
Abstract
Chronic lymphocytic leukemia (CLL) is a heterogeneous disease with alterations in genetic expression and epigenetic modifications. In recent years, the new insight into epigenetics in the pathogenesis of CLL has been developed considerably, including DNA methylation, histone modification, RNA methylation, non-coding RNAs as well as chromatin remodeling. Epigenetic modification regulates various processes such as stem cell biology, cell growth, and tumorigenesis without altering gene sequence. Growing evidence indicates that the disturbance of gene expression profiles which were regulated by epigenetic modifications exerts vital roles in the development and progress in CLL, which provides novel perspectives to explore the etiology of CLL. In addition, the integration with epigenetic therapeutic targets and the in-depth understanding of epigenetic therapy contribute to develop new therapeutic strategies for CLL. Herein, the present review discusses the advances of epigenetic alterations in the pathogenesis, diagnosis, and prognostic assessment of CLL patients and also highlights existing and emerging agents targeting epigenetic regulators.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chronic lymphocytic leukemia (CLL) is the main type of adult leukemia across western countries, which occurs in the elderly for the most part [1,2,3]. The CLL clinical course ranges from months to several years, which makes it a highly heterogeneous disease [4]. In more than 80% of CLL patients, genomic aberrations were detected. These chromosomal aberrations include 13q, 11q and 17p deletions, and trisomy 12 [5]. Among them, the 13q14 deletion is the most frequent abnormality [5]. Although there have been many studies on the pathogenesis of CLL and great achievements have been made in various aspects, relapse/refractory cases and drug resistance are still existing problems.
Epigenetics refers to the heritable changes in gene function with no alterations in DNA sequences, consisting of DNA methylation, histone modification, nucleosome remodeling, and so on [6]. Epigenetic modifications convey the information that plays a key role in regulating DNA-based processes [6]. During the past few decades, epigenetic modifications were found playing significant roles in the occurrence and development of leukemia by some studies and were considered a promising target for treating different types of leukemia and other hematological malignancies [7], and at the same time, it has achieved good clinical effects [8]. For example, the anti-tumor drugs azacitidine and decitabine based on inhibiting DNA methylation have significant effects in treating myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) [9].
Epigenetic researches have also made great progress in CLL, and these promote our comprehension of the pathogenesis of CLL and provide further prospects for diagnosis and treatment strategies. In the present review, we summarize recent advances of CLL in epigenetics such as DNA methylation, histone modification, RNA methylation, non-coding RNAs, and chromatin remodeling and also highlight existing and emerging drugs targeting epigenetic regulators.
DNA methylation in CLL
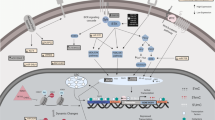
DNA methylation is a form of chemical modification of DNA in which methyl groups are added to the C5 position of the cytosine with s-adenosylmethionine (SAM) as a methyl donor to form 5-methylcytosine under the action of DNA methyltransferases (DNMTs; Fig. 1). DNMT family of enzymes catalyze the transfer of a methyl group from SAM to DNA; three members of which have been reported possess methyltransferase activity in mammals: DNMT1, DNMT3a, and DNMT3b. Cytosine methylation is the most widely studied DNA methylation in humans.

Mechanisms of DNA methylation and histone modification regulating gene expression in chronic lymphocytic leukemia (CLL)
As an essential epigenetic modification, aberrant DNA methylation has been found to be associated with an increasing number of diseases by participating in lots of cellular processes [10]. For example, increased p16INK4a silencing due to DNA methylation affects cell senescence, aging, and cell cycle progression, leading to unlimited cell proliferation [11] in solid tumors such as hepatocellular carcinoma, lung cancer [12], and cervical cancer [13]. Studies suggest that silencing of cell adhesion regulators CDH1 (E-calmodulin) and CDH13 (H-calmodulin) induced by DNA methylation may promote tumor invasion and metastasis [14, 15]. In gastric and colon cancer, suppression of mismatch repair factor MLH1 expression by DNA methylation is related to genomic instability [16, 17]. DNA methylation changes in tumor usually have two aspects: global hypomethylation influencing retroviral elements and the stability of genome, and focal hypermethylation of tumor suppressor gene promoters [18].
Global analysis of DNA methylation in CLL
Compared with healthy control, the DNA of peripheral blood mononuclear cells from patients with CLL is globally hypomethylated. Furthermore, increased mutations and genomic instability are related to less DNA methylation in genome [19]. The emergence of methyl-CpG-binding domain protein-enriched genome-wide sequencing (MBD-Seq) makes it possible to analyze methylated CpG-rich regions in the whole genome. For example, a study displayed the whole methylome of high CpG-rich regions on the basis of MBD-Seq. Compared to normal controls, 5800 hypermethylated and 12,570 hypomethylated CLL-specific differentially methylated genes were identified. Interestingly, non-coding RNA contains 40% hypermethylated genes and 60% hypomethylated genes. In addition, CpG island methylation can influence CLL based on the level of CD38 expression according to a large-scale analysis of DNA methylation [20]. Moreover, for IGHV subgroups grouping by IGHV mutational status, the proportion of common repetitions such as short interspersed elements and long interspersed elements is considerable in CLL-specific differentially methylated regions [21]. Conversely, global DNA methylation/demethylation levels assessment can improve CLL outcome prediction in patients particularly with del (13q) combined 5-methylcytosine (5-mCyt) and conventional cytogenetic approach [22].
In a population-based case–control study, the genome-wide DNA methylation of 48 CLL cases and 28 healthy controls was analyzed using the Infinium HumanMethylation450 BeadChip. In total, 34,797 differentially methylated positions (DMPs) associated with the CLL genome were identified, most of which were hypomethylated and located in gene body regions. Among them, the methylation of ZAP70, FMOD, and ADAMTS17 was significantly different between CLL cases and controls [23]. Their roles in the pathogenesis of CLL deserve further exploration.
As one of the most common mutated genes in CLL, mutations of splicing factor 3b subunit 1 (SF3B1) are associated with adverse prognosis [24]. The study to explore the connection between methylome changes and SF3B1 mutation showed that the methylation levels in 67 genomic regions in CLL patients with SF3B1 mutation were localized declined, mainly near telomeric regions [25].
Hypermethylation and hypomethylation of single-gene promoters in CLL
In CLL, tumor suppressor genes (TSGs) expression is usually silenced by DNA hypermethylation. Wnt signaling is essential for the development of normal B-cell and has been proven to control normal apoptotic process, while abnormal activation of this pathway has been noticed in CLL [26]. Secreted frizzled-related protein 4 (SFRP4) is a member of secreted frizzled-related proteins (SFRPs) family and plays a negative regulatory role in the Wnt signaling pathway, which was observed to be often methylated in samples of CLL. Additionally, silencing of SFRP through CpG island methylation in CLL may activate the Wnt signaling pathway abnormally [27, 28]. In primary CLL samples, abnormal DNA methylation and silencing happened in SFRP4 and other SFRP family members. Through a detailed study of five SFRP family members, SFRP1 was found to be hypermethylated and down-regulated in all samples collecting from CLL patients, indicating that this epigenetic event is a key step in the development of leukemia [29]. What’s more, miR-34b/c, a type of non-coding RNA, was demonstrated as tumor suppressors, the promoter of which is abnormally hypermethylated [30]. TWIST2 is a transcription factor, whose expression is associated with the promoter methylation degree. After the treatment of decitabine, the expression of TWIST2 was increased in a CLL cell line whose promoter is methylated. Studies on 53 CLL patients showed that 72% samples from patients with mutated IGHV demonstrated TWIST2 methylation, while only 16% samples from patients with unmutated IGHV were methylated [31].
