
Abstract
Bitter taste perception is important in preventing animals from ingesting potentially toxic compounds. Whole-genome assembly (WGA) data have revealed that bitter taste receptor genes (TAS2Rs) comprise a multigene family with dozens of intact and disrupted genes in primates. However, publicly available WGA data are often incomplete, especially for multigene families. In this study, we employed a targeted capture (TC) approach specifically probing TAS2Rs for ten species of cercopithecid primates with diverse diets, including eight omnivorous cercopithecine species and two folivorous colobine species. We designed RNA probes for all TAS2Rs that we modeled to be intact in the common ancestor of cercopithecids (“ancestral-cercopithecid TAS2R gene set”). The TC was followed by short-read and high-depth massive-parallel sequencing. TC retrieved more intact TAS2R genes than found in WGA databases. We confirmed a large number of gene “births” at the common ancestor of cercopithecids and found that the colobine common ancestor and the cercopithecine common ancestor had contrasting trajectories: four gene “deaths” and three gene births, respectively. The number of intact TAS2R genes was markedly reduced in colobines (25–28 detected via TC and 20–26 detected via WGA analysis) as compared with cercopithecines (27–36 via TC and 19–30 via WGA). Birth or death events occurred at almost every phylogenetic-tree branch, making the composition of intact genes variable among species. These results show that evolutionary change in intact TAS2R genes is a complex process, refute a simple general prediction that herbivory favors more TAS2R genes, and have implications for understanding dietary adaptations and the evolution of detoxification abilities.
Similar content being viewed by others


Introduction
Gustation (taste perception) is a primary sense of vertebrates, conveying tastant chemical information from the oral cavity to the brain to regulate ingestion (Miura and Barlow 2010). Among the five basic taste senses, bitterness, sweetness, umami, sourness, and saltiness (Kinnamon and Cummings 1992; Lindemann 1996), bitter taste perception is considered to help animals to detect potentially toxic compounds and to prevent animals from ingesting them. Bitter taste perception is mediated by a group of seven-transmembrane G-protein-coupled receptors (GPCRs) known as TAS2Rs (also designated as T2Rs) (Adler et al. 2000). Mammals typically possess dozens of TAS2R genes, which are intronless, typically ~900 base pairs (bp) long and part of the same multigene family (Nei et al. 2008). The multiple TAS2R genes are spread over several chromosomes and are tandemly clustered in each of those chromosomes.
With the advent of massive parallel sequencing [next-generation sequencing (NGS)] technologies, the TAS2R sequences of various species have been accessible through public whole genome assembly (WGA) databases. In the human reference genome database, there are 26 intact and 10 disrupted (pseudogenized) TAS2R genes found (Hayakawa et al. 2014). The number of putative TAS2R genes in vertebrate genomes vary among species from a few to approximately 50 (Li and Zhang 2014). A comparative study of TAS2R genes has been of ongoing research interest given their potential to provide insight into the genetic basis of dietary adaptations and feeding strategies (Dong et al. 2009; Hayakawa et al. 2014; Li and Zhang 2014; Liu et al. 2016; Shi and Zhang 2006).
Glendinning (1994) showed that carnivorous mammals, which rarely encounter bitter and potentially poisonous foods, had a low bitter threshold (i.e., high bitter sensitivity) and low tolerance to ingest toxic compounds, while herbivorous mammals, especially browsers, which commonly encounter bitter and potentially poisonous foods, had a high bitter threshold (i.e., low bitter sensitivity) and high tolerance to ingest toxic compounds. Omnivorous mammals were positioned between the two groups (Glendinning 1994). Based on this observation, we would hypothesize that herbivorous animals have fewer functional TAS2R genes than carnivorous or omnivorous animals, perhaps because they have evolved dietary adaptations to tolerate toxic foods and need not avoid bitter foods. For example, herbivorous ruminants have evolved foregut fermentation by microbes and have higher tolerance for toxic compounds (Freeland and Janzen 1974). However, a later study of 54 vertebrate WGA databases (including 41 mammals, four birds, two reptiles, one amphibian, and six fishes) reported that the number of intact TAS2R genes positively correlated with herbivory, i.e., the fraction of plants in their diet (Li and Zhang 2014). This suggests that herbivores may need to recognize a larger number of bitter compounds than carnivores and omnivores to select the type of plants they eat (Li and Zhang 2014). Nevertheless, this would not necessarily refute the prediction based on Glendinning’s (1994) findings, because effects of ecological factors on TAS2R gene evolution could be complex. As Li and Zhang (2014) suggest, it is imperative to examine a diverse group of species when testing the potential impact of an ecological factor on taste receptor gene evolution.
Primate species belonging to the family Cercopithecidae are widely distributed among diverse habitats in sub-Saharan and northernmost Africa and southern, south-eastern to eastern Asia. They are composed of two subfamilies Cercopithecinae (including macaques, baboons, and guenons) and Colobinae (including colobuses, langurs, lutungs, and surilis). While cercopithecine monkeys are omnivorous with diets including fruits, leaves, seeds, buds, mushrooms, insects, spiders, and smaller vertebrates, the colobine monkeys are specialized for folivory supplemented with flowers, fruits, and occasional insects (Fleagle 2013). Colobine monkeys differ from all other primates in having a foregut-fermentation digestive system (Bauchop and Martucci 1968). To aid in digestion, especially of leaves that are difficult to digest, they possess three or four chambers in their forestomach. Although plant tissues contain more toxic compounds compared with animal tissues, foregut fermenters use symbiont microorganisms (i.e., protists and prokaryotes) to detoxify plant compounds before reaching the intestine, where toxins can be absorbed. Furthermore, the digestive efficiencies of plant fiber are significantly higher in foregut fermenters compared with hindgut fermenters (Edwards and Ullrey 1999). The foregut bacteria are digested by the specialized lysozyme that evolved to be active at low pH, resistant to protease, and expressed in the stomach to recover nutrients (Messier and Stewart 1997). The bacterial RNA molecules are degraded in the small intestine by the specialized RNASE1B to recycle nitrogen efficiently (Guevara et al. 2021a, b; Zhang et al. 2002).
Comparison of the TAS2R gene repertoire among phylogenetically close but ecologically diverse species would facilitate our understanding of ecological (dietary) effect on evolutionary diversity and adaptation of TAS2R genes. Thus, cercopithecid monkeys would be a suitable animal group for testing the potential impact of ecological factors on TAS2R gene evolution. Despite their importance, cercopithecid monkeys have been underrepresented in previous studies of TAS2R gene repertoire: one cercopithecid species (rhesus macaque) in Li and Zhang (2014) and three cercopithecid species (rhesus macaque, crab-eating macaque, and anubis baboon) in Hayakawa et al. (2014). The TAS2R gene repertoire has not yet been investigated for any colobine species.
In addition, dependence of studies of the TAS2R gene repertoire on publicly available WGA databases could be potentially problematic due to their generally low sequencing depth and inherent incompleteness especially for multigene families, such as TAS2Rs. In this study, we applied the targeted capture method and the short-read massive parallel sequencing to achieve high-depth sequencing to reveal TAS2R gene repertoire for five genera, eight species from cercopithecines and two genera, two species from colobines. Combining data from currently available WGA databases, we clarify evolutionary trajectory of TAS2R gene “births” and “deaths” compared between cercopithecines and colobines and among species within each subfamily. We reveal whether folivorous colobine species have TAS2R gene repertoires that differ from omnivorous cercopithecine species and discuss implications for the effect of ecological factors on shaping the evolution of the TAS2R gene repertoire.
Materials and methods
Genomic DNA samples
We targeted and captured TAS2R genes from genomic DNA samples from ten study species (one individual each) in the family Cercopithecidae (Table 1, Fig. 1). Eight species were from the subfamily Cercopithecinae: three species of the genus Macaca (Macaca mulatta, Macaca fuscata, and Macaca nigra), two species of the genus Papio (Papio anubis and Papio hamadryas), three species of the tribe Cercopithecini (Erythrocebus patas, Chlorocebus sabaeus, and Cercopithecus mitis). Two species were from the subfamily Colobinae: Colobus polykomos and Semnopiethecus entellus. The genetic materials from Primate Research Institute, Kyoto University, Japan, were obtained through their Cooperative Research Program (approval numbers 2012-A-11, 2020-A-27, and 2021-A-25). Semen of P. anubis was collected opportunistically by A.M.O. at the time a wild troop census in Kenya and transported to Japan through an export permit no. 0831553 issued by Kenya Wildlife Service, Republic of Kenya. The cellular sources of Erythrocebus patas, Cercopithecus mitis, Colobus polykomos, and Semnopithecus entellus were obtained by T. I. in 1983, 1984, 1988, and 1984, respectively, and cultured and transformed by T. I. (Ishida and Yamamoto 1987).

