
Abstract
Mining of volumetrically small, but relatively enriched (average 0.6% U3O8) breccia pipe uranium (BPU) deposits near the Grand Canyon, Arizona, USA has the potential to affect groundwater and springs in the area. Such deposits also contain base metal sulfides that can oxidize to generate acid mine drainage and release trace metals. In this study, sequential batch experiments were conducted to simulate the geochemistry of local shallow groundwater that contacts BPU ore and then moves downgradient through sedimentary strata. The experiments simulated shallow groundwater in a carbonate aquifer followed by contact with BPU ore. The experiments subsequently simulated contact with sedimentary rocks and changing oxygen availability. Concentrations of several contaminants of potential concern became substantially elevated in the waters exposed to BPU ore, including As, Co, Ni, U, and Zn, and to a lesser extent, Mo. Of these, Co, Mo, Ni, and U were minimally attenuated by downgradient processes, whereas Zn was partially attenuated. Sb and Tl concentrations were more moderately elevated but also generally minimally attenuated. Although the mixture of elements is particular to these BPU ore deposits, sulfide oxidation in the ore and carbonate buffering of pH by sedimentary rocks generates patterns of water chemistry common in acid mine drainage settings. Ultimately, downgradient concentrations of elements sourced from BPU ore will also be strongly influenced by non-geochemical factors such as the quantities of water contacting BPU materials, heterogeneity of materials along flow paths, and mixing with waters that have not contacted BPU materials.
Zusammenfassung
Der Abbau volumetrisch kleiner, aber stark angereicherter Brekzien-Kamin Uranlagerstätten (im Mittel 0,6% U3O8) nahe dem Grand Canyon in Arizona, USA, kann möglicherweise Grundwasser und Quellen in der näheren Umgebung beeinträchtigen. Die Lagerstätten enthalten Nichtedelmetall-Sulfide, die oxidieren und damit zur Bildung saurer Grubenwässern und Freisetzung von Spurenmetallen führen. In dieser Studie wurden sequentielle Batch-Versuche zur Simulation der Geochemie lokaler, oberflächennaher Grundwässern im Kontakt mit den Uranerzen und nachfolgendem Abstrom durch Sedimentschichten durchgeführt. Die Experimente simulierten oberflächennahes Grundwasser in einem karbonatischen Aquifer mit nachfolgendem Kontakt zu den Uranerzen und anschließenden Sedimentgesteinen sowie wechselnder Sauerstoffverfügbarkeit. Die Konzentrationen verschiedener, potentiell gefährlicher Schadstoffe wie As, Co, Ni, U und Zn sowie untergeordnet Mo wurden in den exponierten Wässern deutlich erhöht. Von diesen wurden Co, Mo, Ni und U durch abstromige Prozesse nur minimal verringert, wohingegen Zn teilweise verringert wurde. Die Konzentrationen an Sb und Tl wurden nur moderat erhöht, aber ebenfalls teilweise verringert. Obgleich die Mischung dieser Elemente charakteristisch für diese Uranlagerstätten ist, entstehen durch die Oxidation von Sulfiden im Erz und die karbonatische Pufferung des pH-Werts durch Sedimentgesteine für das Umfeld saurer Grubenwässer typische Signaturen. Schlussendlich wird die abstromige Konzentration der aus dem Uranerz ausgetragenen Elemente jedoch auch stark durch nicht-geochemische Faktoren wie die Wassermenge im Kontakt mit dem Erz, die Heterogenität der Gesteine entlang des Fließpfades und der Vermischung unbelasteter Wässer beeinflusst.
Resumen
La extracción de depósitos de uranio de tubo de brecha (BPU) volumétricamente pequeños, pero relativamente enriquecidos (promedio de 0,6% de U3O8) cerca del Gran Cañón, Arizona, Estados Unidos, tiene el potencial de afectar a las aguas subterráneas y a los manantiales de la zona. Dichos depósitos también contienen sulfuros de metales que pueden oxidarse para generar un drenaje ácido de mina y liberar trazas de metales. En este estudio, se llevaron a cabo experimentos secuenciales por lotes para simular la geoquímica de las aguas subterráneas locales poco profundas que entran en contacto con el mineral de BPU y luego se desplazan hacia abajo a través de los estratos sedimentarios. Los experimentos simularon aguas subterráneas poco profundas en un acuífero carbonatado, seguidas por el contacto con el mineral de BPU. Los experimentos simularon posteriormente el contacto con las rocas sedimentarias y el cambio en la disponibilidad de oxígeno. Las concentraciones de varios contaminantes potencialmente preocupantes se elevaron sustancialmente en las aguas expuestas al mineral de BPU, incluyendo As, Co, Ni, U y Zn, y en menor medida, Mo. De ellos, el Co, el Mo, el Ni y el U fueron atenuados mínimamente por los procesos descendentes, mientras que el Zn fue parcialmente atenuado. Las concentraciones de Sb y Tl fueron más moderadamente elevadas, pero en general también fueron mínimamente atenuadas. Aunque la mezcla de elementos es particular de estos depósitos de mineral de BPU, la oxidación de sulfuro en el mineral y la amortiguación de carbonato del pH por las rocas sedimentarias genera patrones de química del agua comunes en los entornos de drenaje ácido de minas. En última instancia, las concentraciones de elementos procedentes de la mena de BPU también estarán fuertemente influenciadas por factores no geoquímicos como las cantidades de agua que entran en contacto con los materiales de BPU, la heterogeneidad de los materiales a lo largo de las rutas de flujo y la mezcla con aguas que no han entrado en contacto con los materiales de BPU.
