9:557–571. https://doi.org/10.1007/s11295-012-0579-3
Langella O (1999) Populations. CNRS UPR 9034, Gif-sur-Yvette Cedex
Lepais O, Petit RJ, Guichoux E, Lavabre JE, Alberto F, Kremer A, Gerber S (2009) Species relative abundance and direction of introgression in oaks. Mol Ecol 18:2228–2242. https://doi.org/10.1111/j.1365-294X.2009.04137.x
Leroy T, Rougemont Q, Dupouey J-L, Bodénès C, Lalanne C, Belser C, Labadie K, Le Provost G, Aury J-M, Kremer A, Plomion C (2020) Massive postglacial gene flow between European white oaks uncovered genes underlying species barriers. New Phytol 226:1183–1197. https://doi.org/10.1111/nph.16039
Lexer C, Fay MF, Joseph JA, Nica M-S, Heinze B (2005) Barrier to gene flow between two ecologically divergent Populus species, P. alba (white poplar) and P. tremula (European aspen): the role of ecology and life history in gene introgression. Mol Ecol 14:1045–1057. https://doi.org/10.1111/j.1365-294X.2005.02469.x
Lind JF, Gailing O (2013) Genetic structure of Quercus rubra L. and Quercus ellipsoidalis E. J. Hill populations at gene-based EST-SSR and nuclear SSR markers. Tree Genet Genom 9:707–722. https://doi.org/10.1007/s11295-012-0586-4
Müller M, Gailing O (2018) Characterization of 20 new EST-SSR markers for northern red oak (Quercus rubra L.) and their transferability to Fagus sylvatica L. and six oak species of section Lobatae and Quercus. Ann Res 61 211–222 https://doi.org/10.15287/afr.2018.1191
Nei M (1972) Genetic distance between populations. Am Nat 106:283–292
Neophytou C (2015) Does human management affect genetic variation of the oaks in the upper Rhine plain? Forstarchiv 86:183–193. https://doi.org/10.4432/0300-4112-86-183
Nixon KC (1993) Infrageneric classification of Quercus (Fagaceae) and typification of sectional names. Ann Sci for 50:25s–34s. https://doi.org/10.1051/forest:19930701
Oddou-Muratorio S, Vendramin GG, Buiteveld J, Fady B (2009) Population estimators or progeny tests: what is the best method to assess null allele frequencies at SSR loci? Conserv Genet 10:1343–1347. https://doi.org/10.1007/s10592-008-9648-4
Page RDM (1996) Tree View: an application to display phylogenetic trees on personal computers. Bioinformatics 12:357–358. https://doi.org/10.1093/bioinformatics/12.4.357
Peakall R, Smouse PE (2006) genalex 6: genetic analysis in excel. population genetic software for teaching and research. Mol Ecol Notes 6:288–295. https://doi.org/10.1111/j.1471-8286.2005.01155.x
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research – an update. Bioinformatics 28:2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Pérez-Pedraza A, Rodríguez-Correa H, Valencia-Ávalos S, Torres-Miranda CA, Arenas-Navarro M, Oyama K (2021) Effect of hybridization on the morphological differentiation of the red oaks Quercus acutifolia and Quercus grahamii (Fagaceae). Plant Syst Evol 307:37. https://doi.org/10.1007/s00606-021-01757-0
Petit R, Brewer S, Bordács S, Burg K, Cheddadi R, Coart E, Cottrell J, Csaikl U, Dam B, Deans J, Espinel S, Fineschi S, Finkeldey R, Glaz I, Goicoechea P, Jensen J, König A, Lowe A, Søren F, Kremer A (2002a) Identification of refugia and post-glacial colonisation routes of European white oaks based on chloroplast DNA and fossil pollen evidence. For Ecol Manage 156:49–74. https://doi.org/10.1016/S0378-1127(01)00634-X
Petit RJ, Csaikl UM, Bordacs S, Burg K, Coart E, Cottrell J, van Dam B, Deans JD, Dumolin-Lapegue S, Fineschi S, Finkeldey R, Gillies A, Glaz I, Goicoechea PG, Jensen JS, Konig AO, Lowe AJ, Madsen SF, Matyas G, Munro RC, Olalde M, Pemonge M-H, Popescu F, Slade D, Tabbener H, Taurchini D (2002b) Chloroplast DNA variation in European white oaks phylogeography and patterns of diversity based on data from over 2600 populations. For Ecol Manag 156:5–26. https://doi.org/10.1016/S0378-1127(01)00645-4
Petit R, Duminil J, Fineschi S, Hampe A, Salvini D, Giovanni Giuseppe V (2005) Comparative organization of chloroplast, mitochondrial and nuclear diversity in plant populations. Mol Ecol 14:689–701. https://doi.org/10.1111/j.1365-294X.2004.02410.x
Piry S, Luikart G, Cornuet J-M (1999) Computer note. BOTTLENECK: a computer program for detecting recent reductions in the effective size using allele frequency data. J Hered 90:502–503. https://doi.org/10.1093/jhered/90.4.502
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959. https://doi.org/10.1093/genetics/155.2.945
Reif A, Baumgärtel R, Dister E, Schneider E (2016) Zur Natürlichkeit der Stieleiche (Quercus robur L.) in Flussauen Mitteleuropas – eine Fallstudie aus dem Naturschutzgebiet „Kühkopf-Knoblochsaue“ am hessischen Oberrhein. Waldökologie, Landschaftsforschung Und Naturschutz 15:69–92
Rieger N (2018) Die Wiederentdeckung der kroatischen Eiche – Kroatien-Nachrichten. Available from: https://kroatien-nachrichten.de/die-wiederentdeckung-der-kroatischen-eiche/ (October 13, 2021)
Rousset F (2008) genepop’007: a complete re-implementation of the genepop software for Windows and Linux. Mol Ecol Resour 8:103–106. https://doi.org/10.1111/j.1471-8286.2007.01931.x