In addition to hypermethylation of tumor suppressor gene promoters, hypomethylation of oncogene promoters is also of significance in the pathogenesis of CLL. Lipoprotein lipase (LPL) plays a crucial role in pathways associated with fatty acid degradation and signaling in CLL, which might affect the behavior of CLL cells [32]. The overexpression of LPL mRNA is showed to be related to unmutated CLL status and poor clinical outcomes [33]. The reason for the abnormal expression of LPL in unmutated CLL is the demethylation of the LPL gene [34]. In mammalian cells, DNA methyltransferase 3A (DNMT3A) and DNA methyltransferase 3B (DNMT3B), which were members of DNMT3 family, are responsible for establishing DNA methylation dynamically [35]. In CLL, DNMT3A down-regulation was common. A B-cell-restricted Dnmt3a knockout mouse model demonstrated that loss of DNMT3A expression was able to drive the development of CLL and was related to aggressive disease, Notch and Myc signaling activation, and Notch inhibition sensitivity enhancement [36].
DNA methylation profiles in different CLL subgroups
Even in CLL patients with analogous somatic hypermutation status, the DNA methylation profile may be different. Integrated analysis of DNA methylation identified that CLL stereotyped subset #8 (IGHV4-39/IGKV1(D)-39) demonstrated a unique DNA methylome compared with the other U-CLL cases, including subset #6 (IGHV1-69/IGKV3-20) and the hypomethylated and overexpressed TP63 gene becomes a pro-survival factor [37]. Through the whole-epigenome analysis of DNA methylation of CLL patients for the duration of treatment, it was found that enrichment for diverse CLL-specific epigenetic traits responded to chemotherapy that predict patient clinical outcomes, and especially involve epigenetic silencing of HOXA4 in decreasing the therapeutic sensitivity of CLL cells [38]. By comparing the DNA methylation groups of 139 CLL patients with mutated or unmutated IGHV and a couple of mature B-cell subpopulations it can be found that the two subtypes of CLL have different DNA methylation profiles that appear to represent epigenetic imprints from different normal B-cell subpopulations. The most common difference between normal B-cells and the two subtypes of CLL, and between naive B-cells and memory B-cells, is the hypomethylation of DNA in the genome that mainly targets enhancer sites [39]. Additionally, epigenetic burden and recurrent changes are correlated with specific clinical and biological characteristics according to a study of DNA methylation patterns in paired pre-treatment/relapse specimens of 34 CLL patients receiving chemoimmunotherapy, which suggests that DNA methylation responds differently to chemoimmunotherapy in patients with CLL [40].
Histone modification
Histones and DNA together form nucleosomes, which are the main components of chromatin. Histones are proteins with highly conserved sequences in the nucleus, including 5 kinds of H1, H3, H2A, H2B, and H4. The amino acid residues on the amino terminal peptide chain of the core histones can be covalently modified under the modification of the addition or removal of already-existing methyl, acetyl, or phosphate groups by a variety of histone appearance modifying enzymes and the types of modification include methylation, acylation, phosphorylation, and others [41] (Fig. 1). Histone methylation mainly occurs in lysine and arginine residues, and the process is catalyzed by protein arginine N-methyltransferases (PRMTs) and histone lysine N-methyltransferases [42]. The histone acetylation of lysine has been shown highly dynamic and regulated by the opposite effects of two enzyme families, histone acetyltransferases (HATs) and histone deacetylases (HDACs). HAT utilizes acetyl CoA as a cofactor to catalyze the transfer of acetyl groups to the lysine side chain ε-amino group. HDAC enzyme antagonizes HAT and reverses lysine acetylation, which stabilizes the local chromatin structure [43].
Histone modifications alter the transcriptional state of chromatin and transform it into euchromatin with higher transcriptional activity or heterochromatin with lower transcriptional activity. Thus, histone modification affects the development of related diseases by regulating gene transcription and translation. In the CD19 + B-cells of CLL patients, the overexpression of SIRT1 and EZH2, global histone H3/H4 hypoacetylation, and H3 K9 hypermethylation were detected, which indicated that the abnormal histone modification played key roles in the pathogenesis of CLL [44].
Histone methylation
Histone methyltransferases (HMTs) are essential in regulating gene transcription, which can transfer methyl groups to histone proteins from SAM [45]. In solid and hematological malignancies, HMT is destroyed by mechanisms such as chromosomal translocations, genome loss, and/or point mutations [46]. Many HMT aberrations were found in human malignancies. Among them, the repeated deletion and/or inactivating mutations of SETD2, a cancer suppressor gene, originally found in renal clear cell carcinoma [47]. SETD2 is able to catalyze the trimethylation of lysine 36 on histone 3 (H3K36me3), which is one of the main chromatin marks related to active transcription. Evidence supporting the tumor suppressor effect of SETD2 is that its deletion impairs DNA repair and enhances genome precariousness [48, 49]. SETD2 abnormity is a frequent, early loss-of-function event associated with aggressive disease in CLL pathology. Through high-resolution single nucleotide polymorphism (SNP) arrays, repeated loss of the SETD2 locus was identified in 3% of CLL patients. The loss of SETD2 was related to TP53 deletion, genomic complexity, and chromothripsis. In five chemotherapeutic or chemoimmunotherapeutic clinical trials, compared with cases whose three genes are wild type, patients with SETD2 aberrations and wild-type TP53 and ATM had poorer progression-free survival (FPS) and overall survival (OS) [50].
Enhancer of zeste homolog 2 (EZH2) is a human homolog of Drosophila zeste gene enhancer 2, which mainly inhibits the activity of target genes or silences target genes directly through histone modification, thus regulating cell senescence, differentiation, tumorigenesis, and development [51]. Increased expression level of EZH2 relates to an unfavorable prognosis in CLL [52]. A study demonstrated the elevated expression of EZH2, c-Myc, E2F1, and pRb proteins and decreased miR-26a expression in the proliferation centers (PCs) of CLL/small lymphocytic lymphoma (SLL) [53]. A large prospective CLL trial cohort showed that elevated KDM1A and associated gene expression signatures related to aggressive disease and dismal prognosis. Integrated analyses of differential global transcriptomes and H3K4me3 marks in Eµ-TCL1A vs. iKdm1aKD; Eµ-TCL1A mice implied KDM1A as an oncogenic transcriptional repressor in CLL by altering histone methylation patterns with obvious effects on defined cell death and motility pathways [54].