抽象的
开采美国亚利桑那州大峡谷附近体积小而相对富集 (平均0.6% U3O8) 的角砾岩筒型铀矿床 (BPU) 将对地区地下水和泉水产生潜在影响。该类矿床还含有碱金属硫化物, 它们氧化后产生酸性矿山废水和释放微量金属。研究采用序批实验方法模拟了当地浅层地下水与角砾岩筒型铀矿 (BPU) 矿石接触再沿沉积地层流向下游的水化学过程。实验模拟了碳酸盐岩含水层的浅层地下水, 然后地下水与角砾岩筒型铀矿 (BPU) 矿石接触。实验随后模拟了与沉积矿床岩石的接触和获氧条件的变化。与角砾岩筒型铀矿 (BPU) 矿石接触的水的几种潜在受关注污染物的浓度显著提高, 包括As、Co、Ni、U和Zn, 甚至更小浓度级别的Mo。在这些污染物中, Co、Mo、Ni和U在向下游运移过程中衰减最小, Zn部分衰减。Sb和Tl浓度略有增高, 但总体衰减也很少。虽然这些元素的混合方式对角砾岩筒型铀矿床 (BPU) 是特定的, 但是矿石内硫化物的氧化和沉积岩石的碳酸盐pH值缓冲作用产生了常见于酸性矿井废水环境的水化学模式。最终, 源自角砾岩筒型铀矿 (BPU) 矿石的元素在下游的浓度也受非地球化学因素的强烈影响, 例如接触角砾岩筒型铀矿 (BPU) 材料的水量、水流路径上材料的各向异质性、与未接触角砾岩筒型铀矿 (BPU) 材料的水的混合等。
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Breccia pipe uranium (BPU) deposits are an unusual type of uranium (U) resource found in the region surrounding Grand Canyon National Park, Arizona, U.S.A. (Wenrich 1985). Development of BPU deposits has the potential to affect hydrologic, biologic, and cultural resources in the region (Alpine 2010). Beginning in 2012, three parcels of land in the region (≈ 407,344 ha) were withdrawn from new mineral extraction for 20 years so that such potential environmental effects could be studied (U.S. Department of the Interior 2012). The present paper examines potential water-quality effects from mining of BPU deposits via a laboratory study.
Breccia pipes in the Grand Canyon region are narrow, vertical, karst-related collapse features that subsequently propagated upward into overlying strata consisting mainly of siliciclastic rocks. The voids were mainly filled with breccia clasts consisting of sandstones and siltstones from overlying layers and cemented by sulfide-uranium oxide mineralization in ore zones. The nature of the collapse altered their permeability relative to surrounding strata, and mining disturbance seems likely to enhance permeability. Only a small fraction of the breccia pipes contain U-bearing mineral deposits (Van Gosen et al. 2016). The breccia pipes are generally narrow (60–120 m wide) compared to vertical extents of 900 m or greater (Alpine 2010). Where they exist, U mineralization occurs in the pre-existing collapse feature at intermediate depths, relative to the extent of the pipe, and most commonly at the depth of the Hermit Formation (Otton and Van Gosen 2010). Detailed microscopy studies indicate that base-metal sulfide mineralization occurred first and U mineralization followed, with U being reduced by reaction with the sulfides (Van Gosen et al. 2020a). Ore bodies in the 11 known BPU deposits are volumetrically small (average ore volumes of 60,000 m3 and 160,000 metric tons (t), ranging from 12,500 to 172,000 m3 and 33,000 to 454,000 t) and averaging 0.6 wt.% U3O8 (calculated from Mathisen et al. 2017; Otton and Van Gosen 2010). Uranium is present in unoxidized ore mainly as uraninite (Van Gosen et al. 2020a, b; Wenrich 1985). Additionally, BPU deposits are enriched, relative to host rock and local soil, in various trace metals including As, Ba, Cd, Co, Cu, Mo, Ni, Pb, Sb, Se, Th, Zn (Bern et al. 2019; Van Gosen et al. 2020a, b; Wenrich 1985). Such elements in BPU deposits are frequently present in sulfide minerals like pyrite, galena, chalcopyrite, sphalerite, and phases of the colbaltite–gersdorffite series (Van Gosen et al. 2020a, b). Sulfide mineral oxidation is the process that drives acidification and subsequent metal mobilization commonly referred to as acid mine drainage (AMD) and gives BPU deposits the potential to exhibit geochemical patterns similar to AMD (Nordstrom and Alpers 1997; Singer and Stumm 1970; Wenrich et al. 1996).
Environmental concerns with BPU resource development have generally focused on U, but other trace elements like those listed above also pose toxicological concerns (Arnberger and Martin 2018; Cleveland et al. 2021). The U.S. Geological Survey (USGS) has been tasked with studying the potential effects on water, soil, vegetation, and wildlife related to BPU resource development in the Grand Canyon region (Alpine 2010, 2017; Beisner et al. 2017a; Bern et al. 2019; Cleveland et al. 2019; Hinck et al. 2013, 2014; Klymus et al. 2017; Minter et al. 2019; Naftz and Walton-Day 2016). Surface water is scarce in the region, making groundwater and springs crucial resources for humans and wildlife (Alpine and Brown 2010). Species diversity in the vicinity of springs is 100–500 times greater than the surrounding landscape (Grand Canyon Wildlands Council 2004). Groundwater flow is poorly understood in the region because of complexities of multiple, layered aquifers, some of which occur at great depth, and areas of karstic conduit flow (Beisner et al. 2017b, 2020; Solder et al. 2020; Tobin et al. 2018). High costs to install deep monitoring wells to study groundwater near new and pre-existing mines present a challenge, with few or no groundwater data available near BPU deposits. Therefore, study of groundwater emergence at springs, along with focused studies of springs near BPU mines have been used to improve understanding (Beisner et al. 2017a, b). In addition, the current study was undertaken using a laboratory approach to gain insights regarding groundwater chemistry not currently feasible with in situ environmental samples.
The goal of this study was to improve the understanding of the potential chemistry of natural waters that contact BPU ore and the processes that change concentrations of trace elements along potential groundwater flow paths. Particular attention was paid to certain contaminants of potential concern identified by other studies in the region, including As, Cd, Cu, Pb, Ni, Tl, U, and Zn (Hinck et al. 2013, 2014, 2017). Compositional changes in water were explored through a series of sequential batch experiments designed to simulate different subsurface environments using different geologic materials and gas mixtures encountered along potential flow paths. The results are discussed from the perspective of understanding the processes by which mobilization and attenuation of chemical constituents in groundwater may occur. Comparisons of water chemistry evolution are made to the classic patterns of AMD (Blowes et al. 2003; Dzombak and Morel 1990; Smith 1999). Environmental factors not covered by the experiments are also discussed. The study reveals which elements could be mobilized from BPU ore in substantial concentrations and which are subject to geochemical processes that may make them more likely to emerge downgradient, information that is useful for anticipating potential toxicity issues and fingerprinting affected waters.
Materials and Methods
Aquifer-Related Rocks
Two major aquifers exist in the Grand Canyon region (Bills et al. 2007). The upper aquifer is locally perched and sometimes referred to as the Coconino or “C” aquifer; it consists of the Kaibab Formation, Toroweap Formation, and Coconino Sandstone, underlain by the Hermit Formation, which can function as an aquitard (Bills et al. 2007; Tobin et al. 2018). Below is the Supai Group that includes the Esplanade Sandstone, which can also be part of the “C” aquifer, depending on the continuity of shale in the Hermit Formation (Bills et al. 2007). The underlying Redwall aquifer is a regional aquifer consisting of the Redwall Limestone, Temple Butte Formation, and Muav Limestone (Bills et al. 2007; Solder et al. 2020). For reference, BPU deposits are found primarily at the stratigraphic depth of the Hermit Shale but extend up into the Coconino Sandstone and down into the Esplanade Sandstone (Beisner et al. 2020; Otton and Van Gosen 2010).