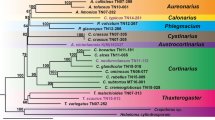
The phylogeny and primary diets of cercopithecid primates which were considered in this study. The phylogenetic relationship and divergence times were generated using TimeTree (Kumar et al. 2017) and modified based on literature (Kuderna et al. 2023; Li et al. 2009; Perelman et al. 2011). The species subjected to the targeted capture are highlighted with gray shadow. The other species were considered for the mapping references and subsequent analyses. Species for which public WGA databases are accessible are given with cloud symbol. Primary diets are symbolized for leaves, insects, fruits, and nuts (Fleagle 2013). Licensed photographs were obtained from Shutterstock (https://www.shutterstock.com) and license-free photographs were from Pixabay (https://pixabay.com). For detailed credit information, see Table S11
TAS2R sequence source information for designing probe
We aim to examine TAS2R genes, which were intact in the common ancestor of cercopithecids (designated as “ancestral-cercopithecid TAS2R genes”). We regarded such TAS2R genes as those that were intact at least one species among the five cercopithecid WGA databases available at the time we started this project (three cercopithecines: Macaca mulatta Mmul_8.0.1 (Zimin et al. 2014), M. fascicularis Macaca_fascicularis_5.0, and Cercocebus atys Caty_1.0 (Palesch et al. 2018) and two colobines: Colobus angolensis Cang.pa_1.0 and Rhinopithecus roxellana Rrox_v1 (Zhou et al. 2014) (Table S1-1). We conducted a Basic Local Alignment Search Tool (BLAST) search (Johnson et al. 2008; NCBI Resource Coordinators 2016) to these databases for TAS2R genes that had been identified by Hayakawa et al. (2014), who studied humans, various other primates, and nonprimate mammals. There were 31 such genes (Table S2-1). Hayakawa et al. (2014) examined three cercopithecid species (rhesus macaque, crab-eating macaque, and anubis baboon) which were available at the time of their study and identified one additional gene, TAS2R418, although the gene was disrupted in all these three species. Their assignment as an intact gene at the cercopithecid ancestor was because the disruptive mutations were different between the macaques and the baboon. However, we excluded this gene from our assignment because its sequence was disrupted in not only in our choice of the three cercopithecine WGA databases but also in the two colobine ones.
As the source of probe DNA sequences (Table S1-1) for cercopithecine TAS2R genes, we chose Mmul_8.0.1 first, Macaca_fascicularis_5.0 second, and Caty_1.0 third. For the colobine TAS2R genes, we first chose Cang.pa_1.0 and second Rrox_v1. If a gene of interest was absent in the WGA database chosen, we searched the lower priority WGA databases for an intact form of the gene. If a gene of interest was disrupted (indicated with a suffix “P” in Table S2-1) or “truncated” (suffix “T”) in the WGA database chosen, we took it as a probe sequence and searched the next WGA databases for an intact form of the gene to add. If a gene of interest was duplicated or multiplicated, including intact genes in the WGA database chosen, we took all these paralogous genes as probe sequences. If duplicated/multiplicated genes were all disrupted or truncated genes, we took the disrupted/truncated ones and searched for the next WGA databases for an intact form of the gene to add (e.g., TAS2R1 in Angolan colobus Cang.pa_1.0 in Table S2-1). For each probe sequence, 100-nucleotides (nt) upstream and downstream flanking regions were included in addition to the entire coding region (approximately 900 bp) (Table S2-1). The probe sets from cercopithecine and from colobine databases were mixed and used in the targeted capture for all the samples.
Neutral reference sequence source information for designing probes
As a control reference to evaluate gene copy number and selective neutrality, a previous study (Akhtar et al. 2022) selected 85 single-locus nonprotein-coding sequences (82 autosomal and three X-nonpseudoautosomal) from the human reference genome hg19 assembly using the Neutral Region Explorer (Arbiza et al. 2012). In brief, the selected regions were without repeat sequences, with a length longer than 1.0 kb, with distance to the nearest gene longer than 0.2 centimorgan (cM) in autosomes or 0.1 cM in the X chromosome, and with a minimum recombination rate of 0.9 cM/Mb under the Neutral Region Explorer. We conducted a BLAST search using these human sequences as queries for the rhesus macaque Mmul_8.0.1 and Angolan colobus Cang.pa_1.0 WGA databases. We took the sequences with a reasonably high similarity (higher than 90%) between human and cercopithecid nonprotein coding regions. When present, there was only one such region detected for each sequence. Although we have not confirmed their orthology to the human sequences by reconstructing the phylogenetic tree, there were no protein-coding genes among them, and these were with a length longer than 1.0 kb. We regarded these regions usable as neutral references in this study. Among the 85 regions, 83 and 79 regions were identified from Mmul_8.0.1 and Cang.pa_1.0 WGA databases, respectively, and used as probes (Table S2-2).
Probe synthesis, targeted capture, and NGS
Probe synthesis was outsourced to the Biodiscovery, LLC (Ann Arbor, MI) as the in-solution biotinylated RNA baits (myBaits®) (Gnirke et al. 2009). Each probe was 120-nt length and was overlapped 60 nt with adjacent probes (i.e., 2× tiling). Construction of the DNA sequencing libraries and targeted capture were performed using myBaits® kit v5 following the manufacturer’s protocol (myBaits® user manual version 5.02). Construction of DNA sequencing library, targeted capture, and NGS were conducted by the Centre for Health Genomics and Informatics and UCDNA Service, University of Calgary, Canada. DNA sequencing libraries were constructed by fragmenting genomic DNA into ~500 bp. Biotinylated RNA baits were introduced and allowed to hybridize to targets at 65 °C. Bait-target hybrids were then pulled out of the solution with streptavidin-coated magnetic beads. The beads were stringently washed at 65 °C to remove nonhybridized and nonspecifically-hybridized molecules. These conditions of hybridization and washing are reported to allow ~10% mismatch between baits and targets according to the manufacturer’s protocol (myBaits® user manual version 5.02). The captured DNA library was released from the beads and amplified by polymerase chain reaction (PCR) targeting the adapter sequence. The massive parallel sequencer used in this study was the Illumina NextSeq with 300 cycle and 150-bp paired end sequencing.
Quality control of NGS reads
We used the PRINSEQ tool for quality control of raw NGS reads (Schmieder and Edwards 2011). For the FASTQ-formatted reads with the base quality score emitted by the Illumina sequencer at each nucleotide position, any nucleotide position with quality score below 20 at either edge of a read was trimmed away. Any read with a mean quality score less than 20 and any read shorter than 10 nucleotides were also filtered out.
Reference sequences for the first-round mapping of NGS reads
For mapping the NGS short reads to reference sequences, we aimed to choose the WGA databases of the phylogenetically closest species to the study species (Table S1-2). At the time we did the mapping, WGA databases of 23 cercopithecid species were publicly accessible. Fifteen species were from Cercopithecinae: six species of Macaca (Macaca mulatta, Macaca fuscata, Macaca fascicularis, Macaca thibetana, Macaca nemestrina, and Macaca nigra), Mandrillus leucophaeus, Cercocebus atys, two species of Papio (Papio anubis and Papio hamadryas), Erythrocebus patas, two species of Chlorocebus (Chlorocebus aethiops and Chlorocebus sabaeus), and two species of Cercopithecus (Cercopithecus albogularis and Cercopithecus mitis). Eight species were from Colobinae: two species of Colobus (Colobus angolensis palliates and Colobus guereza), Piliocolobus tephrosceles, Semnopiethecus entellus, Trachypithecus francoisi, Nasalis larvatus, and two species of Rhinopithecus (Rhinopithecus bieti and Rhinopithecus roxellana) (Jayakumar et al. 2021; Liu et al. 2020; Palesch et al. 2018; Sène et al. 2021; Shao et al. 2023; Simons et al. 2019; Wall et al. 2022; Wang et al. 2019; Warren et al. 2020; Zhang et al. 2022; Zhou et al. 2023).
In addition to the 31 probe-designed TAS2R genes, we examined 22 other TAS2R genes that were absent or disrupted in all these cercopithecid WGA databases but intact in WGA databases of some noncercopithecid primate (Table S3). If the cercopithecid genes were present as a disrupted form, they were used for references. If the cercopithecid genes were truncated or absent, genes from closer outgroup species were used.
Mapping, assemblage, and inference of gene duplications
Our mapping procedure is described below and is illustrated in Fig. S1.
-
1.
First-round mapping
We mapped the filtered reads using the BWA-MEM Tool v. 0.7.17 to the references (Table S3), which were assigned to each TAS2R gene of each study species for the first-round mapping. The tool is specialized for mapping highly similar reads on a reference (Zheng-Bradley et al. 2017), which is the case with TAS2R genes because of the presence of similar paralogous genes. After the mapping, by applying the MarkDuplicates of the Genome Analysis Toolkit (GATK) (DePristo et al. 2011; McKenna et al. 2010; Van der Auwera et al. 2013), we identified the read pairs that were likely to have originated from duplicates of the same original DNA fragments through artefactual processes, such as PCR amplification. Only a single read pair within each set of duplicates was used for the subsequent analyses.
The HaplotypeCaller of GATK (4.2.5.0) was used to assemble the mapped reads, to find insertion/deletion (indel) sites and single-nucleotide polymorphism (SNP) sites, and to determine haplotype phase among these polymorphic sites based on their physical linkage in a sequencing read. We defined an indel site as the site where insertion or deletion of a nucleotide was detected among sequencing reads relative to the reference sequence. We defined an SNP site as the site where two or more nucleotide kinds were present among sequencing reads. We did not count nucleotide sites as SNPs where only one nucleotide kind was detected among reads but the nucleotide kind was different from the one in the reference at the site. Two sequences were generated as putative haplotypes by bcftools consensus v. 1.7. If no read is mapped to a region in a reference sequence, the HaplotypeCaller generates chimeras consisting of the sample’s own sequences for the mapped region and the reference sequences for the unmapped region.