Histone acetylation
Histone acetylation is a reversible dynamic equilibrium process, which is mediated by two enzymes, histone acetyltransferase (HAT) and histone deacetylase (HDAC). Generally, histone acetylation is related to gene expression activation, while histone deacetylation tends to down-regulate gene expression. In CLL leukemia cells, the E-cadherin gene is hypoacetylated. The transcription of this silent gene can be activated by treating with histone deacetylase inhibitor (HDACi) MS-275. Compared with the aberrant exon11 skipped transcripts, the more rightly spliced E-cadherin transcripts expressed by activated genes can inhibit the Wnt signaling pathway [55]. Histone acetylation and other epigenetic modifications jointly promote the occurrence and development of CLL.
RNA methylation
RNA methylation is of great significance in regulating gene expression. N6-methyladenosine (m6A) RNA methylation modification is the main type of mRNA modification [56]. M6A methylation involves methyltransferases (writers) and m6A-binding proteins (readers), which can be reversed by demethylases (erasers; Fig. 2). Methyltransferases mainly include methyltransferase-like 3 (METTL3), methyltransferase-like 14 (METTL14), Wilms tumor 1-associated protein (WTAP), and methyltransferase-like 16 (METTL16) which mediates the process of RNA methylation. The METL3/METTL14/WTAP complex is directly engaged in the methylation regulation of nuclear splicing-related organelles [57]. METTL16 regulates the activity of RNA spliceosome by regulating the methylation level of small nuclear RNA (snRNA) in nucleus [58]. Demethylases include fat mass and obesity-associated protein (FTO) and alkylation repair homolog 5 (ALKBH5), which mediate the procedures of RNA demethylation. Demethylases can directly locate to nuclear speckles, which are rich in splicing factors and closely related to the splicing process of mRNA, and participate in the regulation of methylation. The most common m6A-binding proteins are the YTH domain-containing family 1–3 (YTHDF1-3) and YTH domain-containing protein 1–2 (YTHDC1-2), which can recognize the RNA methylation modification and take part in downstream RNA translation, degradation, splicing, and other processes [59]. Evidence showed that m6A modification is associated with tumorigenesis, tumor multiplication, aggression, and metastases [60, 61].

The mechanism of m6A RNA methylation in eukaryotic cells
There was evidence that FTO, a m6A mRNA demethylase, contributes to the development and progress of AML [62, 63]. Studies have demonstrated that FTO expression was upregulated in CLL patients and was related to a poor prognosis. Moreover, FTO accelerates the survival of CLL cells through DNA damage pathway. A novel inhibitor selectively targeting FTO, FB23-2, has an effective therapeutic potential in eliminating cell survival and inducing cell cycle arrest through m6A methylation [64].
Non-coding RNA
MicroRNA
MicroRNAs (miRNAs) can regulate post-transcriptional silencing of target genes which are short RNA molecules with a size of 19–25 nucleotides. Additionally, a single miRNA usually affects the expression of lots of genes by involving in a functional interacting pathway [65]. The expression and functional characterization of these miRNAs are listed in Table 1.
The loss of miR-15/16 gene on chromosome 13q14 is the most common alteration in CLL. In a small region of chromosome 13q14, miR15 and miR16 locate at the translocation breakpoint that is deleted in more than 65% of CLL. What’s more, in this region, allelic loss is associated with down-regulation of miR-15/16 expression which demonstrate that allelic loss leads to the inactivation of these genes in CLL [66]. Remarkably, the first 9 nucleotides in the 5′-ends of miR-15/16 and bases 3287–3279 in the 3′-end of the BCL2 cDNA are complementary sequences, which was found overexpressed in nearly all CLL patients [67]. Much more, the interaction between these miRNAs and BCL2 is direct [68]. The mechanism by which miR-15/16 regulates the expression of BCL2 and promotes apoptosis is shown in Fig. 3.

The mechanisms of miR-15 and miR-16 regulate cell apoptosis by targeting BCL2 in CLL
MiR-34b and miR-34c at chromosome 11q negatively regulate protein synthesis. Furthermore, the clinical process of the cases with deletion of chromosome 11q usually showed aggressive. The miR-34b/c promoter region was found to have high frequency of abnormal hypermethylation, which was highly in relation to the level of miR-34b/c and the deletion of 11q, confirming miR-34b/c has become a new potential anti-oncogene on chromosome 11q23 [30, 69].
MiR-155 as a critical regulator involved in the B-cells post-transcriptional gene expression is evolutionarily conserved encoded in a region which is called the B-cell integration cluster (BIC, miR155HG) [70]. The high-level miR-155 expression can enhance the sensitivity to BCR ligation. Besides, the cross-talk of the tissue environment can induce the overexpression of miR-155. These findings potentially contribute to connect miR-155 with adverse clinical outcomes in CLL patients [71, 72].
There are also some other miRNAs reported in the past decade, such as miR-29 (proved to have an anti-tumor effect by targeting TRAF4 which can be affected by BCR inhibitors and selectively delivered to CLL cells [73, 74]), miR-125a-5p/miR-34a-5p (as valuable markers to predict Richter syndrome (RS) development in CLL patients [75]), and miR-150 (much abundant expressed in CLL and attached to disease outcome via GAB1 and FOXP1 [76]). A recent research based on forty newly diagnosed CLL patients shows that miRNAs might be the prognostic biomarkers in the process of CLL and endpoint predictors in this disease. Some researchers also found some CLL-delivered exosomes internalized by stromal cells can deliver miRNAs, which can induce an inflammatory phenotype in target cells similar to that of cancer-associated fibroblasts [77].
Long non-coding RNA
Long non-coding RNA (lncRNA) is a kind of RNA with a length of > 200 nucleotides, which is least or no potential possibility to encode proteins [78]. Deletion of chromosome 13q region is considered to be the most common chromosomal abnormal observed in CLL which contains two lncRNAs (DLEU1 and DLEU2). The former is known as the host gene for miR-15 and miR-16 [79]. Thus, deletion of 13q14 leads to inactivation of both collaborating tumor suppressor genes, DLEU7 and miR-15/16. MiR-15/16 and DLEU7 inactivation, respectively, lead to increased BCL2 expression and induction of TNF signaling by TRAFs [80]. There are some lncRNA candidates examined by several studies listed in Table 1, which contains an overview of the functional characterization of these lncRNAs across CLL [81,82,83,84,85].
The association between the p53 and lincRNA-p21
Studies have shown that the deletion of P53, which is related to adverse prognosis in CLL patients, can regulate the expression of some lncRNA. A study showed that in primary CLL cells with wild-type TP53, lincRNA-p21 was up-regulated after radiation, resulting in decreased cell viability, while cells with TP53 mutations or deletions lack this mechanism [86, 87]. The transcription of lincRNA-p21 can be activated by p53 through binding the promoter of lincRNA-p21, and the lincRNA-p21 can also regulate the activity of p53 [88, 89].
