
Abstract
Slavonian oaks (Quercus robur subsp. slavonica (Gáyer) Mátyás) originating from Croatia have been cultivated in Germany mainly in the Münsterland region of North Rhine-Westphalia since the second half of the nineteenth century. Compared to indigenous pedunculate oak stands in Germany, they are characterised by their late bud burst, but also by their excellent bole shape and faster height growth. Previously, Slavonian pedunculate oaks (= late flushing oaks) were mainly studied at chloroplast (cp) DNA markers in order to determine their geographical origin. The origin of the material is probably the Sava lowland between Zagreb and Belgrade. In the present study, the aim was to genetically differentiate between indigenous Quercus robur and Slavonian oak stands using nuclear DNA markers. For this purpose, we used 20 nuclear Simple Sequence Repeats (nSSRs). A total of 37 pedunculate oak stands (mean: 18.6 samples per population with an age of 95 to 210 years) were examined, of which 21 were characterized as Slavonian late flushing oaks and three stands for which the Slavonian origin was not clear. Maternally inherited chloroplast markers were analysed earlier in all 37 stands to validate their geographic origin. We found that the stands of native pedunculate oaks and Slavonian pedunculate oaks are represented by two genetic clusters which are weakly differentiated. Slavonian oaks (Na = 9.85, Ar = 8.689, Ho = 0.490, He = 0.540) showed similar levels of genetic variation as native oak stands (Na = 7.850, Ar = 7.846, Ho = 0.484, He = 0.526). Differences in growth and phenology and low but consistent genetic differentiation between groups suggest that both taxa represent different ecotypes with specific local adaptations, which are perhaps separated by less overlapping flowering phenologies. The nuclear microsatellite markers in combination with the cpDNA markers are suitable to differentiate between Slavonian and local oak stands.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The Slavonian pedunculate oak (Quercus robur L. subsp. slavonica (Gáyer) Mátyás) is an established naturalized variety of the native pedunculate oak (Quercus robur L.) in Germany and occupies a special position within this species. Slavonian oaks have been introduced into the western part of Germany, especially in the region around Münster, in the second half of the nineteenth century with the beginning of extensive seed trade through steam engines (Wachter 2001; Gailing et al. 2007a). According to historical documents and analysis with cpDNA markers, they have their geographic origin in the forest areas of the lowlands of the rivers Sava and Drava between Zagreb and Belgrade in the eastern region of Croatia (Wachter 2001; Gailing et al. 2007a, b). In Germany, Slavonian oaks are characterized by their late bud burst compared to indigenous oaks and are therefore significantly less affected by the European oak leaf roller (Tortrix viridana) and late spring frost (Wachter 2001). From a yield point of view, they have a high growth rate compared to indigenous pedunculate oaks and are characterized by their straight and long stem as well as by their fine branches (Wachter 2001; Gailing et al. 2003). The Slavonian oak stands are first-generation stands in Germany which were established between 1870 and 1912 from seeds collected in the Eastern part of today’s Croatia. A genetic characterization of putative Slavonian stands is important in order to identify seed production areas, certify reproductive material, identify mixed stands and detect gene flow between both taxa and later generation stands.
In previous studies (Petit et al. 2002; Bordács et al. 2002) on the post-glacial recolonisation of pedunculate oak species in Europe and especially the Balkan region, haplotypes HP2, HP5, HP6, HP7-26 and HP17 were found in the area of origin of the Slavonian pedunculate oak (Croatia) and a glacial refugium of populations with these haplotypes in the Balkan region was suggested (Petit et al. 2002; Bordács et al. 2002). Furthermore, in cpDNA marker studies of Slavonian oak populations in western Germany, the haplotypes HP2, HP5 and HP17 were found to be common Slavonian haplotypes (Gailing et al. 2003; Gailing et al. 2007a, b, 2009). However, only haplotype 2 does not occur naturally in Germany (Petit et al. 2002).
Expressed Sequence Tag (EST)-SSR markers (Durand et al. 2010; Burger et al. 2018; Müller and Gailing 2018) used are located in expressed genes and can be derived from publicly available genomic resources, so-called EST libraries, and are located either in coding regions or in 5′ or 3′ untranslated regions (UTRs) (Ellis and Burke 2007). In addition, they can be transferred across taxonomic boundaries because of their location in regions of the DNA that are strongly conserved within phylogenetically related species (Ellis and Burke 2007; Burger et al. 2018). Microsatellites, especially EST-SSRs, are important genetic markers widely used in population genetic analysis of forest tree species, including oaks (Streiff et al. 1998; Dzialuk et al. 2005; Ellis and Burke 2007; Lind and Gailing 2013; Sullivan et al. 2013; Müller and Gailing 2018). The genetic structure of a population is characterized by the number of subpopulations in it, the frequency of alleles in each subpopulation and the degree of genetic isolation of the subpopulation (Chakraborty 1993). Population genetic structure can be analysed through F-statistics (Wright 1965) and/or analysis of molecular variance (AMOVA) (Excoffier et al. 1992) or inferred by clustering individuals into groups (Greenbaum et al. 2016). Clustering of individuals into subpopulations based on genetic data from microsatellite analysis is an often used method (Greenbaum et al. 2016). Cluster analyses can be divided into two methods: I) model-based approaches as implemented in the program STRUCTURE and II) distance-based approaches like principal coordinate analysis (Pritchard et al. 2000; Alexander et al. 2009; Greenbaum et al. 2016).
Based on these kinds of analyses, the aim of the study is to distinguish genetically between indigenous oak stands and stands known as late flushing oaks.
The research questions of our study were: (a) Are the stands known as late flushing oak stands genetically differentiated from the indigenous oak stands in North Rhine-Westphalia? (b) Does the amount of genetic variation vary among varieties and/or are there indications of losses of genetic variation due to bottleneck effects in Slavonian oaks in North Rhine-Westphalia? (c) Are nuclear markers better suited than cpDNA markers to differentiate between indigenous and late flushing oak stands?
Materials and methods
Plant material
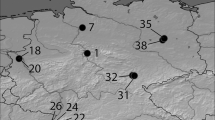
For genotyping Slavonian pedunculate oak and common pedunculate oak populations with nuclear microsatellite markers, extracted DNA samples from 2005, 2006 and 2007 were used (Gailing et al. 2007a, b, 2009) (Table 1). The trees originate from 36 different populations from seven separate regions in North Rhine-Westphalia (Germany) (Fig. 1): the Minden Land (stands planted in 1894), Münsterland (planted between 1826 and 1890), Lower Rhine region (planted in 1878), Lower Rhine bay (planted between 1893 and 1912), Bergisch region (planted between 1887 and 1891) and Sauerland (planted in 1819) (Table 1). Seeds for the establishment of the stand 28 (planted in 2007) were collected directly in Croatia (Vinkovski) (Gailing et al. 2007b). For each of the 37 populations, 16 to 20 samples were used for the genetic characterization with 20 nuclear microsatellite markers. The population Kottenforst 154B, however, consisted of only 4 samples (Table 1).