-
2.
Second-round mapping
Less mismatch is expected between a sequence generated from the first-round mapping and a sequencing read from a gene/genome region. To improve the proportion of mapped regions, we conducted the second-round mapping using the chimeric sequences generated from the first-round mapping. For the second-round mapping references, we used one of the two putative haplotype sequences bearing nucleotides from the sample but not from the reference at SNP sites. Mapping was conducted as in the first mapping. Variant calling for each site was conducted with samtools mpileup v. 1.7 (Li 2011) to identify unmapped regions. Two sequences were generated as putative haplotypes by bcftools consensus v. 1.7. In the generated sequences, we exchanged nucleotides in these unmapped regions for “N”s. We evaluated the sequencing depth of the TAS2R genes and of the neutral references using the samtools bedcov (Li 2011).
-
3.
Third-round mapping
For the third-round mapping, we used the sequences generated from the second-round mapping including “N” regions. There, we used one of the two putative haplotype sequences bearing a smaller number of deletions relative to the reference sequence. We regarded genes/genome regions with low depth (< ten per site) after the second-round mapping as being absent and excluded them from the third-round mapping to improve its specificity. Mapping was conducted as in the first mapping. The HaplotypeCaller was used to assemble the mapped reads and determine haplotype phase among SNPs and indels as in the first mapping. We further refined variant calls using VariantFiltration of GATK. Any variant sites with a genotype quality score less than 20 were filtered out. Two sequences were generated as putative haplotypes by bcftools consensus v. 1.7. The International Union of Pure and Applied Chemistry (IUPAC) degenerate nucleotide codes were used to designate unphased SNP sites with more than one nucleotide kinds. We removed the “N” sites from the generated sequences, which makes them consisting of only the sample’s own sequences.
After the third-round mapping, we evaluated again the sequencing depth of the TAS2R genes (Table S4-1) and of the neutral references (Table S4-2) using the samtools bedcov (Li 2011). We regarded the TAS2R genes and the autosomal neutral references (NR-As) with sequencing depth lower than half of the minimum of lower whisker in boxplots (not shown) of the sequencing depth as being possibly absent and excluded from further analyses. We also excluded the TAS2R genes as possibly absent of which the length of mapped sequence was less than 750 bp (see the section “Classification of TAS2R sequences into intact and disrupted types”). We excluded the neutral references shorter than 1000 bp from the subsequent analyses. We also excluded from further analyses the NR-As with sequencing depth 1.5 times higher than the maximum of upper whisker in the boxplots as being potentially duplicated in at least one allele.
Boxplots of sequencing depths of TAS2R genes and of neutral references after these treatments are shown in Fig. S2. Using the same sequence data set, we evaluated the number of SNP sites (not including indels) per 1000 bp (SNP density) of the TAS2R genes (Table S5-1) and of the NR-As (Table S5-2) using vcftools v 0.1.13 (Danecek et al. 2011). Boxplots of the SNP density of TAS2R genes and NR-As are shown in Fig. S3. We regarded the TAS2R genes as single-locus genes, of which both sequencing depth (Fig. S2) and SNP density (Fig. S3) were below the maximum of the upper whisker of the corresponding boxplots. We generated two putative haplotype sequences for these genes by the HaplotypeCaller after the third mapping and regarded the two sequences, designated as H1 and H2, as two alleles of these genes.
-
4.
Fourth-round mapping
To separate sequencing reads into paralogous TAS2R genes, we regarded the TAS2R genes as potentially duplicated or multiplicated when either sequencing depth (Fig. S2) or SNP density (Fig. S3) was above the maximum of the upper whisker (“upper outlier”) of the corresponding boxplots. To tease apart the sequencing reads into potentially paralogous genes, we generated two putative haplotype sequences (H1 and H2) using the HaplotypeCaller after the third-round mapping and used both of the two putative haplotype sequences as the references of the fourth-round mapping. The genes were classified into the following four categories (Table 2)
.
-
(1)
TAS2R genes not separable to single-locus genes at the fourth mapping: If the same reads were mapped to the two references, H1 and H2, from a source gene and if the proportion of such region was more than roughly 10% of a reference length in the Integrative Genomics Viewer (IGV) (Robinson et al. 2011), we regarded the source gene sequence at the third mapping (i.e., H1 + H2 congregated sequence) as mixture of reads of paralogous and allelic origins, which were not separable to single-locus genes. We regarded the depth and SNP density results at the third-round mapping as the final data for this congregated gene. When evaluating open-reading frame (ORF) and seven transmembrane (7-TM) domain (see the section “Classification of TAS2R sequences into intact and disrupted types”), we tentatively used their H1 and H2 sequences. We displaced to category 4 the genes in which the SNP density was in the upper outlier but the sequencing depth was below the maximum of the upper whisker at the third mapping.
-
(2)
TAS2R genes separated to single-locus genes at the fourth mapping: If the proportion of the same reads mapped to the two references, H1 and H2, from a source gene was less than roughly 10% of a reference length in the IGV, we redesignated the H1 and H2 with suffix “− 1” and “− 2”, respectively, as paralogous genes. If both sequencing depth and SNP density after the fourth mapping were below the maximum of the upper whisker, we regarded such a gene as a single-locus gene. We further generated two haplotype sequences from it by the HaplotypeCaller after the fourth mapping and designated the two sequences with H1 and H2 as its two alleles.
-
(3)
TAS2R genes not separable to single-locus genes at the fifth mapping: The same situation with category 2, but, if sequencing depth and/or SNP density after the fourth mapping were above the maximum of the upper whisker, we regarded such a gene (with suffix “− 1” or “− 2”) still as potentially duplicated or multiplicated. We further generated two putative haplotype sequences (H1 and H2) by the HaplotypeCaller after the fourth-round mapping and used both two putative haplotype sequences as the references of the fifth-round mapping. The TAS2R genes of category 3 turned out to fall in category 1 in the fifth-round mapping. We regarded the source gene sequence at the fourth mapping (i.e., H1 + H2 congregated sequence) as mixture of reads of paralogous and allelic origins, which were not separable to a single-locus gene. We regarded the depth and SNP density results at the fourth-round mapping as the final data for this congregated gene. Only for evaluating ORF and 7-TM domain (see the section “Classification of TAS2R sequences into intact and disrupted types”), we tentatively used their H1 and H2 sequences. We displaced the genes to category 4 of which the SNP density was in the upper outlier, but the sequencing depth was below the maximum of the upper whisker at the fourth mapping.
-
(4)
The TAS2R genes with high SNP density but not with high sequencing depth at the fourth or fifth mapping: This category of TAS2R genes were displaced from category 1 or 3. The reason for this high SNP density remains to be elucidated. We waived decision on whether these genes were duplicated/multiplicated or single-locus.
The final per-site sequencing depth values of the TAS2R genes and the neutral references are listed in Tables S6-1 and S6-2, respectively. The per-site sequencing depth data of the single-locus and ancestral-cercopithecid TAS2R genes averaged in each species were summarized in Table 3. The range of the final sequencing depth of TAS2R genes in the ancestral-cercopithecid TAS2R gene set, that of the NR-As and that of the X-chromosomal neutral references (NR-Xs) in each cercopithecid species are indicated with boxplots (Fig. 2). The final SNP density values of intact, disrupted, and segregating pseudogene categories (see the section “Classification of TAS2R sequences into intact and disrupted types” and Table S7) of ancestral-cercopithecid TAS2R genes in each species are listed in Tables S8-1–10. Among the data, those of the single-locus TAS2R genes are box-plotted with SNP density of the single-locus NR-As in each species (Fig. 3).
Classification of TAS2R sequences into intact and disrupted types
We used the EMBOSS getorf to find ORF (Rice et al. 2000). We regarded any TAS2R sequence with maximum ORF < 750 bp (encoding < 250 amino acids long) as disrupted because such a sequence is unlikely to encode a functional structure (Hayakawa et al. 2014). We also examined if a TAS2R sequence was capable of forming the 7-TM domain, a hallmark of GPCRs, by using DeepTMHMM (Hallgren et al. 2022). We regarded any sequence without proper 7-TM domain as disrupted. Thus, we regarded only TAS2R sequences with an ORF of at least 750 bp and capable of forming a 7-TM domain as intact.
ORF and 7-TM domain cannot be predicted appropriately for the TAS2R sequences containing IUPAC degenerate nucleotide codes assigned to SNP sites among which haplotype phases were not determinable. In such cases, for a pair of H1 and H2 sequences, we replaced an IUPAC degenerate code (e.g., R for A and G) in H1 with a nucleotide code chosen in alphabetical order (A in this example) (H1-1) and that in H2 with another nucleotide (G) (H2-1). We also made a replacement in the opposite combination, G in H1 (H1-2) and A in H2 (H2-2), in this example because ORF and 7-TM domain can be disturbed by one amino acid site irrespective of haplotype phases. The combination of nucleotides between IUPAC degenerate code sites increases exponentially as the number of such sites increases. Rather than testing all the possible combinations exhaustively, we tested the two pairs (H1-1/H2-1 and H1-2/H2-2) throughout all degenerate sites in the same way (for example, suppose there are two degenerate code sites, R (A and G) and Y (C and T), one combination between two sites is in alphabetical order, i.e., A and C in H1 allele for the R and Y sites, respectively, and G and T in H2 allele. Another combination was in the opposite order, i.e., G and T in H1 allele and A and C in H2 allele).