Seventeen samples of sedimentary rocks that host aquifers or that groundwater might contact were obtained from surface exposures along the South Kaibab Trail in Grand Canyon National Park in January of 2017 (Fig. 1). To homogenize and expose fresh surfaces for the experiments, the rocks were crushed in a jaw crusher and ground in a disc mill with ceramic plates to < 1 mm. Splits for elemental and mineralogical analyses were then ground to < 100 µm. Elemental analysis was conducted by AGAT Laboratories (Calgary, Alberta, Canada) by decomposing the sample using a mixture of hydrochloric, nitric, perchloric, and hydrofluoric acids at low temperature and then analyzing the resulting solution by inductively coupled plasma-optical emission spectroscopy (ICP-OES) and inductively coupled plasma-mass spectrometry (ICP-MS) (Briggs 2002; Lamothe et al. 2002) (method ICPOES_MS-49). Blind reference standards constituted 10% of submissions. Total carbon concentrations were measured at AGAT Laboratories using LECO equipment (St. Joseph, MI), and carbonate carbon was measured by coulometric titration (Brown and Curry 2002; Brown et al. 2002). Data were deemed acceptable if recovery of each element in standard reference samples was ± 15% at five times the lower limit of determination and the calculated relative standard deviation (RSD) of laboratory duplicate samples was no greater than 15%. All data collected as part of this study are available via a USGS data release (Bern et al. 2021).

Map showing Grand Canyon National Park, parcels temporarily withdrawn from new mineral extraction, and locations from which samples and data used in the paper were obtained
Semi-quantitative mineralogical analysis was performed at USGS Laboratories (Boulder, Colorado, USA) using x-ray diffraction. Powdered samples were homogenized with 20% corundum and analyzed using a Siemens D500 x-ray diffractometer from 5 to 65° two theta using Cu Kα X-ray radiation, with a step size of 0.02° and a dwell time of 2 s per step. Mineralogical composition was determined using an updated version of the quantitative mineralogy software RockJock (Eberl 2003), utilizing full-pattern fitting with natural reference materials. The full pattern degree-of-fit for most samples ranged from 0.08 to 0.12 and indicated good confidence in the results; one sample had a value of 0.18 and indicated moderate confidence in that result (Table 1).
Composite Ore Samples
Hand specimens of BPU ore collected in 1984 and 1985 and then stored in USGS archives were used to make two unique composites of BPU ore for use in the experiments. Hand specimens of BPU ore originated from the Hack II Mine (14 samples) and Pigeon Mine (2 samples) (Fig. 1). Van Gosen et al. (2020a, b) noted that some samples were more enriched in U and others more enriched in sulfide-related elements (Van Gosen et al. 2020b). Based on these categories, new pieces (individually 42–230 g) were broken off hand specimens to create a U-enriched, composite 1 (C1; total 1633 g) and a sulfide-enriched, composite 2 (C2; total 773 g). Materials were blended by crushing to < 1 mm in a jaw crusher then riffle-split to create portions for batch experiments and for elemental analysis. Splits for analysis were ground to < 100 µm. Elemental analysis was conducted at AGAT Laboratories. Powdered sample was fused at 750 °C with sodium peroxide, and the fusion cake was dissolved in dilute nitric acid and analyzed by ICP-OES and ICP-MS (Briggs and Meier 2002; Meier and Slowik 2002). Mineralogy of the two composites was not analyzed but sulfides and uranium-bearing minerals can be assumed to be the same chalcopyrite, galena, pyrite, sphalerite, and uraninite present in the samples used to create the composites (Van Gosen et al. 2020a, b).
Perspective on the composition of the BPU ore composites was gained by comparing to the compositions of samples from which they were derived and to the compositions of 27 samples of mineralized breccia from the Pinyon Plain Mine (formerly Canyon Mine), and BPU ore data obtained from the National Geochemical Database (Bern et al. 2019; Van Gosen et al. 2020a) (Fig. 1). The latter included data on samples from the following deposits/mines: Pinyon Plain Mine (formerly Canyon Mine (n = 7), EZ-2 Deposit (n = 1), Hack Canyon Mine (n = 23), Hermit Mine (n = 6), Kanab North Mine (n = 9), Orphan Mine (n = 13), Pigeon Mine (n = 9), Ridenour Mine (n = 4), and Sage Deposit (n = 42). A classical principal component analysis (PCA) was conducted using the software R (R Core Team 2021) that focused on patterns and variability in mineralization by restricting it to the elements Ag, As, Cd, Co, Cu, Fe, La, Mo, Ni, P, Pb, S, Sb, U, V, Y, and Zn. First, missing data (1.9% of total) were imputed using the function “impCoda,” and then the PCA was conducted using the function “pcaCoDa”, both from the “robCompositions” package (Filzmoser et al. 2009; Hron et al. 2010). This method applied an isometric log-ratio transformation to the data, performed a robust principal component analysis, and then back-transformed the output of the analysis to centered log-ratio space to generate a more easily interpreted biplot.
Assessment of Groundwater CO2
Carbon dioxide plays a critical role in carbonate equilibria and thereby groundwater chemistry generally, but is especially important for U because of the formation of U-carbonate aqueous complexes (Dong and Brooks 2006; Stumm and Morgan 1995). To determine an appropriate CO2 concentration for the experiments, most probable concentration values of major ions were used from waters obtained from wells at the Pinenut Mine and the Pinyon Plain (formerly Canyon Mine) (Fig. 1) sampled in 2009 (Bills et al. 2010). Saturation indices for CO2 were calculated using the PHREEQC software (Parkhurst and Appelo 1999) and converted to CO2 concentrations. The Pinenut and Pinyon Plain wells had CO2 concentrations of 1.16% and 2.06%, respectively; thus, an average value of 1.6% CO2 was used in the experiments.