Circular RNA
As important members of the gene regulatory environment, circular RNAs (circRNAs) are another kind of non-coding RNA. CircRNA-miR combination has been demonstrated to regulate transcriptome distribution and cell function in physiological and pathological processes; however, the role of circRNAs in cells remains to be explored [90]. To date, five main circRNAs circ-RPL15 [91], circ-CBFB [92], circ_0132266 [93], circZNF91 [94], and circ_0002078 [95], as well as mitochondrial genome-derived (mt)-circ-RNAs mc-COX2 [96] have been focused in CLL (Table 1).
For CLL patients without IGHV mutation, CircRPL15 is considered as a latent biomarker for the diagnosis in plasma [91]. The high expression level of circRPL15 is supposed to increase RAF1 protein levels by sponging miR-146b-3p. In proliferative RAS pathway, RAF1 is an effector which can promote cell growth by phosphorylating and activating mitogen-activated protein kinase (MAPK) signal. [97]. Through the research of its mechanism, it was found that circ-CBFB-activated Wnt/ β-Catenin pathway inhibits the production of Wnt receptor frizzled 3 (FZD3). Circ_00002078 was found highly expressed in CLL and can inhibit cell apoptosis, promote cell proliferation and cell cycle arrest through miR-185-3p/TCF7L1 axis [95]. Besides, recently a study showed that the expression of mc-cox2 in mitochondria is up-regulated in plasma and exosomes of CLL patients and may be involved in disease progression [96].
Chromatin remodeling
Chromatin remodelers consist of four families: SWI/SNF (SWItch/Sucrose Non-Fermentable), ISWI (Imitation of SWItch), CHD (Chromodomain Helicase DNA binding), and INO (INOsitol), which are subdivided by the core ATPase subunit and involved in many pathological processes in CLL. A couple of mutations involving chromatin remodeling have been found to affect the ARID1A and CHD2 genes in hematological malignancies, including CLL [98].
ARID1A is a tumor suppressor, which can contribute to the SWI/SNF chromatin remodeling complex formation and has been indicated to interact directly with p53 [99]. In addition, CHD2 is a member of the SNF2-adenosine triphosphate (ATP-dependent) chromatin remodeling factor CHD family. CHD2 is a complex multi-domain protein consisting of n-terminal tandem chromogenic domains (chromatin tissue modification domains) followed by DEXDc (death-like spirase superfamily) domains and HELIC (C-terminal of the spirase superfamily) domains, both spanning SNF2 (N-terminal of the SNF2 family) domains. CHD2 also contains a putative DNA binding domain (DBD) and a C-terminal domain (DUF4208) of unknown function [100]. Studies have confirmed that the C-terminal part of CHD2 is a functional DBD, which is selective for double-stranded DNA and is necessary to stimulate the ATPase and chromatin remodeling activities of the protein [101].
Epigenetic-targeted therapy
Demethylation agents
DNA methylation is catalyzed by DNMTs, the inhibitors of which can inhibit that process [102]. Azacitidine, a DNMT inhibitor, is not therapeutically effective in CLL, and a phase II clinical trial of azacitidine in fludarabine-refractory CLL was terminated prematurely due to lack of response and slow recruitment [103]. CLL cells can cause abnormal immune regulatory mechanisms that favor T-cell dysfunction and immunosuppression, with an inability to present antigens to the T-cell arm of the immune system. One study combining two epigenetic modifiers 5-aza-2'-deoxycytidine and histone deacetylase inhibitors (HDACis) LAQ824 was effective in restoring immunogenicity in CLL cell lines as well as in primary cells obtained from CLL patients [104].
HDACis
As a kind of promising noval epigenetic anticancer agent histone deacetylase inhibitor (HDACi) can induce a variety of biological process in cancer cells including gene expression regulation, G1/S or G2/M cell cycle arrest, differentiation, and apoptosis [105, 106]. AR-42 (Arno Therapeutics) targeting Class I and IIB HDAC enzymes is an orally bioavailable small molecule, which has anti-tumor activity in solid tumor models in vitro and in vivo [107] and many B-cell malignancies [108]. In CLL cells, AR-42 can not only increase the sensitivity of CLL cells to TNF-related apoptosis inducing ligand (TRAIL) by reducing the expression of cellular FLICE (FADD-like IL-1β-converting enzyme)-like inhibitory protein (c-FLIP), but also engender dose and time-dependent acetylation of histone, thereby inducing apoptosis from dependence on caspase [109].
According to an analysis of apoptosis regulatory genes in CLL, Kendine 92, and SAHA, HDACis have been confirmed that both of them can induce dose-, time- and caspase-dependent apoptosis via the mitochondrial pathway [110]. Ms-275, an HDACi, has been reported to mediate its cytotoxic effects by producing reactive oxygen species (ROS) in proliferating hematopoietic cell lines [111]. Another one is the basic DEPSIPTED (FR901228) clinical trial of DEPSIPTED (FR901228) CLL that is an early observation that CLL has selective in vitro activity in cultured CLL cells (0.038-micron DEPsipeptide). DEPsipeptide induces acetylation of histone H3 and H4 and inhibits deacetylation at concentrations comparable to LC50 [112]. The acetylation of histone occurs to H4 K5, H4 K12, and H3 K9 most commonly, and then H4 K8, but no H4 K16 or H3 K14, which are lysine specific.
However, not all HDACis exhibit efficient anti-tumor activity. MGCD0103 as an orally available class I HDACi, has limited activity in monotherapy in high-risk CLL patients [113]. Much more, preclinical studies of the HDACi DEPsipeptide (FK228) in CLL demonstrated that it can effectively induce apoptosis and at concentrations where HDACi occurs. Although FK288 effectively inhibits HDAC in CLL patients, its use in the current administration plan is limited because of progressive physical symptoms [114].
DAPK inhibitor
The death-associated protein kinase 3 (DAPK3), which mediates histone phosphorylation and responds to the BCR signaling pathway activation, is recruited to RNA polymerase II in an anti-IgM-dependent manner. DAPK inhibitors do not inhibit transcription on its own, but affect mRNA processing, and have a wider antitumor activity than ibrutinib through inhibiting anti-IgM and CD40L-dependent activation [115]. Targeting DAPK3 is a promising alternative for the treatment of CLL with BTK inhibitors.
Conclusions
Altogether, we summarized the recent advances in epigenetics mechanisms and targeted therapies of CLL. Better and earlier identification of the exact function of epigenetic alterations including DNA methylation, histone modification, RNA methylation, non-coding RNAs, and chromatin remodeling can provide new insights into the etiology of CLL. Although there are few mature epigenetic regulators, targeting epigenetics is still one of the directions for the future treatment of CLL.
Availability of data and materials
Not applicable.
References
Burger JA. Treatment of chronic lymphocytic leukemia. N Engl J Med. 2020;383(5):460–73.
Nabhan C, Rosen ST. Chronic lymphocytic leukemia: a clinical review. JAMA. 2014;312(21):2265–76.
Stevenson FK, Forconi F, Kipps TJ. Exploring the pathways to chronic lymphocytic leukemia. Blood. 2021;138(10):827–35.
Dighiero G, Hamblin TJ. Chronic lymphocytic leukaemia. The Lancet. 2008;371(9617):1017–29.