Table S7 summarizes the ORF length and the predicted number of TM domains of H1-1, H2-1, H1-2, and H2-2 sequences for the TAS2R genes that we considered to exist based on sequencing depth in the mapping procedures (Table S6-1). If there was no site with IUPAC degenerate nucleotide codes and if H1 and H2 sequences were identical, ORF length and the predicted number of TM domains were given only to the H1-1 column. If there was no site with IUPAC degenerate nucleotide codes and if H1 and H2 sequences were different, ORF length and the predicted number of TM domains were given only to the H1-1 and H2-1 columns.
If all sequences of a gene were intact, we regarded the gene as intact (Tables S7-1–10). If all sequences were disrupted, we regarded the gene as disrupted (Tables S7-1–10). Regarding the single-locus genes, if both intact and disrupted forms were found, we regarded the gene as heterozygous with intact and disrupted alleles and we designated such TAS2R gene as a segregating pseudogene (Tables S7-1–10). Regarding categories 1 and 3 duplicated/multiplicated TAS2R genes and category 4 genes with high SNP density but not with high sequencing depth (Table 2). “Intact” and “disrupted” mean that the congregated sequence of potentially paralogous and allelic origins is comprised of only intact and disrupted sequences, respectively. On the other hand, “intact and disrupted” means that the congregated sequence is composed of intact and disrupted sequences but it remains uncertain whether intact and disrupted sequences are alleles of a locus or paralogous genes.
Reconstruction of phylogenetic trees
We aligned nucleotide sequences using MUSCLE (Edgar 2004). We estimated the number of nucleotide substitutions per nucleotide site between two sequences using the Tamura-Nei model (Tamura and Nei 1993) with gap sites excluded in pairwise fashion. We reconstructed phylogenetic trees using the neighbor-joining method (Saitou and Nei 1987) and evaluated the reliability of the trees by bootstrap resampling with 1000 replications (Felsenstein 1985) (Figs. 4 and S4). These procedures were conducted under the platform of MEGA11 (Tamura et al. 2021).
Counting gene birth and gene death events
Based on Fig. S4 (1) ~ (30), the evolutionary timing of gene “births” (gain by gene duplication) and gene “deaths” (disruption or loss) was inferred (Fig. 5 and Table S9). The births were considered only for the genes listed in Table 2 as categories 1, 2, and 3 (TAS2R10, TAS2R14, TAS2R50, TAS2R60, TAS2R413, TAS2R414, TAS2R415, and TAS2R419) for which gene duplications/multiplications were inferred based on the sequencing depth and the SNP density. Among the other TAS2R genes, there were genes (TAS2R1, TAS2R2, TAS2R4, TAS2R8, TAS2R9, TAS2R13, TAS2R38, TAS2R42, TAS2R403, TAS2R412, and 418 in Fig. S4) for which the tree topology was not congruent with the phylogenetic relationship among species (Fig. 1). Because incongruency with the species tree could arise due to various causes such as incomplete lineage sorting or low statistical support for the tree topology, we did not assume “hidden” gene duplications after which one of paralogous genes were lost. The timing of gene duplication was inferred based on the maximum parsimony principle so that the sum of the number of gene birth and death events was the minimum under the assumption of frequent occurrence of homogenization between duplicated genes by gene conversion (Hiwatashi et al. 2011). For example, in the TAS2R415 gene tree [Fig. S4 (28)], we assumed a gene duplication once at the common ancestor of cercopithecines (Table S9) and subsequent gene conversions at the common ancestor of Macaca species, at that of Papio species and at that of the tribe Cercopithecini species (patas, green, and blue monkeys) rather than gene duplications at each of these three lineages.
At each tree branch of Fig. 5, the number of intact genes gained or lost/disrupted is indicated with plus (+) or minus (−) symbol, respectively. Segregating-pseudogenizations of single-locus genes were not regarded as gene deaths and were not included as losses. Thus, in Fig. 5 the number of intact genes at tree nodes could include segregating pseudogenes. Gene duplication/multiplication events (categories 1, 2, and 3) were each counted as +1. Category 4 genes were not counted as gains. Disruption (see Table S7) or loss of single-locus genes, category 2 genes, or category 4 genes were each counted as −1. If category 1 or 3 genes had only disrupted sequences (see Table S7), disruptions were counted as −2. If category 1 or 3 genes had both intact and disrupted sequences (see Table S7), disruptions were counted as −1. If category 4 genes had both intact and disrupted sequences (see Table S7), these were not regarded as gene deaths and were not counted as losses. Thus, these were included in the total numbers of intact genes and segregating pseudogenes of the study species, indicated beside their common names in Fig. 5 and in segregating pseudogenes in Fig. 6.
Results
Inference of gene duplications and disruptions for ancestral-cercopithecid TAS2R gene set
Using the probes designed for the TAS2R genes that were inferred to be intact at the last common ancestor of cercopithecid primates (designated as “ancestral-cercopithecid TAS2R gene set”; see Materials and Methods: Table S2-1) and the probes designed for neutral references (Table S2-2), we conducted the targeted capture for ten cercopithecid (eight cercopithecine and two colobine) species (Table 1, Fig. 1). Using up to five rounds of mapping and assemblage of NGS short reads, we retrieved nucleotide sequences of TAS2R genes and neutral references for the ten study species [Supplementary Data 1 (ancestral-cercopithecid TAS2R genes) and 2 (neutral references)].
Gene duplications/multiplications were inferred for eight genes (TAS2R10, TAS2R14, TAS2R50, TAS2R60, TAS2R413, TAS2R414, TAS2R415, and TAS2R419) as category 1, 2, or 3 (Table 2) based on the evaluation of sequencing depth and SNP density (see Materials and Methods). However, there were genes for which SNP density was high but sequencing depth was not, after the final-round of mapping (category 4 in Table 2). With the current dataset, we waived the decision on whether these genes were duplicated/multiplicated or single locus.
We classified the single-locus TAS2R genes into not only intact and disrupted genes but also segregating pseudogenes (genes heterozygous with intact and disrupted alleles) in each species (Tables S7-1–10). The latter is not reliably retrievable from raw NGS short-read data in many public WGA databases due to their low sequencing depth. The congregated sequences of categories 1, 3, and 4 genes were also classified into intact, disrupted, or both intact and disrupted with paralogous and allelic origins mixed (Tables S7-1–10).
Regarding TAS2R genes for which probes were prepared (Table S2-1), there was no gene for which the intact sequences were not retrieved from any of the ten study species (Table S7). Regarding TAS2R genes for which probes were not prepared (Table S3), all genes but TAS2R418 were either absent or disrupted (Tables S6, S7). TAS2R418 was found to be intact in anubis baboon (Table S7-4), hamadryas baboon (Table S7-5), and patas monkey (Table S7-6) samples, evidencing that this gene was intact at the last common ancestor of cercopithecid primates as Hayakawa et al. (2014) assumed. All nonprobed TAS2R genes except TAS2R418 were absent or disrupted in all of cercopithecid WGA databases (Table S1-2) currently available. These results support that all probed TAS2R genes and TAS2R418, but not other TAS2R genes, compose the ancestral-cercopithecid TAS2R gene set.
Sequencing depth and SNP density
The per-site sequencing depth values of the TAS2R genes and the neutral references are listed in Tables S6-1 and S6-2, respectively. Sequencing depth of TAS2R genes (all autosomal) in the ancestral-cercopithecid TAS2R gene set was largely similar to that of the autosomal neutral references (NR-As) (assumed to be single-locus; see Materials and Methods) if excluding the upper outliers of TAS2R genes, which we regarded as duplicated or multiplicated (see Materials and Methods) (Fig. 2). The sequencing depth of the X-chromosomal neutral references (NR-Xs) was roughly half of the NR-As in the samples from male individuals (rhesus macaque, Japanese macaque, Celebes crested macaque, anubis baboon, green monkey, patas monkey, and Hanuman langur) whereas that of the NR-X was similar with that of the NR-As in the samples from female individuals (blue monkey, hamadryas baboon, and king colobus) (Fig. 2). These observations supports the supposition that sequencing depth can reflect the gene ploidy difference in our samples.

The range of the final per-site sequencing depth of the ancestral-cercopithecid TAS2R genes, that of autosomal neutral references (NR-A) and that of the X-chromosomal neutral references (NR-Xs) in each cercopithecid study species, depicted by boxplots. A–H are cercopithecine species: A rhesus macaque, B Japanese macaque, C Celebes crested macaque, D anubis baboon, E hamadryas baboon, F patas monkey, G green monkey, and H blue monkey. I and J are colobine species: I king colobus and J Hanuman langur. The medians are indicated as the horizontal line in the box. The means are indicated as the symbol “X.” The upper whisker value represents the largest within 1.5 times interquartile range above the third quartile, and the lower whisker value represents the smallest within 1.5 times interquartile range below the first quartile. The outliers are indicated by dots. The median was included in the calculation to determine quartile values. The number of data points are indicated with n
The average per-site sequencing depth values of the single-locus and ancestral-cercopithecid TAS2R genes in each species (353 in the patas monkey sample, 1224–1696 in the others, and 1286 on average) were overall much higher than those available in the public WGA database (29–83, with 54 on average) (Table 3). Among SNP density values of NR-As and the single-locus and ancestral-cercopithecid TAS2R genes, SNP density was generally highest in NR-As in each species, which is consistent with expectation given the difference in functional constraint (Fig. 3, Tables S8-1–10).