Sequential Batch Experiments
To simulate potential pathways and evolution of water involving contact with BPU ore, sequential batch experiments were performed consisting of five steps (Figs. 2, 3). Step 1 created artificial shallow groundwater. To do this, 17 L of deionized (18.2 MΩ) water was placed in an acid-washed, 20 L, high density polyethylene (HDPE) carboy, and 850 g of rock was added consisting of a 1:1 mixture of aquifer-related rock samples SK2 (Kaibab Formation) and SK4 (Toroweap Formation) crushed to < 1 mm (Table 1). Dissolution of calcite and dolomite from these rocks in the perched aquifer zone helped to create a solution in general equilibrium with carbonate. A humidified stream of 1.6% CO2 and air was bubbled through the solution. The solution was constantly recirculated using a peristaltic pump for one week and stirred twice daily for the rest of the equilibration. Stirring or shaking during each of the experimental steps increases contact between solutions and geologic materials and helps speed equilibration and simulate flow of solutions through geologic materials. After two weeks, 12 g of reagent grade gypsum (CaSO4·2H2O) and 0.45 g of NaCl were added to the reactor. Addition of these reagents was necessary to mimic natural groundwater (Bills et al. 2010) because gypsum and other salts have generally been leached from surface exposures where the aquifer-related rocks were collected. After four weeks, the solution had reached a stable specific conductance (SC) of 1,065 µS/cm; this solution was then filtered first to < 0.45 µm and then to < 0.2 µm. Aliquots were taken for analysis and acidified to pH < 2 using ultrapure nitric acid.

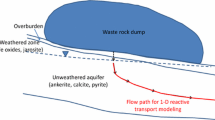
Conceptual cross section of a breccia pipe uranium (BPU) mine and generalized sedimentary rock stratigraphy showing hypothetical flow paths and locations of waters that may correspond to the different steps in the sequential batch experiments. Labeled water types/experimental steps are: (1) shallow groundwater, (2) mine water, (3) anoxic downgradient groundwater, (4) oxic downgradient water, (5) aged, oxic, downgradient water. Figure is not to scale or representative of stratigraphy

Conceptual diagram depicting the sequential batch experiments. Sample identifiers listed in Table 1
Step 2 created artificial mine water. Two splits each of 6 L of artificial groundwater were placed in acid-washed, deionized water-rinsed, 10 L HDPE carboys and 300 g of C1 (U-enriched) were added to one carboy and 300 g of C2 (sulfide-enriched) were added to the other carboy. The same humidified stream of 1.6% CO2 and air was bubbled through both solutions. The solutions were stirred daily. After two weeks, the solutions had reached stable SC values of 2,430 (1) and 3,150 µS/cm (2), respectively. The solutions were filtered first to < 0.45 µm and then to < 0.2 µm.
Step 3 simulated artificial mine water moving downgradient into an anoxic environment. The artificial mine water was sparged with a gas mixture of 2.2% H2, 1.6% CO2, and 96.2% N2 inside a glove bag containing the same gas mixture and then stored overnight. In the same glove bag, 400 mL aliquots of the two artificial mine waters were added to five 500 mL HDPE bottles each. To these were added 20 g of the powdered aquifer-related rock samples SK7, SK8, SK9, SK12, and SK14 (Table 1), selected to represent a range of mineralogical compositions described in the “Results and Discussion” section. The solutions in the bottles were agitated by using stir plates and hand shaking inside the glove bag. After two weeks, the samples were removed from the glove bag and aliquots for analysis were quickly syringe filtered to < 0.2 µm to avoid oxidation.
Step 4 simulated downgradient waters entering an oxic environment, but without the influence of additional reaction with the aquifer-related rocks, to separate possible influences on water quality and simulate emergence at a stratigraphically shallow spring. Water remaining from Step 3 was decanted into acid-washed, deionized water-rinsed HDPE bottles and stored at room temperature in the laboratory for nine weeks with air in the headspace of the bottles. Aliquots for analysis were then syringe filtered to < 0.2 µm.
Step 5 simulated increasing length of time for reactions to proceed to equilibrium and taking into account prolonged water–rock interaction. The influence of the oxic environment (Step 4) was maintained for 40 weeks. Then, the influence of the aquifer-related rocks for an additional two weeks was simulated by adding 20 g of new splits of the same crushed, aquifer-related rock used in Step 3 to each respective bottle of water. The water was then stored at room temperature with periodic shaking to agitate the mixture. Aliquots for analysis were then syringe filtered to < 0.2 µm.
Concentrations of 64 dissolved elements in waters from the experiments were measured by ICP-MS and ICP-OES by AGAT Laboratories (Calgary, Alberta, Canada) (Lamothe et al. 2002). Blind standards were submitted for quality control, and results were considered satisfactory if within 15% of expected values. Data among the elements discussed in the paper met those objectives except for one blind standard with low concentrations of Co (1.07 µg/L) and Ni (6.89 µg/L) that failed this criteria. However, in the same analytical batch, contract laboratory standards with higher concentrations performed acceptably. Anions (Cl−, F−, NO3−, Br−) were analyzed by ion chromatography at a USGS laboratory in Boulder, Colorado (Brinton et al. 1995). Alkalinity was determined by titration on samples from Steps 1 and 2 using the inflection point method (U.S. Geological Survey variously dated). Volume limitations prevented alkalinity determinations on subsequent steps. Charge balance and saturation indices for calcite and dolomite were calculated for samples from Steps 1, 2, and 3 using the PHREEQC software and the Minteq.v4 database (Parkhurst and Appelo 1999). Charge balance errors were < 4%, although the redox potential (pe = 1) had to be estimated for Step 3 because it was not measured. Selection of the estimated pe value is discussed in the “Results and Discussion” section. Saturation indices for gypsum, barite, ferrihydrite, goethite, hematite, uraninite, carnotite, tyuyamunite, U3O8, and UO2(OH)2(β) were calculated for all solutions using the PHREEQC software and the Minteq.v4 database (Parkhurst and Appelo 1999). Charge balance and saturation index output are available in the Supplementary Information.
Results and Discussion
Aquifer-Related Rocks
Elemental concentrations of the contaminants of potential concern associated with mineralization in BPU ore material were generally at least an order of magnitude lower in the aquifer-related rocks (samples SK1-14) compared to concentrations in BPU ore (Bern et al. 2021). Samples of the Kaibab (SK2) and Toroweap (SK4) Formations contained abundant dolomite and calcite, respectively, making a blend of the two suitable for creating a carbonate-equilibrated groundwater in the experiments (Table 1). The five deeper, aquifer-related rocks selected for use in Steps 3 and 5 were chosen to represent different mineralogical types. Coconino Sandstone (SK7) was quartz-dominated with little carbonate buffering capacity and moderate clay content. The Hermit Formation (SK8) contained more abundant clay and 1.2 wt.% of hematite. Esplanade Sandstone (SK9) had moderate clay content and carbonate buffering capacity, primarily from dolomite. Redwall Limestone (SK12) and Muav Limestone (SK14) consisted almost completely of calcite and dolomite, respectively (Table 1).