Jones L, McCalmont H, Evans K, Mayoh C, Kurmasheva RT, Billups CA, Houghton PJ, Smith MA, Lock RB. Preclinical activity of the antibody-drug conjugate denintuzumab mafodotin (SGN-CD19A) against pediatric acute lymphoblastic leukemia xenografts. Pediatr Blood Cancer. 2019;66(8): e27765.
Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150(1):12–27.
Chen J, Odenike O, Rowley JD. Leukaemogenesis: more than mutant genes. Nat Rev Cancer. 2010;10(1):23–36.
Wingelhofer B, Somervaille TCP. Emerging epigenetic therapeutic targets in acute myeloid leukemia. Front Oncol. 2019;9:850.
Santini V, Ossenkoppele GJ. Hypomethylating agents in the treatment of acute myeloid leukemia: a guide to optimal use. Crit Rev Oncol Hematol. 2019;140:1–7.
Robertson KD. DNA methylation and human disease. Nat Rev Genet. 2005;6(8):597–610.
Li J, Poi MJ, Tsai MD. Regulatory mechanisms of tumor suppressor P16(INK4A) and their relevance to cancer. Biochemistry. 2011;50(25):5566–82.
Bearzatto A, Conte D, Frattini M, Zaffaroni N, Andriani F, Balestra D, Tavecchio L, Daidone MG, Sozzi G. p16(INK4A) Hypermethylation detected by fluorescent methylation-specific PCR in plasmas from non-small cell lung cancer. Clin Cancer Res. 2002;8(12):3782–7.
Esteller M, Corn PG, Baylin SB, Herman JG. A gene hypermethylation profile of human cancer. Cancer Res. 2001;61(8):3225–9.
Kim JS, Han J, Shim YM, Park J, Kim DH. Aberrant methylation of H-cadherin (CDH13) promoter is associated with tumor progression in primary nonsmall cell lung carcinoma. Cancer. 2005;104(9):1825–33.
Toyooka KO, Toyooka S, Virmani AK, Sathyanarayana UG, Euhus DM, Gilcrease M, Minna JD, Gazdar AF. Loss of expression and aberrant methylation of the CDH13 (H-cadherin) gene in breast and lung carcinomas. Cancer Res. 2001;61(11):4556–60.
Fleisher AS, Esteller M, Tamura G, Rashid A, Stine OC, Yin J, Zou TT, Abraham JM, Kong D, Nishizuka S, et al. Hypermethylation of the hMLH1 gene promoter is associated with microsatellite instability in early human gastric neoplasia. Oncogene. 2001;20(3):329–35.
Cunningham JM, Christensen ER, Tester DJ, Kim CY, Roche PC, Burgart LJ, Thibodeau SN. Hypermethylation of the hMLH1 promoter in colon cancer with microsatellite instability. Cancer Res. 1998;58(15):3455–60.
Baylin SB, Jones PA. Epigenetic determinants of cancer. Cold Spring Harb Perspect Biol. 2016;8(9):a019505.
Rani L, Mathur N, Gupta R, Gogia A, Kaur G, Dhanjal JK, Sundar D, Kumar L, Sharma A. Genome-wide DNA methylation profiling integrated with gene expression profiling identifies PAX9 as a novel prognostic marker in chronic lymphocytic leukemia. Clin Epigenetics. 2017;9:57.
Rahmatpanah FB, Carstens S, Hooshmand SI, Welsh EC, Sjahputera O, Taylor KH, Bennett LB, Shi H, Davis JW, Arthur GL, et al. Large-scale analysis of DNA methylation in chronic lymphocytic leukemia. Epigenomics. 2009;1(1):39–61.
Subhash S, Andersson PO, Kosalai ST, Kanduri C, Kanduri M. Global DNA methylation profiling reveals new insights into epigenetically deregulated protein coding and long noncoding RNAs in CLL. Clin Epigenetics. 2016;8:106.
Bagacean C, Le Dantec C, Berthou C, Tempescul A, Saad H, Bordron A, Zdrenghea M, Cristea V, Douet-Guilbert N, Renaudineau Y. Combining cytogenetic and epigenetic approaches in chronic lymphocytic leukemia improves prognosis prediction for patients with isolated 13q deletion. Clin Epigenetics. 2017;9:122.
Zhang Q, Gao Y, Lin S, Goldin LR, Wang Y, Stevenson H, Edelman DC, Killian K, Marti G, Meltzer PS, et al. Genome-wide DNA methylation profiling in chronic lymphocytic leukaemia. Front Genet. 2022;13:1056043.
Quesada V, Conde L, Villamor N, Ordóñez GR, Jares P, Bassaganyas L, Ramsay AJ, Beà S, Pinyol M, Martínez-Trillos A, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet. 2011;44(1):47–52.
Pacholewska A, Grimm C, Herling CD, Lienhard M, Königs A, Timmermann B, Altmüller J, Mücke O, Reinhardt HC, Plass C, et al. Altered DNA methylation profiles in SF3B1 mutated CLL patients. Int J Mol Sci. 2021;22(17):9337.
Zhang Y, Wang X. Targeting the Wnt/beta-catenin signaling pathway in cancer. J Hematol Oncol. 2020;13(1):165.
Li H, Zhao J, Sun J, Tian C, Jiang Q, Ding C, Gan Q, Shu P, Wang X, Qin J, et al. Demethylation of the SFRP4 promoter drives gastric cancer progression via the Wnt pathway. Molecul cancer res MCR. 2021;19(9):1454–64.
Pawar NM, Rao P. Secreted frizzled related protein 4 (sFRP4) update: a brief review. Cell Signal. 2018;45:63–70.
Liu TH, Raval A, Chen SS, Matkovic JJ, Byrd JC, Plass C. CpG island methylation and expression of the secreted frizzled-related protein gene family in chronic lymphocytic leukemia. Cancer Res. 2006;66(2):653–8.
Deneberg S, Kanduri M, Ali D, Bengtzen S, Karimi M, Qu Y, Kimby E, Mansouri L, Rosenquist R, Lennartsson A, et al. microRNA-34b/c on chromosome 11q23 is aberrantly methylated in chronic lymphocytic leukemia. Epigenetics. 2014;9(6):910–7.
Raval A, Lucas DM, Matkovic JJ, Bennett KL, Liyanarachchi S, Young DC, Rassenti L, Kipps TJ, Grever MR, Byrd JC, et al. TWIST2 demonstrates differential methylation in immunoglobulin variable heavy chain mutated and unmutated chronic lymphocytic leukemia. J Clin Oncol. 2005;23(17):3877–85.
Bilban M, Heintel D, Scharl T, Woelfel T, Auer MM, Porpaczy E, Kainz B, Krober A, Carey VJ, Shehata M, et al. Deregulated expression of fat and muscle genes in B-cell chronic lymphocytic leukemia with high lipoprotein lipase expression. Leukemia. 2006;20(6):1080–8.