Rungis D, Bérubé Y, Zhang J, Ralph S, Ritland CE, Ellis BE, Douglas C, Bohlmann J, Ritland K (2004) Robust simple sequence repeat markers for spruce (Picea spp.) from expressed sequence tags. Theor Appl Genet 109:1283–1294. https://doi.org/10.1007/s00122-004-1742-5
Rushton BS (1993) Natural hybridization within the genus Quercus L. Ann Sci for 50:73s–90s. https://doi.org/10.1051/forest:19930707
Sandurska E, Ulaszewski B, Burczyk J (2017) Genetic insights into ecological succession from oak- (Quercus robur L.) to beech- (Fagus sylvatica L.) dominated forest stands. Acta Biol Cracov Bot 59:23–33. https://doi.org/10.1515/ABCSB-2017-0002
Schirmer R (2017) Amt für forstliche Saat- und Pflanzenzucht. Slawonische Eiche im Fokus. LWF aktuell: 36
Schuelke M (2000) An economic method for the fluorescent labeling of PCR fragments. Nat Biotechnol 18:233–234. https://doi.org/10.1038/72708
Shi X, Wen Q, Cao M, Guo X, Xu L-a (2017) Genetic Diversity and structure of natural Quercus variabilis population in China as revealed by microsatellites markers. Forests 8(12):495. https://doi.org/10.3390/f8120495
Smouse PE, Banks SC, Peakall R (2017) Converting quadratic entropy to diversity: Both animals and alleles are diverse, but some are more diverse than others. PLoS ONE 12:e0185499. https://doi.org/10.1371/journal.pone.0185499
Spence SE, Fant JB, Gailing O, Griffith MP, Havens K, Hipp AL, Kadav P, Kramer A, Thompson P, Toppila R, Westwood M, Wood J, Zumwalde BA, Hoban S (2021) Comparing genetic diversity in three threatened oaks. Forests 12(5):561. https://doi.org/10.3390/f12050561
Steinkellner H, Fluch S, Turetschek E, Lexer C, Streiff R, Kremer A, Burg K, Glössl J (1997) Identification and characterization of (GA/CT)n-microsatellite loci from Quercus petraea. Plant Mol Biol 33:1093–1096. https://doi.org/10.1023/A:1005736722794
Sullivan AR, Lind JF, McCleary TS, Romero-Severson J, Gailing O (2013) Development and characterization of genomic and gene-based microsatellite markers in North American red oak species. Plant Mol Biol Rep 31:231–239. https://doi.org/10.1007/s11105-012-0495-6
Tanaka K, Tsumura Y, Nakamura T (1999) Development and polymorphism of microsatellite markers for Fagus crenata and the closely related species. F Japonica Theor Appl Genet 99:11–15. https://doi.org/10.1007/s001220051203
The UniProt Consortium (2021) UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res 49:D480–489. https://doi.org/10.1093/nar/gkaa1100
Van Oosterhout CV, Hutchinson WF, Wills DPM, Shipley P (2004) micro-checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538. https://doi.org/10.1111/j.1471-8286.2004.00684.x
Vornam B, Decarli N, Gailing O (2004) Spatial distribution of genetic variation in a natural beech stand (Fagus sylvatica L.) based on microsatellite markers. Conserv Genet 5:561–570. https://doi.org/10.1023/B:COGE.0000041025.82917.ac
Vranckx G, Jacquemyn H, Mergeay J, Cox K, Kint V, Muys B, Honnay O (2014) Transmission of genetic variation from the adult generation to naturally established seedling cohorts in small forest stands of pedunculate oak (Quercus robur L.). For Ecol Manage 312:19–27. https://doi.org/10.1016/j.foreco.2013.10.027
Wachter H (2001) Untersuchungen zum Eichensterben in NRW, Teil II (1951–2000). Schriftenreihe Der Landesforstverwaltung NRW 13:1–112
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370. https://doi.org/10.2307/2408641
Weising K, Gardner RC (1999) A set of conserved PCR primers for the analysis of simple sequence repeat polymorphisms in chloroplast genomes of dicotyledonous angiosperms. Genome 42:9–19. PMID:10207998
Acknowledgements
We are grateful and thankful to Alexandra Dolynska and Christine Radler for their valuable technical assistance. In addition, our thanks go to ISOGEN GmbH & Co. KG and especially Dr. Bernhard Hosius for their help in collecting the leaf samples. Special thanks also to the owner, the forestry company Frhr. von Kettele-Boeselager, for his willingness and agreement to enable sampling in his stands. The study was supported by the German Federal Ministry of Food and Agriculture represented by the Agency for renewable resources (FNR; Reference number: 22011317).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
KB wrote the main manuscript text and prepared all figures and tables. OG revised it critically for important intellectual content. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Communicated by Ruediger Grote.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.





Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Burger, K., Gailing, O. Genetic variability of indigenous (Quercus robur L.) and late flushing oak (Quercus robur L. subsp. slavonica (Gáyer) Mátyás) in adult stands compared with their natural regeneration. Eur J Forest Res 141, 1073–1088 (2022). https://doi.org/10.1007/s10342-022-01491-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10342-022-01491-3