The range of final SNP density values of intact (TAS2R-I), segregating pseudogene (TAS2R-S), and disrupted (TAS2R-D) ancestral-cercopithecid and single-locus TAS2R genes and that of autosomal neutral references (NR-A) in each cercopithecid study species, depicted by boxplots. A–H are cercopithecine species: A rhesus macaque, B Japanese macaque, C Celebes crested macaque, D anubis baboon, E hamadryas baboon, F patas monkey, G green monkey, and H blue monkey. I and J are colobine species: I king colobus and J Hanuman langur. The medians are indicated as the horizontal line in the box. The means are indicated as the symbol “X.” The upper whisker value represents the largest within 1.5 times interquartile range above the third quartile, and the lower whisker value represents the smallest within 1.5 times interquartile range below the first quartile. The outliers are indicated by dots. The median was included in the calculation to determine quartile values. The number of data points are indicated with n
Evolutionary birth and death process of TAS2R genes in cercopithecid primates
Figure 4A shows a phylogenetic tree reconstructed for entire ancestral-cercopithecid TAS2R gene set and Fig. 4B provides an expanded view of the “TAS2R405 group” while Fig. S4 provides subtrees for each gene group. We observed clustering of the TAS2R genes retrieved in this study with their known orthologous outgroups, supporting their orthology identification (Fig. 4A). Based on the subtrees [Fig. S4 (1) ~ (30)], evolutionary timing of gene “birth” (gain by gene duplication) and gene “death” (disruption or loss) was inferred. Among the TAS2R405 group genes (Fig. 4), TAS2R416 and TAS2R417 were previously considered to have arisen in the common ancestor of cercopithecids in Hayakawa et al. (2014), where WGA databases of two macaque and one baboon species were studied. Including more diverse cercopithecid species in this targeted-capture study, we found that these two genes arose within the TAS2R415 at the common ancestor of cercopithecines, and we renamed TAS2R416 as TAS2R415-1 and TAS2R417 as TAS2R415-2 [Fig. S4 (28)]. On the other hand, we found that TAS2R10 and TAS2R413 were duplicated to TAS2R10-1 and TAS2R10-2 [Fig. S4 (9)] and TAS2R413-1 and TAS2R413-2 [Fig. S4 (26) and Fig. 4B], respectively, at the common ancestor of cercopithecids. These duplications were not detected by the previous WGA-based study on the three cercopithecine species (Hayakawa et al. 2014).

Phylogenetic trees reconstructed for the ancestral-cercopithecid TAS2R gene set. Regarding the single-locus TAS2R genes, only one of the two allele sequences (H1) was used to represent each gene. Regarding categories 1 and 3 duplicated/multiplicated TAS2R genes and category 4 genes high SNP density but not with high sequencing depth (Table 2), the congregated sequence of each gene group was used. A A phylogenetic tree covering entire ancestral-cercopithecid TAS2R gene set. Cercopithecid genes are symbolized with black dots. As outgroups, known orthologous gene sequences (Hayakawa et al. 2014) available from human (red dots) and platyrrhines (common marmoset and squirrel monkey) (purple dots) are used. B An expanded view of the “TAS2R405” group phylogenetic tree. The nine orthologous genes (TAS2R410, TAS2R411, TAS2R412, TAS2R413-1, TAS2R413-2, TAS2R414, TAS2R415, TAS2R418, and TAS2R419) are differently colored for easier distinction. Note that human and platyrrhine out-group sequences are not included in (B) because the nine genes occurred at the common ancestor of cercopithecids after its separation from the hominoid ancestor (and thus after separation from platyrrhine ancestor), and there is no corresponding human and platyrrhine sequences to each of the nine genes. A sequence name consists of the gene name followed by the species name abbreviation (e.g., TAS2R410 Mmul; see Table 1 for abbreviation). The disrupted genes are suffixed with “D” after the gene name. Segregating pseudogenes are suffixed with “S” after the gene name. The congregated sequences for which both intact and disrupted sequences were inferred (Table S7) are labeled ± after the sequence names
Figure 5 depicts the birth and death process of the ancestral-cercopithecid TAS2R gene set. Table S9 summarizes the genes gained by gene duplications and the genes disrupted or lost at every branch in Fig. 5. Gene “birth” or “death” events occurred at almost every branch (Fig. 5), making the composition of intact genes variable among species. At the common ancestor of cercopithecids (Branch 1 in Fig. 5), ten births were inferred (Table S9). Eight of the ten were from the TAS2R405 group, including TAS2R413-1 and TAS2R413-2 as noted above. Another was from TAS2R10, giving rise of TAS2R10-1 and TAS2R10-2, as also noted above. The last one was according to Hayakawa et al. (2014) and was from TAS2R409, giving rise of TAS2R403 and TAS2R404 with the latter being lost in the same branch (Table S9). Thus, the number of births in branch 1 happened to be the same with the inference by Hayakawa et al. (2014). At this branch, another death occurred (TAS2R15), according to Hayakawa et al. (2014). The number of intact genes at the common ancestor of cercopithecids were inferred to be 32, as Hayakawa et al. (2014) inferred.

Evolutionary birth and death process of ancestral-cercopithecid TAS2R gene set. The total numbers of intact genes and segregating pseudogenes of the study species are indicated beside their common names. The numbers of intact genes inferred to common ancestors are boxed. The numbers of gene births and gene deaths at every branch are indicated with plus (+) and minus (−) codes, respectively. The numbers in parentheses represent the branch IDs shared with Table S9. The phylogenetic relationship and divergence times were given by using TimeTree (Kumar et al. 2017) and modified based on literature (Li et al. 2009; Perelman et al. 2011)

The numbers of intact, segregating pseudogene, disrupted, and lost TAS2R genes in cercopithecid primates relative to the 32 ancestral-cercopithecid TAS2R genes. The numbers in ten study species are highlighted with gray shadow. The number of intact, disrupted, and absent (lost) TAS2R genes searched from the public WGA databases are also shown
At the common ancestor of cercopithecines (branch 2 in Fig. 5), three gene births from TAS2R50, TAS2R414, and TAS2R415 were inferred, whereas at the common ancestor of colobines (branch 3 in Fig. 5), four gene deaths [one disruption (TAS2R403) and three losses (TAS2R10-2, TAS2R413-2, and TAS2R418)] were inferred (Table S9). Overall, the total number of intact genes and segregating pseudogenes were smaller in colobines (26–29) than in cercopithecids (27–36) (Fig. 5).
In Fig. 6, the numbers of intact, segregating pseudogene, disrupted, and lost TAS2R genes relative to the 32 ancestral-cercopithecid TAS2R genes are band-graphed for cercopithecid species examined in this study. The number of intact genes were 27–36 in cercopithecids and 25–28 in colobines (Fig. 6). Figure 6 also indicates the number of intact, disrupted, and absent (lost) TAS2R genes searched from the public WGA databases. The trend of less intact TAS2R genes in colobines than in cercopithecines was also observed in the public WGA databases: 20–26 in colobines and 19–29 in cercopithecines. Relative to the 32 intact genes at the cercopithecid common ancestor, the anubis baboon increased to 36 with no segregating pseudogenes, and the green monkey retained 32, including segregating pseudogenes. The others have decreased the total number of intact genes and segregating pseudogenes in both cercopithecines and colobines. The decrease appeared to be more evident in colobines.
TAS2R gene retrieval compared with WGA-database search
The “intact” genes of our study represent only genes homozygous with intact alleles whereas those of WGA databases could not only represent them but also include intact alleles from segregating pseudogenes. Nevertheless, the number of intact genes were more in our targeted-capture-based study (25–36) than in WGA-based search (19–29) (Fig. 6).
A gene-by-gene comparison of intact/disrupted/segregating pseudogene/lost (absence) statuses of TAS2R genes between WGA and targeted-capture is summarized in Table S10. There were ten genes that were disrupted in the WGA databases but were intact or duplicated with intact and disrupted genes in our targeted capture [one in Japanese macaque (TAS2R1), four in Celebes crested macaque (TAS2R8, TAS2R38, TAS2R42, TAS2R419), one in hamadryas baboon (TAS2R3), one in patas monkey (TAS2R413), three in Hanuman langur (TAS2R14, TAS2R42, and TAS2R60)]. There were nine genes that were absent in the WGA database but were intact in our targeted capture [two in patas monkey (TAS2R414 and TAS2R418), seven in blue monkey (TAS2R1, TAS2R3, TAS2R4, TAS2R7, TAS2R8, TAS2R9, and TAS2R10)]. There was one gene that was absent in WGA database but was multiplicated with intact and disrupted genes in our targeted capture [green monkey (TAS2R414)]. There were five genes which were intact in the WGA databases and were segregating pseudogenes in our targeted capture [two in rhesus macaque (TAS2R5 and TAS2R413), one in green monkey (TAS2R60), one in blue monkey (TAS2R414), and one in Hanuman langur (TAS2R419)], whereas there was no gene which was intact in the WGA databases but was lost or disrupted in our targeted capture. These results indicate that our targeted capture retrieved more TAS2R genes and characterized their intact/disrupted and duplication statuses better than WGA databases.