Geographical location of the sampled populations (black dots) in North Rhine-Westphalia. Map created in ArcGIS Online (Esri, California, USA). (Color figure online)
All trees of the stands (20 trees per stand) from Gailing et al. (2007a) are phenotypically (straight long bole, fast growth) as well as phenologically (late flushing) characterized as of Slavonian origin showing, based on a combination of PCR-RFLPs and cpSSRs, either haplotype 2 or haplotype 5, both of which are frequent in the Balkan region, but only haplotype 2 does not occur naturally in Germany (Gailing et al. 2007a). Most trees (20 trees per stand) from Gailing et al. (2007b) were also characterized as Slavonian pedunculate oaks due to their growth behaviour, late flushing and historical documents before characterized at cpDNA markers (Gailing et al. 2007b). The predominant haplotypes in most populations are haplotype 2 and haplotype 5 (Table S. 1). In addition, the seeds collected in Vinkovsi (Croatia) for the establishment of stand 28 show haplotype 5 (Gailing et al. 2007b). Besides the Slavonian stands, indigenous stands were also selected in order to be able to compare these with each other. Therefore, populations (mean 18.2 trees per stand) were also selected from Gailing et al. (2007b) defined as common pedunculated oaks (haplotypes 1, 4 and 10). The remaining DNA samples representing indigenous oaks were taken from Gailing et al. (2009). These stands were all established before 1850 and were owned by smallholders, indicating that native plant material was used (Gailing et al. 2009). Haplotype 1 was the most common haplotype in indigenous stands, and haplotype 12 occurred in only one population. Table S. 1 and Fig. 2 give an overview on relative haplotype frequencies and distribution for each population in North Rhine-Westphalia.

a Distribution of Quercus robur and Quercus robur subsp. slavonica (with black circle) chloroplast haplotypes in North Rhine-Westphalia. Populations’ haplotypes were identified in earlier studies (Gailing et al. 2007a,b, 2009). HP1: Italy-Scandinavia line (lineage C), HP2: Croatia-Sicily line (lineage C), HP4: central Europe line (lineage A), HP5: Italy-Eastern Balkan-Germany line (lineage A), HP7-26: Croatia-Catalonia line (lineage A), HP10,: Western Europe-Portugal line (lineage B), HP11, 12: Western Europe line (lineage B), HP17: Italy-Balkan line (lineage E) (as described in Petit et al. 2002). b Distribution of Quercus robur (with white circle) and Quercus robur subsp. slavonica proportion in STRUCTURE clusters in North Rhine-Westphalia. A higher proportion of ancestry in cluster 1 = common oaks, a higher proportion in cluster 2 = Slavonian oaks. The small section on the upper left shows the location of the stand Vinkovsi in Croatia. Maps generated with ArcGIS Online. (Color figure online)
Some Slavonian oak stands, such as Nagel-Doornick 50B, Hamm-Osttünnen 1B1b, Tomberg 10B2, Stadt Viersen 36B/38, Freiherr von der Leyen 17C, Plettenberg 104G, Steprath 3H and Estermann 116A1, also have a low relative frequency (0.05–0.125) of indigenous haplotypes (HP1 and HP10) (see Table S.1, Fig. 2). Conversely, the two indigenous oak stands, Kottenforst 134A&C and Gut Ulenburg 4C, also show the Slavonian haplotype 2 with a relative frequency of 0.05–0.1. The other 27 populations have either only indigenous haplotypes (HP 1, 4, 10, 12) or only haplotypes which are characteristic of Slavonian oaks (HP 2, 5, 7–26) (Table S.1, Fig. 2).
Microsatellite analysis
A total of three genomic simple sequence repeats (gSSRs) (Sullivan et al. 2013) and 17 gene-based expressed sequence tag–simple sequence repeats (EST-SSRs) were used (Table 2). Due to the high transferability of EST-SSRs among species in Quercus and the availability of a large number of nSSRs and EST-SSRs developed for Q. robur, Q. petraea and related species (Steinkellner et al. 1997; Barreneche et al. 2004; Durand et al. 2010), we selected these 20 markers with reliable amplification in multiplex reactions. Thereof, 10 EST-SSR primer pairs were originally developed for Quercus robur L. (Durand et al. 2010). Seven EST-SSRs (Qr0057, Qr0332, Qr1423, FS_C2361, FS_C2660, FS_C2791 and FS_C8183) originally developed in Q. rubra were tested successfully in Müller and Gailing (2018) for their transferability to Q. robur based on primer sequences published by the ‘Hardwood Genomics Project’ (https://www.hardwoodgenomics.org/Transcriptomeassembly/1963023?tripal_pane=group_description_download). The annotation of the sequences was obtained by searching the individual primer sequences in the respective contigs to identify the complete contig sequences for similarity searches against the UniProt Viridiplantae database (The UniProt Consortium 2017) using BLASTx (Basic Local Alignment Search Tool) (Altschul et al. 1990).
Genomic DNA from each of the 689 individual tree samples was amplified with six different multiplexes in a 13 µl PCR mix. The PCR mix of multiplex 1 (2P24, 3A05, 3D15) and multiplex 2 (FIR013, FIR028, FIR035) consisted of 1.5 μl reaction buffer (containing 0.8 M Tris–HCl and 0.2 M (NH4)2SO4), 1.5 μl MgCl2 (25 mM), 1 μl dNTPs (2.5 mM of each dNTP), 0.2 μl HOTFIREPol Taq polymerase (Solis BioDyne, Estonia) (5 units/μl), 5.8 µl H2O, 0.5 µl of each forward primer (5 picomol/µl), 0.5 µl of each reverse primer (5 picomol/µl) and 1 µl DNA (ca. 0.6 ng/μl). The PCR mix of multiplex 3 (PIE125, GOT040, VIT023, VIT107) consisted of 6.5 µl Multiplex Taq PCR Master Mix Kit (QIAGEN, Germantown, Maryland, USA, providing a final concentration of 3 mM MgCl2), 2.8 µl H2O, 0.4 µl of each forward and reverse primer PIE125 (5 picomol/µl), 0.7 µl of each forward and reverse primer VIT107 (5 picomol/µl), 0.25 µl of each forward and reverse primer VIT023 (5 picomol/µl), 0.5 µl of each forward and reverse primer GOT040 (5 picomol/µl) and 1 µl DNA (ca. 0.6 ng/μl). For multiplex 4 (PIE102, FIR104, PIE267), the PCR mix consisted of 1.5 μl reaction buffer (containing 0.8 M Tris–HCl and 0.2 M (NH4)2SO4), 1.5 μl MgCl2 (25 mM), 1 μl dNTPs (2.5 mM of each dNTP), 0.2 μl HOTFIREPol Taq polymerase (Solis BioDyne, Estonia) (5 units/μl), 6.2 µl H2O, each 0.5 µl forward and reverse primer PIE102 (5 picomol/µl) and FIR104 (5 picomol/µl), 0.3 µl of each forward and reverse primer PIE267 (5 picomol/µl) and 1 µl DNA (ca. 0.6 ng/μl). For PCR amplifications of multiplex 5 (FS_C032, FS_C2660, FS_C2791, FS_C8183) and 6 (Qr0057, Qr1423, FS_C2361) a cost-effective tailed-primer approach was used (Schuelke 2000; Kubisiak et al. 2009) consisting of 1.5 μl reaction buffer (containing 0.8 M Tris–HCl and 0.2 M (NH4)2SO4), 1.5 μl MgCl2 (25 mM), 1 μl dNTPs (2.5 mM of each dNTP), 0.2 μl HOTFIREPol Taq polymerase (Solis BioDyne, Estonia) (5 units/μl), 5.5 μl H2O, 0.2 μl M13 (5’-CACGACGTTGTAAAACGAC-3 ́) (Kubisiak et al. 2009) tailed forward primer (5 pmol/μl), 0.5 μl PIG-tailed reverse primer (5ʹ-GTTTCTT-3ʹ) (5 pmol/μl) (Brownstein et al. 1996; Schuelke 2000; Kubisiak et al. 2009), 1 μl M13 (6-FAM/HEX) primer (5 pmol/μl), 5 µl H2O (5.7 µl H2O for multiplex 6) and 1 µl DNA (ca. 0.6 ng/μl).