Kaderi MA, Kanduri M, Buhl AM, Sevov M, Cahill N, Gunnarsson R, Jansson M, Smedby KE, Hjalgrim H, Jurlander J, et al. LPL is the strongest prognostic factor in a comparative analysis of RNA-based markers in early chronic lymphocytic leukemia. Haematologica. 2011;96(8):1153–60.
Abreu C, Moreno P, Palacios F, Borge M, Morande P, Landoni AI, Gabus R, Dighiero G, Giordano M, Gamberale R, et al. Methylation status regulates lipoprotein lipase expression in chronic lymphocytic leukemia. Leuk Lymphoma. 2013;54(8):1844–8.
Lyko F. The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat Rev Genet. 2018;19(2):81–92.
Biran A, Yin S, Kretzmer H, Ten Hacken E, Parvin S, Lucas F, Uduman M, Gutierrez C, Dangle N, Billington L, et al. Activation of notch and myc signaling via B-cell-restricted depletion of dnmt3a generates a consistent murine model of chronic lymphocytic leukemia. Cancer Res. 2021;81(24):6117–30.
Papakonstantinou N, Ntoufa S, Tsagiopoulou M, Moysiadis T, Bhoi S, Malousi A, Psomopoulos F, Mansouri L, Laidou S, Papazoglou D, et al. Integrated epigenomic and transcriptomic analysis reveals TP63 as a novel player in clinically aggressive chronic lymphocytic leukemia. Int J Cancer. 2019;144(11):2695–706.
Barrow TM, Nakjang S, Lafta F, Bilotkach K, Woodhouse L, Junge G, Tudhope SJ, Wallis JP, Marr H, Marshall S, et al. Epigenome-wide analysis reveals functional modulators of drug sensitivity and post-treatment survival in chronic lymphocytic leukaemia. Br J Cancer. 2021;124(2):474–83.
Kulis M, Heath S, Bibikova M, Queiros AC, Navarro A, Clot G, Martinez-Trillos A, Castellano G, Brun-Heath I, Pinyol M, et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat Genet. 2012;44(11):1236–42.
Tsagiopoulou M, Papakonstantinou N, Moysiadis T, Mansouri L, Ljungstrom V, Duran-Ferrer M, Malousi A, Queiros AC, Plevova K, Bhoi S, et al. DNA methylation profiles in chronic lymphocytic leukemia patients treated with chemoimmunotherapy. Clin Epigenetics. 2019;11(1):177.
Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705.
Clarke S. Protein methylation. Curr Opin Cell Biol. 1993;5(6):977–83.
Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21(3):381–95.
Zhou K, Zhang Q, Liu Y, Xiong Y, Wu S, Yang J, Zhou H, Liu X, Wei X, Song Y. Aberrant histone modification in CD19(+) B cells of patients with chronic lymphocytic leukemia. Onco Targets Ther. 2017;10:1173–9.
Albert M, Helin K. Histone methyltransferases in cancer. Semin Cell Dev Biol. 2010;21(2):209–20.
Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 2011;11(10):726–34.
Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, Butler A, Davies H, Edkins S, Hardy C, Latimer C, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463(7279):360–3.
Pfister SX, Ahrabi S, Zalmas LP, Sarkar S, Aymard F, Bachrati CZ, Helleday T, Legube G, La Thangue NB, Porter AC, et al. SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell Rep. 2014;7(6):2006–18.
Carvalho S, Vitor AC, Sridhara SC, Martins FB, Raposo AC, Desterro JM, Ferreira J, de Almeida SF. SETD2 is required for DNA double-strand break repair and activation of the p53-mediated checkpoint. Elife. 2014;3: e02482.
Parker H, Rose-Zerilli MJ, Larrayoz M, Clifford R, Edelmann J, Blakemore S, Gibson J, Wang J, Ljungstrom V, Wojdacz TK, et al. Genomic disruption of the histone methyltransferase SETD2 in chronic lymphocytic leukaemia. Leukemia. 2016;30(11):2179–86.
Yaswen P. HDAC inhibitors conquer Polycomb proteins. Cell Cycle. 2010;9(14):2705.
Rabello Ddo A, Lucena-Araujo AR, Alves-Silva JC, da Eira VB, de Vasconcellos MC, de Oliveira FM, Rego EM, Saldanha-Araujo F, Pittella Silva F. Overexpression of EZH2 associates with a poor prognosis in chronic lymphocytic leukemia. Blood Cells Mol Dis. 2015;54(1):97–102.
Szurian K, Csala I, Marosvari D, Rajnai H, Dezso K, Bodor C, Piurko V, Matolcsy A, Reiniger L. EZH2 is upregulated in the proliferation centers of CLL/SLL lymph nodes. Exp Mol Pathol. 2018;105(2):161–5.
Jiang Q, Stachelscheid J, Bloehdorn J, Pacholewska A, Aszyk CM, Grotenhuijs F, Müller TA, Onder O, Wagle P, Herling CD, et al. Oncogenic role and target properties of the lysine-specific demethylase KDM1A in chronic lymphocytic leukemia. Blood. 2023;142(1):44–61.
Jordaan G, Liao W, Sharma S. E-cadherin gene re-expression in chronic lymphocytic leukemia cells by HDAC inhibitors. BMC Cancer. 2013;13:88.
Zaccara S, Ries RJ, Jaffrey SR. Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol. 2019;20(10):608–24.
Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, Adhikari S, Shi Y, Lv Y, Chen YS, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24(2):177–89.
Pendleton KE, Chen B, Liu K, Hunter OV, Xie Y, Tu BP, Conrad NK. The U6 snRNA m(6)A methyltransferase METTL16 regulates SAM synthetase intron retention. Cell. 2017;169(5):824-835.e814.
Meyer KD, Jaffrey SR. Rethinking m(6)A readers, writers, and erasers. Annu Rev Cell Dev Biol. 2017;33:319–42.
He L, Li H, Wu A, Peng Y, Shu G, Yin G. Functions of N6-methyladenosine and its role in cancer. Mol Cancer. 2019;18(1):176.
Huang H, Weng H, Chen J. m(6)A Modification in Coding and Non-coding RNAs: roles and Therapeutic Implications in Cancer. Cancer Cell. 2020;37(3):270–88.
Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, Huang H, Nachtergaele S, Dong L, Hu C, et al. FTO plays an oncogenic role in acute myeloid leukemia as a N(6)-Methyladenosine RNA Demethylase. Cancer Cell. 2017;31(1):127–41.
Chen J, Du B. Novel positioning from obesity to cancer: FTO, an m(6)A RNA demethylase, regulates tumour progression. J Cancer Res Clin Oncol. 2019;145(1):19–29.
Zhang Y, Hu X, Han Y, Zhou X, Zhang H, Yun X, Wang X. Targeting inhibition of n6-methyladenosine demethylase fto displays potent anti-tumor activities in chronic lymphocytic leukemia. Blood. 2020;136(Supplement 1):35–6.
Lu TX, Rothenberg ME. MicroRNA. J Allergy Clin Immunol. 2018;141(4):1202–7.
Pekarsky Y, Croce CM. Role of miR-15/16 in CLL. Cell Death Differ. 2015;22(1):6–11.
Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA. 2005;102(39):13944–9.