Composite Ore Samples
Compositional patterns in BPU ore and the two composite samples can be observed by examining a biplot of results from the PCA (Fig. 4). One pattern is that the loadings, depicted as rays, organize themselves into four groups: (1) a Co, Fe, Mo, and Ni group; (2) an Sb, As, Cd, S, and Zn group; (3) a La, Pb, P, U, V, and Y group; and (4) a Cu plus Ag group. Such groupings may not represent mineral phases so much as sources or conditions related to mineralization. Another pattern is the broad overlap of compositions from ten different localities. Such overlap indicates that, although there is variation in the composition of samples from within individual BPU deposits, similar compositions can be found in other localities. An exception to this pattern exists for some material from the Pinyon Plain Mine, which exhibits relative enrichment in As, Cu, and Ag. Composite 1 (C1) plots near the center of the data distribution, indicating it is fairly representative of BPU ore material in the region. Based on high U content (Table 2), C1 is referred to as the U-enriched composite. Composite 2 (C2) plots out near one edge of the data distribution, indicating a less common composition with greater enrichment of sulfide and associated trace elements. Based upon this enrichment, C2 is referred to as the sulfide-enriched composite. Note that samples from six other localities plot relatively close to C2, indicating that similar material is not rare among BPU deposits and localities.

Biplot depicting classical principal component analysis of BPU ore compositions along with the two BPU ore composites C1 (U-enriched) and C2 (sulfide-enriched) created from archival material. Data included are 17 elements associated with mineralization: antimony (Sb), arsenic (As), cadmium (Cd), cobalt (Co), copper (Cu), iron (Fe), lanthanum (La), lead (not visible), molybdenum (Mo), nickel (Ni), phosphorus (P), silver (Ag), sulfur (S), uranium (U), vanadium (V), yttrium (Y), and zinc (Zn). Principal component (PC) scores for samples are plotted on the bottom and left axes, and loadings (rays) for elements are plotted on the top and right axes
Sequential Batch Experiments, Major Ions
Substantial changes in chemistry occurred as the sequential batch experiment waters moved through the five experimental steps. An examination of selected trace elements and major ions provides insight to bulk geochemical changes. Artificial shallow groundwater from Step 1 was circumneutral (Table 3, Fig. 5), representing a Ca-HCO3 type composition and was oversaturated relative to calcite (saturation index, SI = 0.2) and barite (SI = 0.2) and undersaturated relative to dolomite (SI = − 0.4) and gypsum (SI = − 0.7). The major ion concentrations generally were similar to those in water that discharges from springs in the Coconino Sandstone (Beisner et al. 2017b).

Concentrations of dissolved major elements and pH in order of steps in the sequential batch experiments: a pH, b aluminum, c iron, d manganese, and e sulfur. Square symbols reflect solutions made from the C1 (uranium-rich) composite and diamond symbols reflect solutions made from the C2 (sulfide-rich) composite
In Step 2, exposure to the U-enriched ore composite (C1) decreased the solution pH slightly from 7.1 to 6.8 (Table 3, Fig. 5a). Elemental Ca and Mg concentrations (Table 2) indicated potential carbonate buffering of the C1-derived solution as did saturation relative to calcite (SI = 0.2) and moderate undersaturation relative to dolomite (SI = − 0.9). Carbonate minerals can be intimately mixed with sulfide minerals in BPU ore (Van Gosen et al. 2020a, b). Exposure to the sulfide-enriched ore composite (C2) decreased the pH substantially to 3.8 and the absence of measurable alkalinity indicated a lack of carbonate buffering. Concentrations of total sulfur (S) increased substantially in both Step 2 solutions, indicating oxidation of sulfide minerals in the ore as the source of the acidity, although much greater concentrations of S occurred in the C2-derived solution. Both solutions were saturated relative to gypsum (SI = 0) and barite (SI > 0.5 and 0.6), indicating secondary precipitation, particularly of barite, as a limiting factor on S concentrations. Dissolved Al and Fe increased substantially in the C2-derived artificial mine water (Table 3, Fig. 5). Iron was likely released by pyrite oxidation and the drop in pH helped sustain both elements in solution. Concentrations of Mn increased slightly in both Step 2 solutions (Table 3, Fig. 5).
In the artificial anoxic, downgradient water created in Step 3, the pH increased in most samples, likely in response to carbonate buffering from the aquifer-related rocks (Fig. 5a). All the C1-derived solutions were oversaturated relative to calcite, except for the solution exposed to the Coconino Sandstone (SI = − 0.5). In the C2-derived solutions, pH buffering was incomplete as indicated by undersaturation relative to calcite (SI = − 3.4 to − 0.7) and dolomite (SI = − 7.4 to − 2.2). The partial buffering appeared to drive dissolved Al concentrations in the C2-derived samples dramatically lower (Fig. 5b), presumably by precipitating aluminum minerals. Using the reporting limit concentration of Al (1 µg/L), C2-derived solutions were indicated to be undersaturated relative to the Al(OH)3 minerals amorphous aluminum hydroxide and gibbsite. Nevertheless, precipitation was the likely loss mechanism, because concentrations of Al in Step 2 (17.8 mg/L) and pH conditions in Step 3, would have caused solutions to be oversaturated relative to both phases during the transition from Step 2 to Step 3 conditions. Based on the Al concentration decrease, ≈ 51.5 mg/L of Al(OH)3 would have precipitated from each C2-derived solution in this step.
In Step 3, iron concentrations decreased slightly in the C2-derived solutions, indicating precipitation of hydrous-ferric-oxide (HFO) minerals, likely goethite but possibly also ferrihydrite (Blowes et al. 2003). The precipitation of some but not all of the dissolved iron reflects moderately reducing conditions that favor retention in solution and increased pH that favors precipitation. Because pe was not measured, it was estimated in the PHREEQC calculations. A pe value of 1.0 was about the lowest pe that would indicate saturation for either goethite or ferrihydrite in the C2-derived solution exposed to Coconino Sandstone (SK7) and therefore reflect the modest decrease in solution Fe (Fig. 5c). Using that pe value, all C2-derived solutions were oversaturated relative to goethite (SI = 0.0–3.5) and results for ferrihydrite were mixed (SI = − 2.7 to 0.8). A pe value of 1.0 does not indicate strongly reducing conditions, but moderately reducing conditions are indicated by maintenance of substantial Fe in solution, as well as mobilization of Mn from the aquifer-related rocks (Fig. 5c, d). Based on Fe concentration decreases, 13–92 mg/L of HFO minerals would have precipitated from the different batches. The precipitation of HFO and Al(OH)3 indicate the formation of secondary minerals with fresh surfaces for sorption of trace elements.