All PCR reactions were performed in a Biometra Thermal Cycler (MJ Research PTC 200, Analytik Jena, Germany) with a touchdown program. The PCR protocol for each marker was as follows: 15 min initial denaturation at 95 °C followed by 10 touchdown cycles at 94 °C for 1 min, 1 min at 60 °C (decreasing 1 °C each cycle) and 1 min at 72 °C, followed by 25 cycles at 94 °C for 1 min, annealing at 50 °C for 1 min and elongation at 72 °C for 1 min, and a final extension step at 72 °C for 20 min. The PCR amplification was tested on 1.5% agarose gels in 1 × TAE buffer. Amplification products were resolved on an ABI 3130xl Genetic Analyzer (Applied Biosystems, Foster City, USA) using the GeneScan™ Rox-500 and Liz-500 (only for multiplex 3) size markers. For the fragment length analysis, multiplexes 5 and 6 were run together. Scoring of alleles was conducted using GeneMapper® version 4.1 (Applied Biosystems, Foster City, USA).
Statistical data analysis
Genetic variation in populations was calculated as the number of alleles per locus (Na), observed heterozygosity (Ho) and expected heterozygosity (He) in GenAlEx version 6.51b2 (Peakall and Smouse 2006, 2012; Smouse et al. 2017). Inbreeding coefficients (FIS) and their significance were determined using the Fstat version 2.9.4 software (Goudet 2003). Significant deviations from zero were determined after Bonferroni correction (α = 0.05, p < 0.00007) implemented in the software Fstat (Goudet 2003) to compensate for type I errors. Allelic richness (Ar) was also calculated using Fstat. In addition, linkage disequilibrium (LD) was calculated for each pair of loci in the 37 populations using Genepop version 4.7.2 (Rousset 2008) based on the following settings: dememorization 10,000, batches 100 and iterations per batch 5000.
The software BOTTLENECK version 1.2.02 (Piry et al. 1999) was used to detect signatures of recent genetic bottlenecks. Therefore, we performed a Wilcoxon signed-rank test (one-tailed) for heterozygosity excess for ‘the infinite alleles model’ (IAM) and ‘the stepwise mutation model’ (SMM) and a mode-shift analysis to test for a distortion in the allele frequency distribution.
To measure the genetic variation among populations, an analysis of molecular variance (AMOVA) was performed with GenAlEx using 9999 permutations. The genetic differentiation among populations was also calculated as the fixation index FST, GST and Hedrick’s standardized GST (G’ST(Hed)) for individual markers and across all markers in GenAlEx. Besides, a principal coordinate analysis (PCoA) was performed in GenAlEx based on the genetic distance implemented in GenAlEx between populations in order to find and plot the major patterns within this dataset (Peakall and Smouse 2006, 2012).
The Windows®-based software MicroChecker version 2.2.3 (van Oosterhout et al. 2004) was used to identify genotyping errors due to non-amplified alleles (null alleles) which can lead to overestimates of the inbreeding coefficient (FIS). Arlequin version 3.5.2.2 (Excoffier and Lischer 2010) was run with 50,000 simulations of 100 demes per group with the infinite island model based on FST in order to detect outliers which deviate significantly from the variation and differentiation expected under neutrality.
The calculation of population structure was performed using STRUCTURE version 2.3.4 (Pritchard et al. 2000) to identify possible subpopulations based on the microsatellite dataset. Here, we tested 2–40 possible populations with ten runs per each K. The admixture model and correlated allele frequencies were selected initially, where a burn-in period of 50,000, Markov Chain–Monte Carlo (MCMC) repetitions of 100,000 and the LOCPRIOR model were used. However, we used additionally the default setting in STRUCTURE (admixed model without LOCPRIOR) to identify population structure solely based on genetic information. We also used the "no admixture" model in STRUCTURE to test if there is a clearer differentiation between the native and the Slavonian populations. The online program STRUCTURE HARVESTER v. 0.6.94 (Earl and von Holdt 2012) was used to determine the 'Best K' from the logarithmic results and the ΔK method (Evanno et al. 2005). In addition, the CLUMPAK software (Cluster Markov Packager Across K) was used to post-process the results of the model-based population structure analysis (Kopelman et al. 2015).
A small proportion of indigenous haplotypes are also found in populations characterized as Slavonian oaks and vice versa. Therefore, additional STRUCTURE and principal component analysis were performed, for which individuals with the indigenous haplotypes (HP1, 4 and 10) in Slavonian populations (such as Tomberg 10B2, Kanitz 77C, Estermann 116A1) and individuals with the Slavonian haplotypes (HP5 and 7–26) in indigenous oak populations (Gut Ulenburg 4C, Kottenforst 134A&C) were removed (see Table S.1).
The software GeneClass2 was used to assign three populations (Tomberg 10B2, Freiherr v. der Leyen 17C and Kanitz 32H/39A) with unknown origin (see Table 1) to reference populations (here: Q. robur and Q. robur subsp. slavonica stands) based on multilocus data (Piry et al. 2004). These population assignments are modelled on Nei’s standard distance (Nei 1972), Goldstein’s distance (Goldstein et al. 1995) and using the Bayesian method (Baudouin and Lebrun 2001) taking the allele size of the SSRs into account (Piry et al. 2004). GeneClass was also used to perform self-assignment simulations among the individuals using the leave-one-out procedure.
Results
Genetic variation within populations
Inbreeding coefficients across all markers were not significantly different from zero in any population. The mean expected heterozygosity ranged from 0.469 in Graf Merveldt 3A to 0.566 in Plettenberg 106G, and the mean observed heterozygosity ranged from 0.43 in Blix Flur 2/155 to 0.552 in Kottenforst 70D (Kottenforst 154B is not representative due to the low sample size of 4 samples). A summary of the genetic parameters across all loci is given in Table S. 3. The observed and expected heterozygosity and inbreeding coefficient were lower (not significant) in the group of indigenous oaks (mean Ho: 0.484, He:0.526, FIS: 0.082) compared to Slavonian oaks (mean Ho: 0.490, He:0.540, FIS: 0.093).
The mean expected heterozygosity per locus ranged from 0.117 at locus 3A05 to 0.863 at locus VIT107, and the mean observed heterozygosity per locus ranged from 0.109 at locus 2A05 to 0.837 at locus GOT040 (Table S. 7).