Pekarsky Y, Balatti V, Croce CM. BCL2 and miR-15/16: from gene discovery to treatment. Cell Death Differ. 2018;25(1):21–6.
Fabbri M, Bottoni A, Shimizu M, Spizzo R, Nicoloso MS, Rossi S, Barbarotto E, Cimmino A, Adair B, Wojcik SE, et al. Association of a microRNA/TP53 feedback circuitry with pathogenesis and outcome of B-cell chronic lymphocytic leukemia. JAMA. 2011;305(1):59–67.
Leng RX, Pan HF, Qin WZ, Chen GM, Ye DQ. Role of microRNA-155 in autoimmunity. Cytokine Growth Factor Rev. 2011;22(3):141–7.
Cui B, Chen L, Zhang S, Mraz M, Fecteau JF, Yu J, Ghia EM, Zhang L, Bao L, Rassenti LZ, et al. MicroRNA-155 influences B-cell receptor signaling and associates with aggressive disease in chronic lymphocytic leukemia. Blood. 2014;124(4):546–54.
Ferrajoli A, Shanafelt TD, Ivan C, Shimizu M, Rabe KG, Nouraee N, Ikuo M, Ghosh AK, Lerner S, Rassenti LZ, et al. Prognostic value of miR-155 in individuals with monoclonal B-cell lymphocytosis and patients with B chronic lymphocytic leukemia. Blood. 2013;122(11):1891–9.
Chiang CL, Goswami S, Frissora FW, Xie Z, Yan PS, Bundschuh R, Walker LA, Huang X, Mani R, Mo XM, et al. ROR1-targeted delivery of miR-29b induces cell cycle arrest and therapeutic benefit in vivo in a CLL mouse model. Blood. 2019;134(5):432–44.
Sharma S, Pavlasova GM, Seda V, Cerna KA, Vojackova E, Filip D, Ondrisova L, Sandova V, Kostalova L, Zeni PF, et al. miR-29 modulates CD40 signaling in chronic lymphocytic leukemia by targeting TRAF4: an axis affected by BCR inhibitors. Blood. 2021;137(18):2481–94.
Balatti V, Tomasello L, Rassenti LZ, Veneziano D, Nigita G, Wang HY, Thorson JA, Kipps TJ, Pekarsky Y, Croce CM. miR-125a and miR-34a expression predicts Richter syndrome in chronic lymphocytic leukemia patients. Blood. 2018;132(20):2179–82.
Mraz M, Chen L, Rassenti LZ, Ghia EM, Li H, Jepsen K, Smith EN, Messer K, Frazer KA, Kipps TJ. miR-150 influences B-cell receptor signaling in chronic lymphocytic leukemia by regulating expression of GAB1 and FOXP1. Blood. 2014;124(1):84–95.
Paggetti J, Haderk F, Seiffert M, Janji B, Distler U, Ammerlaan W, Kim YJ, Adam J, Lichter P, Solary E, et al. Exosomes released by chronic lymphocytic leukemia cells induce the transition of stromal cells into cancer-associated fibroblasts. Blood. 2015;126(9):1106–17.
Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet. 2016;17(1):47–62.
Garding A, Bhattacharya N, Claus R, Ruppel M, Tschuch C, Filarsky K, Idler I, Zucknick M, Caudron-Herger M, Oakes C, et al. Epigenetic upregulation of lncRNAs at 13q14.3 in leukemia is linked to the In Cis downregulation of a gene cluster that targets NF-kB. PLoS Genet. 2013;9(4):e1003373.
Pekarsky Y, Zanesi N, Croce CM. Molecular basis of CLL. Semin Cancer Biol. 2010;20(6):370–6.
Dahl M, Kristensen LS, Gronbaek K. Long non-coding RNAs guide the fine-tuning of gene regulation in B-cell development and malignancy. Int J Mol Sci. 2018;19(9):2475.
Ni J, Hong J, Li Q, Zeng Q, Xia R. Long non-coding RNA CRNDE suppressing cell proliferation is regulated by DNA methylation in chronic lymphocytic leukemia. Leuk Res. 2021;105: 106564.
Du X, Liu H, Yang C, Shi X, Cao L, Zhao X, Miao Y, Zhu H, Wang L, Xu W, et al. LncRNA landscape analysis identified LncRNA LEF-AS1 as an oncogene that upregulates LEF1 and promotes survival in chronic lymphocytic leukemia. Leuk Res. 2021;110: 106706.
Bomben R, Roisman A, D’Agaro T, Castellano G, Baumann T, Delgado J, López-Guillermo A, Zucchetto A, Dal-Bo M, Bravin V, et al. Expression of the transcribed ultraconserved region 70 and the related long non-coding RNA AC092652.2–202 has prognostic value in chronic lymphocytic leukaemia. Br J Haematol. 2019;184(6):1045–50.
Ronchetti D, Manzoni M, Agnelli L, Vinci C, Fabris S, Cutrona G, Matis S, Colombo M, Galletti S, Taiana E, et al. lncRNA profiling in early-stage chronic lymphocytic leukemia identifies transcriptional fingerprints with relevance in clinical outcome. Blood Cancer J. 2016;6(9): e468.
Isin M, Ozgur E, Cetin G, Erten N, Aktan M, Gezer U, Dalay N. Investigation of circulating lncRNAs in B-cell neoplasms. Clin Chim Acta. 2014;431:255–9.
Blume CJ, Hotz-Wagenblatt A, Hüllein J, Sellner L, Jethwa A, Stolz T, Slabicki M, Lee K, Sharathchandra A, Benner A, et al. p53-dependent non-coding RNA networks in chronic lymphocytic leukemia. Leukemia. 2015;29(10):2015–23.
Baldassarre A, Masotti A. Long non-coding RNAs and p53 regulation. Int J Mol Sci. 2012;13(12):16708–17.
Chen S, Liang H, Yang H, Zhou K, Xu L, Liu J, Lai B, Song L, Luo H, Peng J, et al. LincRNa-p21: function and mechanism in cancer. Med Oncol. 2017;34(5):98.
Ebbesen KK, Hansen TB, Kjems J. Insights into circular RNA biology. RNA Biol. 2017;14(8):1035–45.
Wu Z, Sun H, Liu W, Zhu H, Fu J, Yang C, Fan L, Wang L, Liu Y, Xu W, et al. Circ-RPL15: a plasma circular RNA as novel oncogenic driver to promote progression of chronic lymphocytic leukemia. Leukemia. 2020;34(3):919–23.
Xia L, Wu L, Bao J, Li Q, Chen X, Xia H, Xia R. Circular RNA circ-CBFB promotes proliferation and inhibits apoptosis in chronic lymphocytic leukemia through regulating miR-607/FZD3/Wnt/beta-catenin pathway. Biochem Biophys Res Commun. 2018;503(1):385–90.
Wu W, Wu Z, Xia Y, Qin S, Li Y, Wu J, Liang J, Wang L, Zhu H, Fan L, et al. Downregulation of circ_0132266 in chronic lymphocytic leukemia promoted cell viability through miR-337-3p/PML axis. Aging. 2019;11(11):3561–73.