When the waters were exposed to oxygen in Step 4, but without any buffering from aquifer-related rocks, pH increased about 1 standard unit from ≈ pH 7 to ≈ pH 8 in the C1-derived solutions (Fig. 5a). In contrast, pH decreased almost two standard units from ≈ pH 6 to ≈ pH 4 in the C2-derived samples, presumably as acidity was increased as a result of oxidation of Fe2+ to Fe3+, hydrolysis of the Fe(III), and subsequent precipitation as HFO. In Step 4, 43–158 mg/L of HFO and associated sorption potential was potentially generated among the C2-derived solutions. Both Al and Mn concentrations remained essentially unchanged (Fig. 5b, 5=d). Total S concentrations decreased in the C2-derived solutions in Step 4 (Fig. 5e). Although all solutions remained saturated relative to gypsum (SI ≈ 0), the S decrease was attributed to barite precipitation. Barium concentrations had increased in all solutions in Step 3 in response to aquifer-related rock additions (Fig. 6a) and all solutions were oversaturated relative to barite (SI = 0.7–1.1). However, the Step 4 pH increase for C2-derived solutions created conditions more favorable for barite precipitation. Both Ba and total S concentrations decreased for C2-derived solutions (Figs. 5e, 6a), and both C1 and C2-derived solutions remained oversaturated relative to barite (SI = 0.7–1.1).

Concentrations of dissolved barium (a) and vanadium (b) in the sequential batch experiments. Square symbols reflect solutions made from the C1 (uranium-rich) composite and diamond symbols reflect solutions made from the C2 (sulfide-rich) composite
With the addition of aquifer-related rocks again in Step 5, carbonate buffering shifted most C1- and C2-derived solutions to a circumneutral pH (Fig. 5a). Greater pH caused most of the remaining dissolved Fe in C2-derived solutions to precipitate. Saturation indices indicated oversaturation for goethite (SI = 3.3–4.3) and ferrihydrite (SI = 0.6–1.6) and 107–250 mg/L of HFO and associated sorption capacity were potentially generated. With more Ba available for release by re-addition of the aquifer-related rocks (Fig. 6a), additional barite precipitation appeared to draw most dissolved S concentrations down further across both C1- and C2-derived samples (barite SI = 0.8–1.1) (Fig. 5e). Aluminum concentrations remained relatively unchanged and Mn showed relatively small effects.
Sequential Batch Experiments, Contaminants of Potential Concern
Mobilization of trace elements from BPU ore was assessed by taking the artificial shallow groundwater from Step 1 and exposing it to the ore composites in Step 2. Concentrations of trace elements were generally low in Step 1 and increased substantially in Step 2 (Table 3, Fig. 7). More than 20% of the U in each composite was mobilized in Step 2 (Table 2) indicating a high potential for mobilization. Uranium increased from 0.5 µg/L in Step 1 to 201,000 µg/L in the C1-derived artificial mine water in Step 2 (Table 3). Most U in BPU ore is present as uraninite (UO2) (Van Gosen et al. 2020a, b; Wenrich 1985), but it is plausible that over geologic time some has oxidized to U3O8. The C1-derived artificial mine water (Step 2) was substantially undersaturated relative to uraninite (SI = − 2.8), moderately undersaturated relative to UO2(OH)2(β) (SI = − 0.6), and barely undersaturated relative to U3O8 (SI = − 0.1). In contrast, the C1-derived water was oversaturated relative to carnotite (SI = 0.5) and tyuyamunite (SI = 2.9). Thus, although U concentration reached 201,000 µg/L, a higher concentration may have been limited by reaching near equilibrium conditions and/or secondary precipitation as suggested by decreases in dissolved V concentrations (Fig. 6b). In comparison, the C2-derived artificial mine water (Step 2) had a U concentration of 1,980 µg/L and was moderately undersaturated relative to uraninite (SI = − 0.4) and substantially undersaturated relative to UO2(OH)2(β) (SI = − 4.2), U3O8 (SI = − 4.9), carnotite (SI = − 9.9), and tyuyamunite (SI = − 18.5). Thus, the relatively small amounts of U-bearing material in the C2 ore composite likely limited U concentrations in the solution.

Concentrations of dissolved trace elements in the sequential batch experiments: a antimony, b arsenic, c cadmium, d cobalt, e copper, f lead, g molybdenum, h nickel, i selenium, j thallium, k uranium, and l zinc. Square symbols reflect solutions made from the C1 (uranium-rich) composite and diamond symbols reflect solutions made from the C2 (sulfide-rich) composite. Symbol colours are the same as in Fig. 5. Note logarithmic scales
Similar to how U was mobilized in Step 2, the fractions of Co and Ni leached from the two ore composites were large and relatively similar, thus indicating presence in relatively labile forms (Table 2). Concentrations of such elements in solution are thus controlled more by ore composition than water chemistry. For Sb, Se, Tl, and Zn, percent mobilizations were more similar, but abundance in ore still influenced relative concentrations in solution (Table 2, Fig. 7). Percent Mo mobilization was much greater from the C1 composite, but the higher concentration in the C2 composite appeared to help generate similar concentrations in solution. In contrast, percent mobilizations of As, Cd, Cu, and Pb were an order of magnitude or more greater from the sulfide-enriched, C2 ore composite compared to the C1 composite (Table 2). As a result, concentrations of most of those elements were much greater in the C2-derived water (Fig. 7). In some cases, concentrations of a trace element were only moderately greater in the C2 ore composite itself (Table 2), and the lower pH of the C2-derived water (Fig. 5a) drove the magnitude of mobilization. Acidification of waters by sulfide oxidation and consequent trace element mobilization is one of the predominant patterns of AMD (Blowes et al. 2003; Nordstrom and Alpers 1997). By comparison, the carbonate buffered, circumneutral pH of Step 2 waters created using the C1 ore composite somewhat limited mobilization of As, Cd, Cu, and Pb, and perhaps Tl and Zn. Thus, carbonate buffering can limit some element mobilization if the carbonates are in relatively close contact with mineralized, acid-generating material. However, relatively high concentrations (> 1000 µg/L) of Co, Ni, U, and Zn, along with notable concentration of Mo (420–500 µg/L) occurred in both the acidic (C2) and circumneutral (C1) artificial mine waters, thus indicating high potential for mobilization under most conditions (Fig. 7d, g, h, k, l).