The genetic differentiation (FST) between the 37 populations was relatively low at most loci ranging from 0.025 for VIT023 to 0.062 for PIE125. Overall, the mean differentiation among all populations was 0.041 (GST = 0.011, G’ST(Hed) = 0.027) (Online-Resource 1). The mean pairwise FST between indigenous oaks is 0.014 (GST = 0.003, G’ST(Hed) = 0.008), between Slavonian oaks 0.018 (GST = 0.003, G’ST(Hed) = 0.010) and between Slavonian and indigenous oaks 0.023 (GST = 0.010, G’ST(Hed) = 0.034). Despite the low mean pairwise FST value (0.023) between Slavonian and local oaks, these two groups were distinguished in the PCoA (Fig. 3).

Principal coordinate analysis (PCoA) based on nSSRs for all populations. HP = haplotype. Symbols square = phenologically characterized as Slavonian oak, circle = common oak, triangle = not clearly defined. The different colours stand for the most common haplotypes (Petit et al. 2002): red = HP 1, yellow = HP 10, orange = HP 12, blue = HP7-26, purple = HP5, pink = HP 2. The colour light purple means that both haplotype 2 and haplotype 5 (Schulze-Becking 54B1/B2) occur. The same applies to the colour turquoise, where both haplotype 5 and haplotype 17 (Kottenforst 70D) occur. The colour light orange stands for the occurrence of haplotypes 1, 4 and 10 (Kottenforst 134A&C, Kottenforst 85B). (Color figure online)
Genetic variation among populations
The outlier tests with Arlequin showed that there are no markers with signatures of selection among populations and between groups (Slavonian vs. common oak) (Fig. S. 3).
There were no loci in LD in any population. Null alleles were detected in 31 populations at least at one of the markers 3D15, FIR028 (highest null allele frequency = 0.3015 in Kanitz 19A), FIR035, PIE125, VIT023, VIT107, FIR104, PIE102, PIE267, Qr0057, Qr0332 (highest null allele frequency = 0.3079 in Kanitz 77), Qr1423 and FS_C8183 (Table S. 2). However, in all populations except Graf Merveldt 3A, Kottenforst 85B, Gut Ulenburg 4C, Königsforst 127c, Kottenforst 70D and Kottenforst 154B null alleles only occurred with a frequency between 0.51% and 5.51% over all loci (Table S. 2). Null alleles across all populations per locus ranged from 0.005 at loci FS_C1423 and FS_C8183 to 0.064 at locus FIR035 (Table S. 7). Furthermore, comparison of null alleles between marker types showed that only one of the three gSSRs (3D15) showed null alleles with a low frequency (0.045), the null allele frequencies of EST-SSRs developed for Q. robur ranged from 0.006 at locus VIT023 to 0.126 at locus FIR028 and the null allele frequencies of EST SSRs developed for Q. rubra ranged from 0.005 at loci FS_C8183 and FS_C1423 to 0.028 at locus Qr0057 (Table S. 7). Across all loci, EST-SSRs developed for Q. robur have the highest null allele frequency (0.029), followed by gSSRs with 0.015 and EST-SSRs developed for Q. rubra (0.007).
The results of the program BOTTLENECK show that the allele frequencies of all populations except Kottenforst 154B followed a normal L-shaped distribution (Table S. 8). The AMOVA showed that 99% of the molecular variance was within populations (9% between individuals and 90% within individuals) and only 1% among populations.
Structure analysis of populations
Using the Evanno method and STRUCTURE HARVESTER an optimal K (ΔK) = 2 was determined (Fig. S. 1, Table S. 2, Table S. 4). The diagram calculated by STRUCTURE using the admixed model and LOCPRIOR with the population assumption of K = 2 is presented in Fig. 5. The results of the STRUCTURE analysis for K = 3 and K = 4 are presented in the supplementary material (Fig. S. 7, 8). In addition, Fig. 2 and Table S. 1 show the proportion of membership of each sampled population in the 2 clusters, but there is no specific cluster for each origin, since both clusters occur in both origins. A proportion of ancestry > 0.5 or < 0.5 in cluster 1 is characteristic for indigenous oak and Slavonian stands, respectively. The results of the PCoA (Fig. 3) show that the stands of common oaks and Slavonian oaks are represented by two clusters which are weakly differentiated. In addition, Fig. 4 shows a clearer distinction between Slavonian and local populations, as individuals with local haplotypes in Slavonian populations and individuals with Slavonian haplotypes in local populations (Table S. 1) were excluded (see chapter ‘plant material’). However, after the exclusion of these individuals, the results in STRUCTURE were similar (Fig. S. 4). The results using the default setting in STRUCTURE without LOCPRIOR also show genetic differentiation of native and Slavonian populations, but less pronounced (Fig. S. 9, 10, 11). Besides, the results of the STRUCTURE “no admixture” model (K = 2, 3 and 4) provided an even clearer distinction between native and Slavonian populations (Fig. S. 12, 13, 14). A further analysis in STRUCTURE shows that the Slavonian and native haplotypes are differentiated at nSSRs (Fig. S. 5).

Principal coordinate analysis (PCoA) based on nSSRs for all populations without Slavonian haplotypes in common oaks and common haplotypes in Slavonian oaks. HP = haplotype. Squares = phenologically characterized as Slavonian oak, circles = common oak, triangles = not clearly defined. The different colours stand for the most common haplotypes (Petit et al. 2002): red = HP 1, yellow = HP 10, orange = HP 12, blue = HP7-26, purple = HP5, pink = HP 2. The colour light purple means that both haplotype 2 and haplotype 5 (Schulze-Becking 54B1/B2) occur. The same applies to the colour turquoise, where both haplotype 5 and haplotype 17 (Kottenforst 70D) occur. The colour light orange stands for the occurrence of haplotypes 1, 4 and 10 (Kottenforst 134A&C, Kottenforst 85B). (Color figure online)
The GeneClass analysis provided similar results as STRUCTURE and PCoA (see Table S. 1 and Table S. 5). According to the PCoA all three populations (Tomberg 10B2, Freiherr v. der Leyen 17C and Kanitz 32H/39A) cluster with Slavonian oak stands. However, using STRUCTURE Tomberg 10B2 and Freiherr v. der Leyen 17C show an assignment of 50% per cluster, only Kanitz 32H/39A belongs to Cluster 2 with more than 60% which is characteristic for Slavonian stands. Based on Nei’s distance and the Bayesian approach, GeneClass assigns all three populations to Slavonian oak, whereas based on Goldstein’s distance Tomberg is assigned to indigenous oak (see Table S. 5). The self-assignment test of GeneClass based on the Bayesian method showed that 83.5% of the individuals were assigned to the correct group (Slavonian or native oak) (Online-Resource 2).
Discussion
The stands of oaks known as late flushing oaks in North Rhine-Westphalia are well studied in terms of chloroplast DNA markers (Gailing et al. 2007a, b, 2009). To our knowledge, however, this is the first study on genetic differentiation between indigenous and Slavonian oak stands using nuclear microsatellites. The high transferability of EST-SSRs among species in Quercus and the availability of a large number of nSSRs and EST-SSRs (Steinkellner et al. 1997; Barreneche et al. 2004), made it possible to study 13 indigenous, 21 oak stands phenotypically described as Slavonian oak and 3 oak stands, for which it was not certain whether they are Slavonian oaks, based on a set of 20 nuclear microsatellite markers (Table S. 7 and Table 2).