Discussion
In this study we employed the targeted capture to specifically probe the TAS2R genes and the autosomal and X-chromosomal neutral reference regions from genomic DNA samples of ten Cercopithecidae primates including eight omnivorous Cercopithecinae and two folivorous Colobinae species. The targeted capture was followed by short-read and high-depth massive-parallel sequencing. We confirmed that the sequencing depth well reflected the ploidy difference of X-chromosomal neutral references between males and females relative to autosomal regions, validating the utility of sequencing depth as an indicator of gene number differences in each sample. The sequencing depth achieved by the targeted capture was roughly 20 times higher than that of publicly available cercopithecid WGA databases, consolidating reliability of our sequencing results. Consistently, our targeted capture retrieved more intact TAS2R genes and characterized their intact/disrupted and duplication statuses better than WGA databases.
We designed probes for all TAS2R genes that were inferred to be intact at the common ancestor of cercopithecids (designated as “ancestral-cercopithecid TAS2R gene set”). By using this phylogeny-based approach, we aimed to depict the evolutionary trajectory of the gene set, i.e., which genes increased their numbers by gene duplications (gene birth) and which genes were disrupted or lost (gene death). Disrupted genes (pseudogenes) already present in the common ancestor of cercopithecid primates were not considered in this study. This was because they should remain inactive thereafter and be irrelevant to the evolution and diversification of taste receptor function in cercopithecid primates. Inclusion of such pseudogenes at the common ancestor of study species, which previous WGA-based studies have done (Li and Zhang 2014; Liu et al. 2016; Shi and Zhang 2006), could ambiguate the significance of the interspecies comparison of pseudogene numbers.
We were surprised to find intact TAS2R418 genes in our samples (anubis baboon, hamadryas baboon, and patas monkey). We did not include the TAS2R418 gene in our probe set because the TAS2R418 gene was previously found disrupted in the three cercopithecine and the two colobine WGA databases we used for designing probes (Hayakawa et al. 2014). It was our fortune that the gene was captured by other gene probes, possibly the TAS2R419 probe designed from rhesus macaque, for which the sequence difference was only 7–8% different from TASR418, well within the range of ~10% mismatch allowed between baits and targets. This supports Hayakawa et al’s (2014) hypothesis that it was intact (or at least a segregating pseudogene) in the last common ancestor of Cercopithicidae.
Li and Zhang (2014) reported that the number of TAS2R genes in vertebrates was positively correlated with the fraction of dietary plants by studying 54 WGA databases. This led to a general prediction that herbivores may need more TAS2R genes to recognize a larger number of bitter compounds than carnivores and omnivores to select the type of plants they eat (Li and Zhang 2014). This appears to be consistent with the large number of gene birth at the common ancestor of cercopithecids. As Hayakawa et al. (2014) and Toda et al. (2021) discussed, progressive increase in body size of ancestral cercopithecid primates during the Oligocene epoch may have been associated with a change of protein source from insects to leaves although they were not committed folivores (Kay et al. 1997; Williams et al. 2010).
While cercopithecid primates have maintained folivory as a part of their omnivorous diets, colobine primates are specialized for folivory as their main diet. However, opposed to the herbivory prediction, the number of intact TAS2R genes was overall smaller in folivorous colobines than in omnivorous cercopithecines. Glendinning (1994) showed that herbivorous mammals, especially browsers, which commonly encounter bitter and potentially poisonous foods, had a high bitter threshold (i.e., low bitter sensitivity) and high tolerance to ingest toxic compounds. This was explained as herbivores may have evolved detoxification mechanisms, such as the foregut fermentation by microbes in ruminants, to tolerate toxic compounds (Freeland and Janzen 1974). Colobine monkeys have also independently evolved a foregut-fermentation digestive system and use symbiont microorganisms to detoxify plant compounds (Bauchop and Martucci 1968). The foregut bacteria are digested by the specialized lysozyme expressed in the stomach to recover nutrients (Messier and Stewart 1997). The bacterial RNA molecules are degraded in the small intestine by the specialized RNASE1B to recycle nitrogen efficiently (Zhang et al. 2002). A fewer number of intact TAS2R genes in colobines than in cercopithecines appears to be congruent with the Glendinning’s (1994) observation. Previous research (Purba et al. 2017) identified four nonsynonymous nucleotide substitutions in the colobine TAS2R38 gene that were responsible for the decreased sensitivity of the TAS2R38 to phenylthiocarbamide (PTC). Furthermore, compared with macaque monkeys, colobines have lower sensitivities to PTC, which can be recognized by TAS2R38 in behavioral and in vitro functional analyses (Purba et al. 2020). These findings support the hypothesis that colobines have developed a tolerance to bitterness as an adaptation to their leaf-eating dietary specialization. Overall, our results show that the evolutionary change of number of intact TAS2R genes is a complex process including evolutionary acquisition of detoxication ability and refute a simple general prediction that herbivores need more TAS2R genes.
Our study has revealed far greater variation and complexity in the composition of TAS2R genes among primates. Indeed, we found that birth or death events occurred at almost every phylogenetic-tree branch in the species we studied. Such a discovery welcomes additional research to understand this dynamic evolutionary process. Bitter compounds have been identified for various TAS2Rs by heterologous expression of their genes in cultured cells and by behavioral tests (Itoigawa et al. 2021; Meyerhof et al. 2010; Purba et al. 2020; Tsutsui et al. 2016). Further clarification of the TAS2R gene repertoire, ligands of the receptors, and bitterness perception compared between cercopithecines and colobines and among species aligned with their distinct dietary strategies will further elucidate the evolution of bitter taste receptors in relation to feeding ecology.
Data availability
This is not applicable.
References
Adler E, Hoon MA, Mueller KL, Chandrashekar J, Ryba NJ, Zuker CS (2000) A novel family of mammalian taste receptors. Cell 100:693–702. https://doi.org/10.1016/s0092-8674(00)80705-9
Akhtar MS, Ashino R, Oota H, Ishida H, Niimura Y, Touhara K, Melin AD, Kawamura S (2022) Genetic variation of olfactory receptor gene family in a Japanese population. Anthropol Sci 130:93–106. https://doi.org/10.1537/ase.211024
Arbiza L, Zhong E, Keinan A (2012) NRE: a tool for exploring neutral loci in the human genome. BMC Bioinform 13:301. https://doi.org/10.1186/1471-2105-13-301
Bauchop T, Martucci RW (1968) Ruminant-like digestion of the langur monkey. Science 161:698–700. https://doi.org/10.1126/science.161.3842.698
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, McVean G, Durbin R, the 1000 Genomes Project Analysis Group (2011) The variant call format and VCFtools. Bioinformatics 27:2156–2158. https://doi.org/10.1093/bioinformatics/btr330
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43:491–498. https://doi.org/10.1038/ng.806
Dong D, Jones G, Zhang S (2009) Dynamic evolution of bitter taste receptor genes in vertebrates. BMC Evol Biol 9:12. https://doi.org/10.1186/1471-2148-9-12
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucl Acids Res 32:1792–1797. https://doi.org/10.1093/nar/gkh340
Edwards MS, Ullrey DE (1999) Effect of dietary fiber concentration on apparent digestibility and digesta passage in non-human primates. II. Hindgut- and foregut-fermenting folivores. Zoo Biol 18:537–549. https://doi.org/10.1002/(SICI)1098-2361(1999)18:6%3c537::AID-ZOO8%3e3.0.CO;2-F