Li S, Chen J, Fan Y, Xu X, Xiong M, Qi Y, Wu W, Zhao Y. circZNF91 promotes the malignant phenotype of chronic lymphocytic leukemia cells by targeting the miR-1283/WEE1 axis. Biomed Res Int. 2022;2022:2855394.
Zhang X, Han Y, Hu X, Wang H, Tian Z, Zhang Y, Wang X. Competing endogenous RNA networks related to prognosis in chronic lymphocytic leukemia: comprehensive analyses and construction of a novel risk score model. Biomarker research. 2022;10(1):75.
Wu Z, Sun H, Wang C, Liu W, Liu M, Zhu Y, Xu W, Jin H, Li J. Mitochondrial genome-derived circRNA mc-cox2 functions as an oncogene in chronic lymphocytic leukemia. Molecular therapy Nucleic acids. 2020;20:801–11.
de Perez AO, Rossi M, Gorospe M. Circular RNAs in blood malignancies. Front Mol Biosci. 2020;7:109.
Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem. 2009;78:273–304.
Guan B, Wang TL, Shih Ie M. ARID1A, a factor that promotes formation of SWI/SNF-mediated chromatin remodeling, is a tumor suppressor in gynecologic cancers. Cancer Res. 2011;71(21):6718–27.
Rodríguez D, Bretones G, Quesada V, Villamor N, Arango JR, López-Guillermo A, Ramsay AJ, Baumann T, Quirós PM, Navarro A, et al. Mutations in CHD2 cause defective association with active chromatin in chronic lymphocytic leukemia. Blood. 2015;126(2):195–202.
Liu JC, Ferreira CG, Yusufzai T. Human CHD2 is a chromatin assembly ATPase regulated by its chromo- and DNA-binding domains. J Biol Chem. 2015;290(1):25–34.
Kim M, Costello J. DNA methylation: an epigenetic mark of cellular memory. Exp Mol Med. 2017;49(4): e322.
Malik A, Shoukier M, Garcia-Manero G, Wierda W, Cortes J, Bickel S, Keating MJ, Estrov Z. Azacitidine in fludarabine-refractory chronic lymphocytic leukemia: a phase II study. Clin Lymphoma Myeloma Leuk. 2013;13(3):292–5.
Dubovsky JA, Wang D, Powers JJ, Berchmans E, Smith MA, Wright KL, Sotomayor EM, Pinilla-Ibarz JA. Restoring the functional immunogenicity of chronic lymphocytic leukemia using epigenetic modifiers. Leuk Res. 2011;35(3):394–404.
Lane AA, Chabner BA. Histone deacetylase inhibitors in cancer therapy. J clin oncol off j Am Soc Clin Oncol. 2009;27(32):5459–68.
Rodríguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med. 2011;17(3):330–9.
Burns SS, Akhmametyeva EM, Oblinger JL, Bush ML, Huang J, Senner V, Chen CS, Jacob A, Welling DB, Chang LS. Histone deacetylase inhibitor AR-42 differentially affects cell-cycle transit in meningeal and meningioma cells, potently inhibiting NF2-deficient meningioma growth. Can Res. 2013;73(2):792–803.
Zhang S, Suvannasankha A, Crean CD, White VL, Chen CS, Farag SS. The novel histone deacetylase inhibitor, AR-42, inhibits gp130/Stat3 pathway and induces apoptosis and cell cycle arrest in multiple myeloma cells. Int J Cancer. 2011;129(1):204–13.
Lucas DM, Alinari L, West DA, Davis ME, Edwards RB, Johnson AJ, Blum KA, Hofmeister CC, Freitas MA, Parthun MR, et al. The novel deacetylase inhibitor AR-42 demonstrates pre-clinical activity in B-cell malignancies in vitro and in vivo. PLoS ONE. 2010;5(6): e10941.
Perez-Perarnau A, Coll-Mulet L, Rubio-Patino C, Iglesias-Serret D, Cosialls AM, Gonzalez-Girones DM, de Frias M, de Sevilla AF, de la Banda E, Pons G, et al. Analysis of apoptosis regulatory genes altered by histone deacetylase inhibitors in chronic lymphocytic leukemia cells. Epigenetics. 2011;6(10):1228–35.
Lucas DM, Davis ME, Parthun MR, Mone AP, Kitada S, Cunningham KD, Flax EL, Wickham J, Reed JC, Byrd JC, et al. The histone deacetylase inhibitor MS-275 induces caspase-dependent apoptosis in B-cell chronic lymphocytic leukemia cells. Leukemia. 2004;18(7):1207–14.
Aron JL, Parthun MR, Marcucci G, Kitada S, Mone AP, Davis ME, Shen T, Murphy T, Wickham J, Kanakry C, et al. Depsipeptide (FR901228) induces histone acetylation and inhibition of histone deacetylase in chronic lymphocytic leukemia cells concurrent with activation of caspase 8-mediated apoptosis and down-regulation of c-FLIP protein. Blood. 2003;102(2):652–8.
Blum KA, Advani A, Fernandez L, Van Der Jagt R, Brandwein J, Kambhampati S, Kassis J, Davis M, Bonfils C, Dubay M, et al. Phase II study of the histone deacetylase inhibitor MGCD0103 in patients with previously treated chronic lymphocytic leukaemia. Br J Haematol. 2009;147(4):507–14.
Byrd JC, Marcucci G, Parthun MR, Xiao JJ, Klisovic RB, Moran M, Lin TS, Liu S, Sklenar AR, Davis ME, et al. A phase 1 and pharmacodynamic study of depsipeptide (FK228) in chronic lymphocytic leukemia and acute myeloid leukemia. Blood. 2005;105(3):959–67.
Thomas F, Holmes KB, Kreuz S, Hillmen P, Lefevre PF. DAPK3 participates in the mRNA processing of immediate early genes in chronic lymphocytic leukaemia. Mol Oncol. 2020;14(6):1268–81.
Acknowledgements
Not applicable.
Funding
This study was funded by National Natural Science Foundation (No. 82270200, No. 82000195, No. 82070203, and No. 81770210); China Postdoctoral Science Foundation (No. 2022M721981); Key Research and Development Program of Shandong Province (No. 2018CXGC1213); Taishan Scholars Program of Shandong Province (No. tspd20230610, tsqn201909184); Translational Research Grant of NCRCH (No. 2021WWB02, No. 2020ZKMB01); Shandong Provincial Natural Science Foundation (No. ZR2020QH094); Shandong Provincial Engineering Research Center of Lymphoma; Academic Promotion Programme of Shandong First Medical University (No. 2019QL018).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. XW and YZ designed the scheme; XZ and HW wrote the manuscript and drew the figures and tables; XW and YZ amended the article; all authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethics approval
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, X., Wang, H., Zhang, Y. et al. Advances in epigenetic alterations of chronic lymphocytic leukemia: from pathogenesis to treatment. Clin Exp Med 24, 54 (2024). https://doi.org/10.1007/s10238-023-01268-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10238-023-01268-x