Amount of genetic variation
Our dataset exhibits high levels of genetic diversity at the SSR loci examined (see Table S. 3) for all populations including the introduced Slavonian stands (e.g. He ranged from 0.450 to 0.566). This relatively high level of genetic variation, as measured by basic statistics, was very similar across populations (see Table S.3) and is common for woody species (Hamrick et al. 1992). In addition, similar diversity values of the introduced plantations as compared to native stands, suggest that Slavonian stands were established with seed material that was sampled in Croatia from a representative number of trees per population.
The relatively high heterozygosity and allelic diversity allow for a good resolution of the underlying population genetic structures. Null alleles occurred with a frequency between 0.51% and 5.51% across all loci (Table S. 2). According to Oddou-Muratorio et al. (2009), however, null allele frequencies between 5 and 8% on average across loci are not expected to have effects on population genetic analyses. The markers selected for this study are also located on different linkage groups (see Table 2) and dispersed rather regularly across the genome (Durand et al. 2010). Also, the absence of linkage disequilibrium between marker pairs suggests that we have adapted a representative marker set.
Furthermore, the 10 EST-SSR markers developed for Q. robur (Durand et al. 2010) (see Table S. 3) revealed similar levels of genetic variation as in Bodénès et al. (2012) (Ar = 4.20, Ho = 0.51, He = 0.74 and Ar = 2.96, Ho = 0.41, He = 0.53). However, higher Na, He and Ho values on average were revealed by Crăciunesc et al. (2011) (Na = 13.43, Ho = 0.723 and He = 0.769), Streiff et al (1998) (Ho = 0.81 and He = 0.87) and Neophytou et al. (2010) (Ho = 0.741 and He = 0.814) at potentially neutral gSSRs which are generally more variable than EST-SSRs (Ellis and Burke 2007; Buonaccorsi et al. 2012; Harmon et al. 2017). The total variation of all oaks in the present study (Na = 10.55, Ar = 10.54, Ho = 0.488, He = 0.539) corresponds to the total variation of the Slavonian oaks (Na = 9.85, Ar = 8.689, Ho = 0.490, He = 0.540) (see Table S.3). In addition, both the mean within stand variation and total variation are similar in Slavonian and indigenous stands (see Table S.3). Since neither the expected heterozygosity nor the number of alleles per locus and allelic richness, both for variation within stands and total variation, is significantly lower in the Slavonian populations (Table S. 3), there is no evidence of losses in allelic richness and thus no evidence of a bottleneck effect (Nei et al. 1975). In addition, no evidence of a recent genetic bottleneck was identified in the microsatellite data using the infinite alleles and stepwise mutation models (Table S. 8). The fact that Kottenforst 154B shows a shifted mode in the allele frequency distribution is probably due to the low sample size of only 4 individuals. Therefore, there is no indication that the seed transfer of Slavonian oaks to Germany through humans reduced the variation within stands. Furthermore, the mean variation across all populations is almost equal to the total variation (see Table S. 3) and also the pairwise FST values between Slavonian and indigenous oaks are low (0.023) (see Table S. 7), providing no evidence for fixation or genetic drift. A hierarchical division of the total genetic diversity for the two taxa also reveals that most of the variation is within populations (99%), of which a comparatively high amount is within individuals (90%). Differentiation among populations is low (1%). However, pedunculate oaks almost always show low genetic differentiation between populations and high variation within populations (Zanetto et al. 1994; Gömöry et al. 2001; Mariette et al. 2002; Neophytou 2015).
Differentiation of stands
The differentiation between Slavonian and indigenous pedunculate oaks with nuclear microsatellite markers worked well at the population level, but less well at the individual level. This can be seen, on the one hand, in the PCoA based on populations that classifies the populations into two groups (Fig. 4). Only the two populations Kottenforst 154B and Vinkovsi are slightly separated within the Slavonian group. For population Kottenforst 154B, this is probably due to the small sample size of only 4 individuals. For population Vinkovsi, the larger differences could be due to the fact that seeds were sampled that came directly from Vinkovsi in Croatia (Gailing et al. 2007b) and not from old growth first-generation forest stands (Gailing et al. 2007a, b, 2009).
On the other hand, the differentiation between taxa is also shown in STRUCTURE results based on individuals (Pritchard et al. 2000; Evanno et al. 2005; Kalinowski 2011), but the differentiation is not as pronounced as in the PCoA (see Fig. 5 and Fig. S. 4). It shows that the assignment of individuals to clusters as in STRUCTURE is not suitable for genetically slightly differentiated taxa. In addition to these two methods (PCoA and STRUCTURE), there are a few more methods for population structure analysis such as LAMP (Sankararaman et al. 2008; Paşaniuc et al. 2009; Baran et al. 2012), ADMIXTURE (Alexander et al. 2009; Alexander and Lange 2011; Zhou et al. 2011) and FRAPPE (Tang et al. 2005; Alexander et al. 2009). However, these programs also have some disadvantages like no explicit account for LD between markers (ADMIXTURE), applicability only for SNP data (LAMP) or the estimation can be slightly inaccurate (FRAPPE); hence, STRUCTURE and PCoA were chosen as the most common methods for the analysis (Porras-Hurtado et al. 2013).

STRUCTURE diagram with admixed model and LOCPRIOR for K = 2. Blue = Slavonian oak, orange = common oak. The red line divides the indigenous oaks (left) from the Slavonian oaks (right). (Color figure online)
In addition to these nuclear marker results, the inclusion of cpDNA haplotypes from Gailing et al. (2007a, b, 2009) shows that Slavonian oak stands are not always pure stands (see Table S.1). After the exclusion of indigenous haplotypes in Slavonian populations, the separation between the two groups detected by the PCoA was more pronounced (Fig. 4). A possible reason for the occurrence of indigenous haplotypes in stands established with Slavonian seeds is that indigenous seeds may have been used for replanting in these stands. On the other hand, an additional natural regeneration of the common oak by the Eurasian jay (Garrulus glandarius) cannot be excluded. According to Hafer and Bauer (1993), this bird can transport acorns over 5–8 km distances. For example, in nearby stands such as Kanitz 77C (90% HP5 and 10% HP1) and Kanitz 76A (100% HP1) (Fig. 1: population number 10 and 26; Fig. 2) transport of seeds could be a reason for the occurrence of haplotype 1 in the Slavonian population Kanitz 77C.
Furthermore, there were three populations, Kanitz 32H/39A, Tomberg 10B2 and Freiherr v. d. Leyen 17C (Table 1, Table S. 1), which show predominantly haplotype 5; however, the Slavonian origin was unclear. Due to the phylogeographic variation patterns of the cpDNA haplotypes, cpDNA markers are particularly useful to infer the origin of oak trees (Petit et al. 2002; Gailing et al. 2003; Finkeldey et al. 2010; Finkeldey and Hattemer 2010). However, the exact geographic origin of populations with haplotype 5 is unclear, as it occurs naturally in Germany and Croatia. In addition to haplotype 5, Tomberg 10B2 and Freiherr v. d. Leyen 17C reveal a relatively high frequency (15% and 20%) of non-Slavonian haplotypes 1 and 10 (Table S. 1) suggesting a mixture of reproductive material or later replanting.