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791. https://doi.org/10.1111/j.1558-5646.1985.tb00420.x
Fleagle JG (2013) Chapter 6 - old world monkeys. In: Fleagle JG (ed) Primate adaptation and evolution, 3rd edn. Academic Press, San Diego, pp 119–150. https://doi.org/10.1016/B978-0-12-378632-6.00006-9
Freeland WJ, Janzen DH (1974) Strategies in herbivory by mammals - role of plant secondary compounds. Am Nat 108:269–289. https://doi.org/10.1086/282907
Glendinning JI (1994) Is the bitter rejection response always adaptive? Physiol Behav 56:1217–1227. https://doi.org/10.1016/0031-9384(94)90369-7
Gnirke A, Melnikov A, Maguire J, Rogov P, LeProust EM, Brockman W, Fennell T, Giannoukos G, Fisher S, Russ C, Gabriel S, Jaffe DB, Lander ES, Nusbaum C (2009) Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol 27:182–189. https://doi.org/10.1038/nbt.1523
Guevara EE, Greene LK, Blanco MB, Farmer C, Ranaivonasy J, Ratsirarson J, Mahefarisoa KL, Rajaonarivelo T, Rakotondrainibe HH, Junge RE, Williams CV, Rambeloson E, Rasoanaivo HA, Rahalinarivo V, Andrianandrianina LH, Clayton JB, Rothman RS, Lawler RR, Bradley BJ, Yoder AD (2021a) Molecular adaptation to folivory and the conservation implications for Madagascar’s lemurs. Front Ecol Evol 9:736741. https://doi.org/10.3389/fevo.2021.736741
Guevara EE, Webster TH, Lawler RR, Bradley BJ, Greene LK, Ranaivonasy J, Ratsirarson J, Harris RA, Liu Y, Murali S, Raveendran M, Hughes DST, Muzny DM, Yoder AD, Worley KC, Rogers J (2021b) Comparative genomic analysis of sifakas (Propithecus) reveals selection for folivory and high heterozygosity despite endangered status. Sci Adv 7:eabd2274. https://doi.org/10.1126/sciadv.abd2274
Hallgren J, Tsirigos KD, Pedersen MD, Almagro Armenteros JJ, Marcatili P, Nielsen H, Krogh A, Winther O (2022) DeepTMHMM predicts alpha and beta transmembrane proteins using deep neural networks. bioRxiv. https://doi.org/10.1101/2022.04.08.487609
Hayakawa T, Suzuki-Hashido N, Matsui A, Go Y (2014) Frequent expansions of the bitter taste receptor gene repertoire during evolution of mammals in the Euarchontoglires clade. Mol Biol Evol 31:2018–2031. https://doi.org/10.1093/molbev/msu144
Hiwatashi T, Mikami A, Katsumura T, Suryobroto B, Perwitasari-Farajallah D, Malaivijitnond S, Siriaroonrat B, Oota H, Goto S, Kawamura S (2011) Gene conversion and purifying selection shape nucleotide variation in gibbon L/M opsin genes. BMC Evol Biol 11:312. https://doi.org/10.1186/1471-2148-11-312
Ishida T, Yamamoto K (1987) Survey of nonhuman primates for antibodies reactive with Epstein-Barr virus (EBV) antigens and susceptibility of their lymphocytes for immortalization with EBV. J Med Primatol 16:359–371. https://doi.org/10.1111/j.1600-0684.1987.tb00346.x
Itoigawa A, Fierro F, Chaney ME, Lauterbur ME, Hayakawa T, Tosi AJ, Niv MY, Imai H (2021) Lowered sensitivity of bitter taste receptors to β-glucosides in bamboo lemurs: an instance of parallel and adaptive functional decline in TAS2R16? Proc R Soc B 288:20210346. https://doi.org/10.1098/rspb.2021.0346
Jayakumar V, Nishimura O, Kadota M, Hirose N, Sano H, Murakawa Y, Yamamoto Y, Nakaya M, Tsukiyama T, Seita Y, Nakamura S, Kawai J, Sasaki E, Ema M, Kuraku S, Kawaji H, Sakakibara Y (2021) Chromosomal-scale de novo genome assemblies of Cynomolgus Macaque and Common Marmoset. Sci Data 8:159. https://doi.org/10.1038/s41597-021-00935-6
Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden TL (2008) NCBI BLAST: a better web interface. Nucleic Acids Res 36:W5-9. https://doi.org/10.1093/nar/gkn201
Kay RF, Ross C, Williams BA (1997) Anthropoid origins. Science 275:797–804. https://doi.org/10.1126/science.275.5301.797
Kinnamon SC, Cummings TA (1992) Chemosensory transduction mechanisms in taste. Ann Rev Physiol 54:715–731. https://doi.org/10.1146/annurev.ph.54.030192.003435
Kuderna LFK, Gao H, Janiak MC, Kuhlwilm M, Orkin JD, Bataillon T, Manu S, Valenzuela A, Bergman J, Rousselle M, Silva FE, Agueda L, Blanc J, Gut M, de Vries D, Goodhead I, Harris RA, Raveendran M, Jensen A, Chuma IS, Horvath JE, Hvilsom C, Juan D, Frandsen P, Schraiber JG, de Melo FR, Bertuol F, Byrne H, Sampaio I, Farias I, Valsecchi J, Messias M, da Silva MNF, Trivedi M, Rossi R, Hrbek T, Andriaholinirina N, Rabarivola CJ, Zaramody A, Jolly CJ, Phillips-Conroy J, Wilkerson G, Abee C, Simmons JH, Fernandez-Duque E, Kanthaswamy S, Shiferaw F, Wu D, Zhou L, Shao Y, Zhang G, Keyyu JD, Knauf S, Le MD, Lizano E, Merker S, Navarro A, Nadler T, Khor CC, Lee J, Tan P, Lim WK, Kitchener AC, Zinner D, Gut I, Melin AD, Guschanski K, Schierup MH, Beck RMD, Umapathy G, Roos C, Boubli JP, Rogers J, Farh KK, Marques BT (2023) A global catalog of whole-genome diversity from 233 primate species. Science 380:906–913. https://doi.org/10.1126/science.abn7829
Kumar S, Stecher G, Suleski M, Hedges SB (2017) TimeTree: a resource for timelines, timetrees, and divergence times. Mol Biol Evol 34:1812–1819. https://doi.org/10.1093/molbev/msx116
Li H (2011) A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27:2987–2993. https://doi.org/10.1093/bioinformatics/btr509
Li D, Zhang J (2014) Diet shapes the evolution of the vertebrate bitter taste receptor gene repertoire. Mol Biol Evol 31:303–309. https://doi.org/10.1093/molbev/mst219
Li J, Han K, Xing J, Kim H-S, Rogers J, Ryder OA, Disotell T, Yue B, Batzer MA (2009) Phylogeny of the macaques (Cercopithecidae: Macaca) based on Alu elements. Gene 448:242–249. https://doi.org/10.1016/j.gene.2009.05.013
Lindemann B (1996) Taste reception. Physiol Rev 76:719–766. https://doi.org/10.1152/physrev.1996.76.3.719
Liu Z, Liu G, Hailer F, Orozco-terWengel P, Tan X, Tian J, Yan Z, Zhang B, Li M (2016) Dietary specialization drives multiple independent losses and gains in the bitter taste gene repertoire of Laurasiatherian Mammals. Front Zool 13:28. https://doi.org/10.1186/s12983-016-0161-1
Liu Z, Zhang L, Yan Z, Ren Z, Han F, Tan X, Xiang Z, Dong F, Yang Z, Liu G, Wang Z, Zhang J, Que T, Tang C, Li Y, Wang S, Wu J, Li L, Huang C, Roos C, Li M (2020) Genomic mechanisms of physiological and morphological adaptations of limestone langurs to karst habitats. Mol Biol Evol 37:952–968. https://doi.org/10.1093/molbev/msz301
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303. https://doi.org/10.1101/gr.107524.110
Messier W, Stewart CB (1997) Episodic adaptive evolution of primate lysozymes. Nature 385:151–154. https://doi.org/10.1038/385151a0
Meyerhof W, Batram C, Kuhn C, Brockhoff A, Chudoba E, Bufe B, Appendino G, Behrens M (2010) The molecular receptive ranges of human TAS2R bitter taste receptors. Chem Senses 35:157–170. https://doi.org/10.1093/chemse/bjp092
Miura H, Barlow LA (2010) Taste bud regeneration and the search for taste progenitor cells. Arch Ital Biol 148:107–118
NCBI Resource Coordinators (2016) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 44:D7-19. https://doi.org/10.1093/nar/gkv1290
Nei M, Niimura Y, Nozawa M (2008) The evolution of animal chemosensory receptor gene repertoires: roles of chance and necessity. Nat Rev Genet 9:951–963. https://doi.org/10.1038/nrg2480
Palesch D, Bosinger SE, Tharp GK, Vanderford TH, Paiardini M, Chahroudi A, Johnson ZP, Kirchhoff F, Hahn BH, Norgren RB, Patel NB, Sodora DL, Dawoud RA, Stewart CB, Seepo SM, Harris RA, Liu Y, Raveendran M, Han Y, English A, Thomas GWC, Hahn MW, Pipes L, Mason CE, Muzny DM, Gibbs RA, Sauter D, Worley K, Rogers J, Silvestri G (2018) Sooty mangabey genome sequence provides insight into AIDS resistance in a natural SIV host. Nature 553:77–81. https://doi.org/10.1038/nature25140
Perelman P, Johnson WE, Roos C, Seuánez HN, Horvath JE, Moreira MAM, Kessing B, Pontius J, Roelke M, Rumpler Y, Schneider MPC, Silva A, O’Brien SJ, Pecon-Slattery J (2011) A molecular phylogeny of living primates. PLOS Genet 7:e1001342. https://doi.org/10.1371/journal.pgen.1001342