Downgradient attenuation of concentrations of contaminants of potential concern mobilized from the BPU ore was assessed in Steps 3, 4, and 5. Each trace element responded differently, but the similarity in response among steps allows some elements to be grouped together and shed light on processes affecting trace element attenuation. Copper and Pb concentrations were both substantially attenuated in Step 3 (Fig. 7e, f). Neutralization of the C2-derived waters in Step 3 (Fig. 5) and precipitation of HFO minerals and some Al(OH)3 would have provided pH conditions and sorption sites for Cu and Pb removal. Both Cu and Pb were adsorbed early in the shift from acidic to neutral pH and with relatively high sorption selectivity (Dzombak and Morel 1990; Smith 1999). However, Cu and Pb were also removed from C1-derived solutions where absence of fresh HFO or Al(OH)3 made sorption less likely. Instead, precipitation as chalcopyrite (CuFeS2) and galena (PbS) are a possible removal mechanism. Although at the pe of 1.0 estimated based upon goethite saturation indicates the solutions would have been undersaturated relative to chalcopyrite and galena, both the C1- and C2-derived solutions would have been saturated relative to those phases at moderately lower pe values of − 1 to − 4. Some Pb and Cu were apparently remobilized under the acidic and oxidizing conditions in some Step 4 samples, but this is most likely due to to accidental transfer of precipitated minerals during decanting between steps. Cadmium and Zn are expected to sorb to a lesser extent on HFO, as their sorption behaviour starts at slightly higher pH values (Dzombak and Morel 1990; Smith 1999). It is possible that competition for binding sites left substantial fractions of these elements in solution until the circumneutral conditions and additional precipitation of HFO that occurred in Step 5. The experiments do not distinguish between sorption and precipitation, but decreases in dissolved concentrations through Steps 3, 4, and 5 indicate attenuation.
Most of the substantial amounts of As mobilized in the C2-derived waters in Step 2 remained in solution in step 3, but then concentrations decreased by factors of 3–76 in Step 4 and decreased further in Step 5 (Fig. 7b). The retention of As in solution in Step 3 may have resulted from some interplay of potential oxidation of As(III) to As(V) and their contrasting sorption edges in the vicinity of pH 6 (Dixit and Hering 2003). However, relatively high concentrations of other trace elements competing for binding sites in Step 3 might be the best explanation. Greater amounts of HFO precipitation in Step 4, and a decrease to pH ~ 4 by the C2-derived waters would certainly have enhanced As(V) sorption (Dixit and Hering 2003). Availability of binding sites is also likely a factor, as illustrated by similar orders of concentration decrease in As and Fe between Steps 4 and 5 (Figs. 5c, 7b).
Selenium is also redox sensitive and decreased in concentrations in Step 3 (Fig. 7i), likely reflecting sorption of the lower redox-state Se(IV) on HFO under circumneutral conditions, but some sorption of Se(VI) could also have occurred if it was present (Fernándes-Martínez and Charlet 2009). Under oxidative conditions and acidic pH in the C2-derived waters (Step 4), both Se(IV) and Se(VI) would have been sorbed (Balistrieri and Chao 1990). Lack of additional Se removal in Step 5 likely reflects the predominance of Se(VI) and its poor sorption at circumneutral pH.
The iron-oxidation-driven drop in pH for C2-derived waters in Step 4 was possible because of the absence of any carbonates from aquifer-related rocks. This created ideal conditions for removal of Sb and Mo, both of which approach their sorption maxima around pH 4 (Goldberg et al. 1998; Guo et al. 2014). However, the combination of acidic pH and HFO precipitation may be unusual in groundwater flow paths in the Grand Canyon as it would require a portion of the flow path to pass through carbonate-free rock. Among the sedimentary rocks of the Grand Canyon region, only the Coconino Sandstone comes close to this requirement, and it contains about 1.4 wt.% carbonate (Table 1). Thus, the patterns of Sb and Mo mobilization and scant removal demonstrated by the C1 waters seem more likely in the environment.
Certain elements did not show substantial decreases in dissolved concentrations under most conditions encountered in Steps 3, 4, and 5 (Fig. 7). This is broadly true for Co, Ni, Tl, and U. Cobalt, Ni, and Tl concentrations decreased somewhat under the oxic and circumneutral conditions in Step 5. Sorption on minerals present in the aquifer-related rocks appeared to play a role because decreases were most pronounced in the presence of the relatively clay-rich Esplanade Sandstone and Hermit Formation (Table 1, Fig. 7d, h, j).
The lack of change in U concentrations in the C1-derived waters (Fig. 7k) is explained partly by the strong tendency of U to form stable complexes with carbonate anions along with Ca and Mg, thereby decreasing U adsorption (Dong and Brooks 2006). The C1-derived waters were oversaturated relative to various U-bearing phases in Steps 3, 4, and 5, In Step 3, solutions were oversaturated relative to uraninite (SI = 1.0–4.9) and U3O8 (SI = 3.3–7.9) but no decrease in solution U was observed. In Step 4, C1-derived solutions were oversaturated relative to U3O8 (SI = 2.2–2.4), UO2(OH)2(β) (1.0–1.1), carnotite (SI = 0–2.1), and tyuyamunite (SI = 1.5–5.9), but decreases in dissolved U were so minor relative to the high concentrations as to be suspect (Fig. 7k). Solutions were oversaturated relative to the same four phases in Step 5, but U concentrations were once again similar to those in Steps 2 and 3 (Fig. 7k). In contrast, U concentrations decreased for the C2-derived waters by factors of 1.5–9.2, 1.8–2.1, and 1–13 in Steps 3, 4, and 5, respectively (Fig. 7k). Although many of the solutions were oversaturated relative to some U-bearing phases in some steps, these concentration decreases were likely due to sorption of U onto HFO. In Step 3, the C2-derived solutions were oversaturated relative to uraninite (SI = 2.0–4.5) and U3O8 (SI = 0.3–4.7). In Step 4, C2-derived solutions were undersaturated relative to all modelled U-bearing phases. In Step 5 C2-derived solutions were variably saturated relative to carnotite (SI = − 31.1 to 1.4), tyuyamunite (SI = − 5.2 to 3.7), and U3O8 (SI = − 0.9 to 0.1). However, matches between patterns of U and Fe concentration decreases that reflected precipitation of HFO make the argument for sorption (Figs. 5c, 7k). Although U forms stable complexes with carbonate, and U sorption decreases with increasing carbonate concentration, U is still sorbed by HFO in the presence of carbonate (Wazne et al. 2003). In Steps 3, 4, and 5, increasing amounts of Fe from solution were precipitated as HFO and increasing amounts of U were also lost from solution (Figs. 5c, 7k). The pattern of most substantial decreases in U concentrations occurring in Step 5 matched the most substantial decreases in dissolved Fe occurring in that step. The link between U and sorption on HFO is also supported by proportionately smaller U loss from the C2-SK7 batch, which was the only batch still retaining substantial dissolved Fe in Step 5 (Figs. 5c, 7k). In considering the potential for additional attenuation of dissolved U concentrations, notable additional U removal in response to reactions with aquifer-related rocks would not be expected once dissolved Fe to support HFO precipitation was depleted.