GeneClass was used to assign these three populations to one of the two origins indigenous or Slavonian oak (Piry et al. 2004). The population Kanitz 32H/39A can be assigned to a population of Slavonian origin, since all three methods (Nei’s distance with 74%, Goldstein’s distance with 69% and the Bayesian method with 100%) assign this population to Slavonian oak. In addition, Freiherr v. d. Leyen 17 C can also be assigned to Slavonian oak, according to Goldstein’s distance with 67% and the Bayesian method with 99.9%, but regarding Nei’s distance only with 52% (see Table S. 5). The assignment of these two populations also corresponds to the STRUCTURE results (Freiherr v. d. Leyen 17C with 51% and Kanitz 32H/39A with 64% and those of the PCoA (see Table S.1)). Population Tomberg 10B2, however, cannot be clearly assigned, because according to STRUCTURE it reveals 50% ancestry in each cluster; according to PCoA, it is rather assigned to Slavonian oak (see Fig. 3, 4), according to GeneClass based on Nei’s distance (55%) and based on the Bayesian method (100%) to Slavonian oak and based on Goldstein’s distance (51%) again to indigenous oak (see Table S. 1 and Table S.5). According to the results of the cpDNA haplotypes, the stands Tomberg 10B2 and Freiherr v. der Leyen 17C show a low amount of indigenous haplotypes (HP1, HP10) compared to Kanitz 32H/39A consisting only of Slavonian haplotypes (see Table S.1). The occurrence of indigenous haplotypes in both of these stands might lead to the slightly lower population assignment to Slavonian oak (see Table S.5). After exclusion of the indigenous haplotypes HP1 and HP10 in Tomberg 10B2 and Freiherr v. der Leyen 17C GeneClass assigns the two stands as Slavonian oaks based on Nei’s standard distance (Tomberg 10B2: 59%, Leyen 17C: 57%), Goldstein’s distance (Tomberg 10B2: 58%, Leyen 17C: 78%) and also based on Bayesian analysis (Tomberg 10B2: 100%, Leyen 17C: 100%) (see Table S.6).
In conclusion, nuclear markers can be used to validate the geographic origin of populations with haplotypes, which occur both in Germany and in Croatia. The usefulness of nuclear markers to differentiate between individuals and populations of German and Slavonian origin was validated for individual trees and stands with haplotype 2, which does not occur naturally in Germany. A combination of the results of both marker types (cpDNA marker and nuclear SSRs) is necessary to differentiate between taxa that differ only slightly from each other and to identify admixture.
Practical relevance
In the course of climate change and the associated forest restructuring towards stable mixed stands, the Slavonian oak is becoming increasingly important. On the one hand, forest owners are showing interest in planting Slavonian oaks because of their high growth rate, very good quality characteristics, late bud burst and possibly better adaptation to climate change (Schirmer 2017). On the other hand, the Slavonian oak is also of great interest to many tree nurseries, as suitable oak seed is needed due to the decline in oak seedling harvest years and the desired forest restructuring (Schirmer 2017). For practical purposes, seed material with known geographic origin is needed for forest management geared towards the sustainable use of genetic resources ensuring the long-term adaptability of tree species (Geburek and Schüler 2012). Accordingly, the forest owner needs certainty about the origin of the planting material through certification of the origin of reproductive material. For this purpose, the marker set used in this study is suitable to identify mixed stands, to certify the origin of the reproductive material and to differentiate between Slavonian and native oak stands. Wypukol et al. (2008) also successfully tested beech (Fagus sylvatica) for varietal purity and identity using nuclear markers. Neophytou (2012), for instance, used nuclear microsatellites to differentiate between oak species Q. robur and Q. petraea.
Summary and outlook
The nuclear marker set tested in this study is useful for further studies to identify Slavonian stands which are already established in Germany (e.g. stands of late flushing oak Burg-Eltz in Rhineland-Palatinate) in order to investigate whether the Slavonian oak is an alternative variety with regard to climate change. The low differentiation at genetic markers but phenotypic and phenological differences suggest different local adaptations of both taxa. Both taxa are likely differentiated at a few genomic regions which are associated with these differences. Outlier screens and QTL mapping will be used in future studies to identify these regions. Furthermore, the low genetic differences between the two oak taxa suggest that the Slavonian oak is not a separate subspecies of the common oak, but rather a variety adapted to the local microclimatic conditions in the region of origin. In general, sufficient reference samples from all Slavonian oaks in Croatia and Germany would be important to better delineate the origin of individual trees and stands. In addition, the natural rejuvenation of nearby indigenous and Slavonian oaks must be analysed in order to estimate the amount of gene flow between the two taxa.
References
Alexander DH, Lange K (2011) Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinformatics 12:246. https://doi.org/10.1186/1471-2105-12-246
Alexander DH, Novembre J, Lange K (2009) Fast model-based estimation of ancestry in unrelated individuals. Genome Res 19(9):1655–1664. https://doi.org/10.1101/gr.094052.109
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410. https://doi.org/10.1016/S0022-2836(05)80360-2
Baran Y, Pasaniuc B, Sankararaman S, Torgerson DG, Gignoux C, Eng C, Rodriguez-Cintron W, Chapela R, Ford JG, Avila PC, Rodriguez-Santana J, Burchard EG, Halperin E (2012) Fast and accurate inference of local ancestry in Latino populations. Bioinformatics 28(10):1359–1367. https://doi.org/10.1093/bioinformatics/bts144
Barreneche T, Casasoli M, Russell K, Akkak A, Meddour H, Plomion C, Villani F, Kremer A (2004) Comparative mapping between quercus and castanea using simple-sequence repeats (SSRs). TAG Theor Appl Genet 108(3):558–566. https://doi.org/10.1007/s00122-003-1462-2
Baudouin L, Lebrun P (2001) An operational Bayesian approach for the identification of sexually reproduced cross-fertilized populations using molecular markers. Proc Int Symp on molecular markers Eds. Doré, Dosba and Baril Acta Hort. 546: 81–94.