Purba LH, Widayati KA, Tsutsui K, Suzuki-Hashido N, Hayakawa T, Nila S, Suryobroto B, Imai H (2017) Functional characterization of the TAS2R38 bitter taste receptor for phenylthiocarbamide in colobine monkeys. Biol Lett. https://doi.org/10.1098/rsbl.2016.0834
Purba LHPS, Widayati KA, Suzuki-Hashido N, Itoigawa A, Hayakawa T, Nila S, Juliandi B, Suryobroto B, Imai H (2020) Evolution of the bitter taste receptor TAS2R38 in colobines. Primates 61:485–494. https://doi.org/10.1007/s10329-020-00799-1
Rice P, Longden I, Bleasby A (2000) EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 16:276–277. https://doi.org/10.1016/s0168-9525(00)02024-2
Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP (2011) Integrative genomics viewer. Nat Biotechnol 29:24–26. https://doi.org/10.1038/nbt.1754
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425. https://doi.org/10.1093/oxfordjournals.molbev.a040454
Schmieder R, Edwards R (2011) Quality control and preprocessing of metagenomic datasets. Bioinformatics 27:863–864. https://doi.org/10.1093/bioinformatics/btr026
Sène MA, Kiesslich S, Djambazian H, Ragoussis J, Xia Y, Kamen AA (2021) Haplotype-resolved de novo assembly of the Vero cell line genome. NPJ Vaccines 6:106. https://doi.org/10.1038/s41541-021-00358-9
Shao Y, Zhou L, Li F, Zhao L, Zhang BL, Shao F, Chen JW, Chen CY, Bi X, Zhuang XL, Zhu HL, Hu J, Sun Z, Li X, Wang D, Rivas-González I, Wang S, Wang YM, Chen W, Li G, Lu HM, Liu Y, Kuderna LFK, Farh KK, Fan PF, Yu L, Li M, Liu ZJ, Tiley GP, Yoder AD, Roos C, Hayakawa T, Marques-Bonet T, Rogers J, Stenson PD, Cooper DN, Schierup MH, Yao YG, Zhang YP, Wang W, Qi XG, Zhang G, Wu DD (2023) Phylogenomic analyses provide insights into primate evolution. Science 380:913–924. https://doi.org/10.1126/science.abn6919
Shi P, Zhang J (2006) Contrasting modes of evolution between vertebrate sweet/umami receptor genes and bitter receptor genes. Mol Biol Evol 23:292–300. https://doi.org/10.1093/molbev/msj028
Simons ND, Eick GN, Ruiz-Lopez MJ, Hyeroba D, Omeja PA, Weny G, Zheng H, Shankar A, Frost SDW, Jones JH, Chapman CA, Switzer WM, Goldberg TL, Sterner KN, Ting N (2019) Genome-wide patterns of gene expression in a wild primate indicate species-specific mechanisms associated with tolerance to natural simian immunodeficiency virus infection. Genome Biol Evol 11:1630–1643. https://doi.org/10.1093/gbe/evz099
Tamura K, Nei M (1993) Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 10:512–526. https://doi.org/10.1093/oxfordjournals.molbev.a040023
Tamura K, Stecher G, Kumar S (2021) MEGA11: molecular evolutionary genetics analysis version 11. Mol Biol Evol 38:3022–3027. https://doi.org/10.1093/molbev/msab120
Toda Y, Hayakawa T, Itoigawa A, Kurihara Y, Nakagita T, Hayashi M, Ashino R, Melin AD, Ishimaru Y, Kawamura S, Imai H, Misaka T (2021) Evolution of the primate glutamate taste sensor from a nucleotide sensor. Curr Biol 31:4641-4649.e4645. https://doi.org/10.1016/j.cub.2021.08.002
Tsutsui K, Otoh M, Sakurai K, Suzuki-Hashido N, Hayakawa T, Misaka T, Ishimaru Y, Aureli F, Melin AD, Kawamura S, Imai H (2016) Variation in ligand responses of the bitter taste receptors TAS2R1 and TAS2R4 among New World monkeys. BMC Evol Biol 16:208. https://doi.org/10.1186/s12862-016-0783-0
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, Banks E, Garimella KV, Altshuler D, Gabriel S, DePristo MA (2013) From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinform. https://doi.org/10.1002/0471250953.bi1110s43
Wall JD, Robinson JA, Cox LA (2022) High-resolution estimates of crossover and noncrossover recombination from a captive baboon colony. Genome Biol Evol 14:evac040. https://doi.org/10.1093/gbe/evac040
Wang L, Wu J, Liu X, Di D, Liang Y, Feng Y, Zhang S, Li B, Qi XG (2019) A high-quality genome assembly for the endangered golden snub-nosed monkey (Rhinopithecus roxellana). GigaScience 8:giz098. https://doi.org/10.1093/gigascience/giz098
Warren WC, Harris RA, Haukness M, Fiddes IT, Murali SC, Fernandes J, Dishuck PC, Storer JM, Raveendran M, Hillier LW, Porubsky D, Mao Y, Gordon D, Vollger MR, Lewis AP, Munson KM, DeVogelaere E, Armstrong J, Diekhans M, Walker JA, Tomlinson C, Graves-Lindsay TA, Kremitzki M, Salama SR, Audano PA, Escalona M, Maurer NW, Antonacci F, Mercuri L, Maggiolini FAM, Catacchio CR, Underwood JG, O’Connor DH, Sanders AD, Korbel JO, Ferguson B, Kubisch HM, Picker L, Kalin NH, Rosene D, Levine J, Abbott DH, Gray SB, Sanchez MM, Kovacs-Balint ZA, Kemnitz JW, Thomasy SM, Roberts JA, Kinnally EL, Capitanio JP, Skene JHP, Platt M, Cole SA, Green RE, Ventura M, Wiseman RW, Paten B, Batzer MA, Rogers J, Eichler EE (2020) Sequence diversity analyses of an improved rhesus macaque genome enhance its biomedical utility. Science. https://doi.org/10.1126/science.abc6617
Williams BA, Kay RF, Kirk EC (2010) New perspectives on anthropoid origins. Proc Natl Acad Sci USA 107:4797–4804. https://doi.org/10.1073/pnas.0908320107
Zhang JZ, Zhang YP, Rosenberg HF (2002) Adaptive evolution of a duplicated pancreatic ribonuclease gene in a leaf-eating monkey. Nat Genet 30:411–415. https://doi.org/10.1038/Ng852
Zhang RS, Zhou C, Jin XL, Liu KH, Fan ZX, Xing JC, Li J (2022) Chromosome-level genome assembly of Tibetan macaque (Macaca thibetana) and species-specific structural variations. Zool Res 43:880–885. https://doi.org/10.24272/j.issn.2095-8137.2022.149
Zheng-Bradley X, Streeter I, Fairley S, Richardson D, Clarke L, Flicek P, the 1000 Genomes Project Consortium (2017) Alignment of 1000 Genomes Project reads to reference assembly GRCh38. GigaScience 6:1–8. https://doi.org/10.1093/gigascience/gix038
Zhou X, Wang B, Pan Q, Zhang J, Kumar S, Sun X, Liu Z, Pan H, Lin Y, Liu G, Zhan W, Li M, Ren B, Ma X, Ruan H, Cheng C, Wang D, Shi F, Hui Y, Tao Y, Zhang C, Zhu P, Xiang Z, Jiang W, Chang J, Wang H, Cao Z, Jiang Z, Li B, Yang G, Roos C, Garber PA, Bruford MW, Li R, Li M (2014) Whole-genome sequencing of the snub-nosed monkey provides insights into folivory and evolutionary history. Nat Genet 46:1303–1310. https://doi.org/10.1038/ng.3137
Zhou Y, Zhan X, Jin J, Zhou L, Bergman J, Li X, Rousselle MMC, Belles MR, Zhao L, Fang M, Chen J, Fang Q, Kuderna L, Marques-Bonet T, Kitayama H, Hayakawa T, Yao YG, Yang H, Cooper DN, Qi X, Wu DD, Schierup MH, Zhang G (2023) Eighty million years of rapid evolution of the primate Y chromosome. Nat Ecol Evol 7:1114–1130. https://doi.org/10.1038/s41559-022-01974-x
Zimin AV, Cornish AS, Maudhoo MD, Gibbs RM, Zhang X, Pandey S, Meehan DT, Wipfler K, Bosinger SE, Johnson ZP, Tharp GK, Marçais G, Roberts M, Ferguson B, Fox HS, Treangen T, Salzberg SL, Yorke JA, Norgren RB Jr (2014) A new rhesus macaque assembly and annotation for next-generation sequencing analyses. Biol Direct 9:20. https://doi.org/10.1186/1745-6150-9-20
Acknowledgements
We thank the following organizations and researches for making the WGA data available: Genome Institute at Washington University for Macaca_fascicularis_5.0, Baylor College of Medicine Human Genome Sequencing Center for Mnem_1.0, Mleu.le_1.0l, Cercopithecus_mitis_HiC, and Cang.pa_1.0, Broad Institute for SemEnt_v1_BIUU and NasLar_v1_BIUU, Dr. Yasuhiro Go for macFus_1.0, Dr. Tyler Scott Alioto for mMacNir1.1, and Dr. Yaping Zhang and Dr. Yu Li for ASM169854v1.
Funding
Open Access funding provided by The University of Tokyo. This article is funded by Japan Society for the Promotion of Science, 23H02561, Shoji Kawamura, 18H04005, Shoji Kawamura, 15H02421, Shoji Kawamura, 23405016, Akiko Matsumoto-Oda, 22H02674, Hiroo Imai, Canada Research Chairs, 950-231257, Amanda D. Melin, National Sciences and Engineering Research Council of Canada, RGPIN-2017-03782, Amanda D. Melin, Cooperative Research Program of the Primate Research Institute of Kyoto University grants, 2021-A-25, Shoji Kawamura, 2020-A-27, Shoji Kawamura, 2012-A-11, Shoji Kawamura.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Hou, M., Akhtar, M.S., Hayashi, M. et al. Reduction of bitter taste receptor gene family in folivorous colobine primates relative to omnivorous cercopithecine primates. Primates (2024). https://doi.org/10.1007/s10329-024-01124-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10329-024-01124-w