Bodénès C, Chancerel E, Gailing O, Vendramin GG, Bagnoli F, Durand J, Goicoechea PG, Soliani C, Villani F, Mattioni C, Koelewijn HP, Murat F, Salse J, Roussel G, Boury C, Alberto F, Kremer A, Plomion C (2012) Comparative mapping in the Fagaceae and beyond with EST-SSRs. BMC Plant Biol 12:153. https://doi.org/10.1186/1471-2229-12-153
Bordács S, Flaviu P, Slade D, Csaikl U, Lesur I, Borovics A, Kézdy P, König A, Gömöry D, Brewer S, Burg K, Petit R (2002) Chloroplast DNA variation of white oaks in the Northern Balkans and in the Carpathian Basin. For Ecol Manage 156:197–209. https://doi.org/10.1016/S0378-1127(01)00643-0
Brownstein MJ, Carpten JD, Smith JR (1996) Modulation of non-templated nucleotide addition by Taq DNA polymerase: primer modifications that facilitate genotyping. Biotechniques 20(6):1004–1010. https://doi.org/10.2144/96206st01
Buonaccorsi VP, Kimbrell CA, Lynn EA, Hyde JR (2012) Comparative population genetic analysis of bocaccio rockfish Sebastes paucispinis using anonymous and gene-associated simple sequence repeat loci. J Hered 103(3):391–399. https://doi.org/10.1093/jhered/ess002
Burger K, Müller M, Gailing O (2018) Characterization of EST-SSRs for European beech (Fagus sylvatica L.) and their transferability to Fagus orientalis Lipsky, Castanea dentata Bork, and Quercus rubra L. Silvae Genet 67(1):127–132. https://doi.org/10.2478/sg-2018-0019
Chakraborty R (1993) Analysis of genetic structure of populations: meaning, methods, and implications. In: Majumder PP (ed) Human population genetics. Springer, Boston, MA, pp 189–206
Crăciunesc I, Ciocîrlan E, Şofletea N, Curtu AL (2011) Genetic diversity of pedunculate oak (Quercus robur L.) in prejmer natural reserve. Bulletin of the Transilvania University of Braşov, 4 (53) No 1: 15–20
Durand JC, Bodénès C, Chancerel E, Frigerio JM, Vendramin G, Sebastiani F, Buonamici A, Gailing O, Koelewijn HP, Villani F, Mattioni C, Cherubini M, Goicoechea PG, Herrán A, Ikaran Z, Cabané C, Ueno S, Alberto F, Dumoulin PY, Guichoux E, de Daruvar A, Kremer A, Plomion C (2010) A fast and cost-effective approach to develop and map EST-SSR markers: oak as a case study. BMC Genomics 11(1):570. https://doi.org/10.1186/1471-2229-12-153
Dzialuk A, Chybicki I, Burczyk J (2005) PCR multiplexing of nuclear microsatellite loci in Quercus species. Plant Mol Biol Report 23(2):121–128. https://doi.org/10.1007/BF02772702
Earl DA, von Holdt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4(2):359–361. https://doi.org/10.1007/s12686-011-9548-7
Ellis JR, Burke JM (2007) EST-SSRs as a resource for population genetic analyses. Heredity 99(2):125–132. https://doi.org/10.1038/sj.hdy.6801001
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14(8):2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10(3):564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131(2):479–491
Finkeldey R, Hattemer HH (2010) Genetische variation in Wäldern–wo stehen wir? Genetic variation in forests–state of the art. Forstarchiv 81(4):123–129
Finkeldey R, Leinemann L, Gailing O (2010) Molecular genetic tools to infer the origin of forest plants and wood. Appl Microbiol Biotechnol 85(5):1251–1258. https://doi.org/10.1007/s00253-009-2328-6
Gailing O, Wachter H, Leinemann L, Hosius B, Finkeldey R, Schmitt HP, Heyder J (2003) Characterisation of different provenances of late flushing pedunculate oak (Quercus robur L.) with chloroplast markers. Allg Forst u J Ztg 174(12):227–231
Gailing O, Wachter H, Heyder J, Schmitt HP, Finkeldey R (2007a) Chloroplast DNA analysis in oak stand (Quercus robur L.) in North Rhine-Westphalia with presumably Slavonian origin: Is there an association between geographic origin and bud phenology? Appl Bot Food Qual 81:165–171
Gailing O, Wachter H, Schmitt HP, Curtu AL, Finkeldey R (2007b) Characterization of different provenances of Slavonian pedunculate oaks (Quercus robur L.) in Münsterland (Germany) with chloroplast DNA markers: PCR-RFLPs and chloroplast microsatellites. Allg Forst u J Ztg 178:85–90
Gailing O, Wachter H, Heyder J, Rogge M, Finkeldey R (2009) Chloroplast DNA analyses of very old, presumably autochthonous Quercus robur L. stands in North-Rhine Westphalia. Allg Forst u J Ztg 180:221–227
Geburek T, Schüler S (2012) Nachhaltigkeit - ohne Genetik geht es nicht! BFW-Praxisinformation 27:14–16
Goldstein DB, Ruiz Linares A, Cavalli-Sforza LL, Feldman MW (1995) Genetic absolute dating on microsatellites and the origin of modern humans. Proc Natl Acad Sci USA 92:6723–6727. https://doi.org/10.1073/pnas.92.15.6723
Gömöry D, Yakovlev I, Zhelev P, Jedináková J, Paule L (2001) Genetic differentiation of oak populations within the Quercus robur/Quercus petraea complex in Central and Eastern Europe. Heredity 56:190–199. https://doi.org/10.1046/j.1365-2540.2001.00874.x
Goudet J (2003) FSTAT (version 2.9.4), a program (for Windows 95 and above) to estimate and test population genetics parameters. Department of Ecology & Evolution, Lausanne University, Switzerland, pp 1–54
Greenbaum G, Templeton AR, Bar-David S (2016) Inference and analysis of population structure using genetic data and network theory. Genetics 202(4):1299–1312. https://doi.org/10.1534/genetics.115.182626
Hafer J, Bauer M (1993) Corvidae. In: Glutz von Blotzheim UN (ed) Handbuch der Vögel Mitteleuropas. Band 13/III: Passeriformers (Teil 4), Wiesbaden, pp 1375–2022
Hamrick JL, Godt MJW, Sherman-Broyles SL (1992) Factors influencing levels of genetic diversity in woody plant species. New Forest 6(1–4):95–124. https://doi.org/10.1007/978-94-011-2815-5_7
Harmon M, Lane T, Staton M, Coggeshall MV, Best T, Chien CC, Liang H, Zembower N, Drautz-Moses DI, Hwee YZ, Schuster SC, Schlarbaum SE, Carlson JE, Gailing O (2017) Development of novel genic microsatellite markers from transcriptome sequencing in sugar maple (Acer saccharum Marsh.). BMC Res Notes 10(369):1–7. https://doi.org/10.1186/s13104-017-2653-2
Kalinowski ST (2011) The computer program STRUCTURE does not reliably identify the main genetic clusters within species: simulations and implications for human population structure. Heredity 106(4):625–632. https://doi.org/10.1038/hdy.2010.95
Kopelman MM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15(5):1179–1191. https://doi.org/10.1111/1755-0998.12387
Kubisiak T, Carey D, Burdine C, Koch J (2009) Characterization of ten EST-based polymorphic SSR loci isolated from American beech, Fagus grandifolia Ehrh. Permanent Genetic Resources Note Added to Mol Ecol Resour. https://doi.org/10.1111/j.1755-0998.2009.02759.x
Lind JF, Gailing O (2013) Genetic structure of Quercus rubra L. and Quercus ellipsoidalis E. J. Hill populations at gene-based EST-SSR and nuclear SSR markers. Tree Genet Genomes 9(3):707–722. https://doi.org/10.1007/s11295-012-0586-4
Mariette S, Le Corre V, Austerlitz F, Kremer A (2002) Sampling within the genome for measuring within-population diversity: trade-offs between markers. Mol Ecol 11(7):1145–1156. https://doi.org/10.1046/j.1365-294X.2002.01519.x
Müller M, Gailing O (2018) Characterization of 20 new EST-SSR markers for northern red oak (Quercus rubra L.) and their transferability to Fagus sylvatica L. and six oak species of section Lobatae and Quercus. Ann For Res 61(2):211–222. https://doi.org/10.15287/afr.2018.1191
Nei M (1972) Genetic distance between populations. Amer Nat 106(949):283–329. https://doi.org/10.1086/282771
Nei M, Maruyama T, Chackraborty R (1975) The bottleneck effect and genetic variability in populations. Evolution 29(1):1–10. https://doi.org/10.1111/j.1558-5646.1975.tb00807.x
Neophytou C (2012) Genetische Differenzierung innerhalb von und zwischen Beständen der Stiel-und der Traubeneiche im Oberrheingraben: Erste Ergebnisse eines Interreg-Projekts. Forstarchiv 83(1):34–40
Neophytou C (2015) Wirkt sich menschliche Bewirtschaftung auf das Erbgut der Eichen am Oberrhein aus? Does human management affect genetic variation of the oaks in the Upper Rhine plain? Forstarchiv 86:183–193. https://doi.org/10.4432/0300-4112-86-183
Neophytou C, Aravanpopoulus FA, Fink S, Dounavia A (2010) Detecting interspecific and geographic differentiation patterns in two interfertile oak species (Quercus petraea (Matt.) Liebl. and Q. robur L.) using small sets of microsatellite markers. For Ecol Manage 259(10):2026–2035. https://doi.org/10.1016/j.foreco.2010.02.013
Oddou-Muratorio S, Vendramin GG, Buiteveld J, Fady B (2009) Population estimators or progeny tests: what is the best method to assess null allele frequencies at SSR loci? Conserv Genet 10(5):1343–1347. https://doi.org/10.1007/s10592-008-9648-4
Paşaniuc B, Sankararaman S, Kimmel G, Halperin E (2009) Inference of locus-specific ancestry in closely related populations. Bioinformatics (oxford, England) 25(12):i213–i221. https://doi.org/10.1093/bioinformatics/btp197
Peakall ROD, Smouse PE (2006) GenAlEx 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6(1):288–295. https://doi.org/10.1111/j.1471-8286.2005.01155.x
Peakall ROD, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28(19):2537–2539
Petit RJ, Brewer S, Bordács S, Burg K, Cheddadi R, Coart E, Cottrell J, Csaikl UM, van Dam B, Deans JD, Espinel S, Fineschi S, Finkeldey R, Glaz I, Goicoechea PG, Svejgaard Jensen JS, König AO, Lowe AJ, Flemming Madsen S, Mátyás G, Munro RC, Popescu F, Slade D, Tabbener H, de Vries SGM, Ziegenhagen B, de Beaulieu JL, Kremer A (2002) Identification of refugia and post-glacial colonisation routes of European white oaks based on chloroplast DNA and fossil pollen evidence. For Ecol Manage 1526(1–3):49–74. https://doi.org/10.1016/S0378-1127(01)00634-X
Piry S, Luikart G, Cornuet JM (1999) BOTTLENECK: A computerprogram for detecting recent reductions in the effective popu-lation size using allele frequency data. J Hered 90(4):502–503. https://doi.org/10.1093/jhered/90.4.502
Piry S, Alapetite A, Cornuet JM, Paetkau D, Baudouin L, Estoup A (2004) GeneClass2: a software for genetic assignment and first-generation migrant detection. J Hered 95:536–539. https://doi.org/10.1093/jhered/esh074
Porras-Hurtado L, Ruiz Y, Santos C, Phillips C, Carracedo A, Lareu MV (2013) An overview of STRUCTURE: applications, parameter settings, and supporting software. Front Genet 4:98. https://doi.org/10.3389/fgene.2013.00098
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155(2):945–959
Rousset F (2008) Genepop’007: a complete re-implementation of the genepop software for Windows and Linux. Mol Ecol Resour 8(1):103–106. https://doi.org/10.1111/j.1471-8286.2007.01931.x
Sankararaman S, Sridhar S, Kimmel G, Halperin E (2008) Estimating local ancestry in admixed populations. Am J Hum Geneti 82(2):290–303. https://doi.org/10.1016/j.ajhg.2007.09.022
Schirmer R (2017) Amt für forstliche Saat- und Pflanzenzucht. Slawonische Eiche im Fokus. LWF aktuell 3:36
Schuelke M (2000) An economic method for the fluorescent labeling of PCR fragments. Nat Biotechnol 18(2):233–234. https://doi.org/10.1038/72708
Smouse PE, Banks C, Peakall R (2017) Converting quadratic entropy to diversity: both animals and alleles are diverse, but some are more diverse than others. PLoS ONE 12(10):e0185499. https://doi.org/10.1371/journal.pone.0185499
Steinkellner H, Fluch S, Turetschek E, Lexer C, Streiff R, Kremer A, Burg K, Gössl J (1997) Identification and characterization of (GA/CT)n-microsatellite loci from Quercus petraea. Plant Mol Biol 33:1093–1096. https://doi.org/10.1023/a:1005736722794
Streiff R, Labbe T, Bacilieri R, Steinkellner H, Glossl J, Kremer A (1998) Within-population genetic structure in Quercus robur L. and Quercus petraea (Matt.) Liebl. assessed with isozymes and microsatellites. Mol Ecol 7(3):317–328. https://doi.org/10.1046/j.1365-294X.1998.00360.x
Sullivan AR, Lind JF, McCleary TS, Romero-Severson J, Gailing O (2013) Development and characterization of genomic and gene-based microsatellite markers in North American red oak species. Plant Mol Biol Rep 31(1):231–239. https://doi.org/10.1007/s11105-012-0495-6
Tang H, Peng J, Wang P, Risch NJ (2005) Estimation of individual admixture: analytical and study design considerations. Genet Epidemiol 28(4):289–301. https://doi.org/10.1002/gepi.20064
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) Micro-checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4(3):535–538. https://doi.org/10.1111/j.1471-8286.2004.00684.x
Wachter H (2001) Untersuchungen zum Eichensterben in NRW. Schriftenreihe Der Landesforstverwaltung NRW 13:1–112
Wright SE (1965) The Interpretation of population structure by f-statistics with special regard to systems of mating. Evolution 19(3):395–420. https://doi.org/10.1111/j.1558-5646.1965.tb01731.x
Wypukol H, Liepelt S, Ziegenhagen B, Gebhardt K (2008) Genetische methoden zur Abstammungsanalyse und-prüfung von Sortenechtheit und-reinheit. In: Gebhardt K (Ed.) Herkunftskontrolle an forstlichem Vermehrungsgut mit Stabilisotopen und genetischen Methoden. Nordwestdeutsche Forstliche Versuchsanstalt Hann, Münden
Zanetto A, Roussel G, Kremer A (1994) Geographic variation of interspecific differentiation between Quercus robur L. and Quercus petraea (Matt.) Liebl. Forest Genet 1(2):111–123
Zhou H, Alexander D, Lange K (2011) A quasi-Newton acceleration for high-dimensional optimization algorithms. Stat Comput 21(2):261–273. https://doi.org/10.1007/s11222-009-9166-3
Acknowledgements
We thank Alexandra Dolynska for her valuable technical assistance in the laboratory work. The study was supported by the German Federal Ministry of Food and Agriculture represented by the Agency for renewable resources (FNR; Reference number: 22011317).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Gediminas Brazaitis.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Burger, K., Müller, M., Rogge, M. et al. Genetic differentiation of indigenous (Quercus robur L.) and late flushing oak stands (Q. robur L. subsp. slavonica (Gáyer) Mátyás) in western Germany (North Rhine-Westphalia). Eur J Forest Res 140, 1179–1194 (2021). https://doi.org/10.1007/s10342-021-01395-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10342-021-01395-8