With respect to trace element patterns, relatively high concentrations of As, Cu, and Pb were mobilized from the BPU ore, particularly if intimately mixed carbonate minerals were insufficiently abundant to buffer the acidity generated by sulphide oxidation. However, subsequent experimental steps, designed to simulate downgradient aquifer conditions and processes expected in the subsurface environment, largely attenuated As, Cu, and Pb concentrations. In contrast, relatively high concentrations of Co, Mo, Ni, U, and Zn were mobilized from the BPU ore regardless of buffering by carbonates in the ore. Of these, Co, Mo, Ni, and U were not substantially removed by simulated downgradient conditions, and Zn was only partially attenuated. Mobilization of Sb and Tl from BPU ore generated more moderate concentrations in water, but these elements were also poorly attenuated.
Environmental Factors
The experiments conducted here provide insight into potential effects and water compositions resulting from water interacting with BPU deposits, but concentrations of trace elements in the environment will be influenced by additional factors. Heterogeneity in the abundance, composition, and reactivity of both mineralized BPU material and other rock types along flow paths will all be factors, along with heterogeneity in the flow paths themselves (Nordstrom 2011). Trace elements can be sorbed both on static surfaces and fresh precipitates suspended in water (Runkel et al. 1999). Factors such as ratios of water to ore and contact times with different materials are likely to differ from those used in the experiments. The experiments indicate that mobilization of some trace elements will be attenuated by reactions with downgradient carbonate-bearing rock, and mobilization of others will not. Acid-neutralizing carbonate rock commonly attenuates some components of AMD (Blowes et al. 2003). Concentrations of the non-attenuated trace elements are likely to become diluted by mixing with downgradient waters unaffected by mineralized BPU material that likely have lower concentrations of those elements (Beisner et al. 2017b). Mixing ratios and dilution will depend on the relative volumes of water, and therefore relate to the scales of deposits and unaffected waters.
With regards to scale, two other factors are worth considering and comparing to other mineralized settings and mining operations to provide perspective. First, in many mining districts, mineralized rock with acid-generating capacity is semi-continuous across areas of several to tens of km2, and development involves multiple mine adits, extensive lateral tunnel systems, and substantial fractions of acid-generating rock that are left exposed to oxidation underground or on the surface when mining is complete (Cowie et al. 2014; España et al. 2005). By comparison, U-bearing, breccia pipes with narrow diameters (60–120 m) and low ore tonnage (160,000 tons) are rather modest in terms of size and spaced widely across the landscape in large expanses of unmineralized rock (Fig. 1; Mathisen et al. 2017; Otton and Van Gosen 2010; Van Gosen et al. 2016). Second, it is common in many mining operations to create artificial flow paths via lateral tunnels that drain mine water directly or indirectly to emerge nearby at the surface and with minimal opportunity to contact non-mineralized rock (Walton-Day and Mills 2015). By comparison, BPU mines are often vertical features that might facilitate entry of water from the surface, but not necessarily create enhanced vertical or lateral drainage away from the mineralized zone (Fig. 2). Vertical infiltration of water from the surface can be minimal under the generally arid climate in the Grand Canyon region areas (Alpine and Brown 2010). Thus, the relatively small scale of BPU deposits, modest alteration to natural flow paths, and carbonate buffering capacity of surrounding rock, along with potential dilution by mixing with unaffected groundwaters, all indicate smaller potential for environmental issues related to trace element mobilization compared to many other acid-generating deposit types.
Conclusions
In summary, laboratory experiments designed to simulate groundwater environments demonstrated that sulfide-bearing BPU ore has the potential to mobilize contaminants of potential concern in a manner similar to those in AMD produced from other sulfide-bearing ore deposits. Mobilization of certain elements, particularly As, was enhanced if carbonate minerals were not intimately mixed with the oxidizing sulfides to buffer the pH. Regardless of buffering at the source, relatively high concentrations of Co, Mo, Ni, U, and Zn were mobilized from the BPU ore. Of these, Co, Mo, Ni, and U were not substantially removed by downgradient conditions and processes expected in the subsurface environment, and Zn was only partially attenuated. Smaller proportional concentrations of Sb and Tl were mobilized, but these elements were also poorly removed under expected environmental conditions. Such patterns indicate which BPU ore-sourced trace elements are most likely to be present at a downgradient location, like a well or spring, from both a toxicological and fingerprinting perspective. Trace element mobilization in the environment will be determined by the quantities of water contacting the relatively small BPU deposits, and concentrations are likely to be lowered by mixing with other waters that have not contacted the BPU ore.
References
Alpine AE (ed) (2010) Hydrological, geological, and biological site characterization of breccia pipe uranium deposits in Northern Arizona. U.S. Geol. Surv., Sci. Inv. Rep. 2010–5025, https://pubs.usgs.gov/sir/2010/5025/. Accessed 9 Nov 2017
Alpine AE, Brown KM (2010) Introduction. In: Alpine AE (ed) Hydrological, geological, and biological site characterization of breccia pipe uranium deposits in Northern Arizona. U.S. Geol. Surv., Sci. Inv. Rep. 2010–5025, p 1–18, https://pubs.usgs.gov/sir/2010/5025/. Accessed 9 Nov 2017
Arnberger R, Martin S (2018) Grand Canyon is a national treasure, not a place for uranium mining. https://www.cnn.com/2018/01/09/opinions/grand-canyon-mining-opinion-arnberger-martin/index.html. Accessed 3 May 2022
Balistrieri LS, Chao TT (1990) Adsorption of selenium by amorphous iron oxyhydroxide and manganese dioxide. Geochim Cosmochim Acta 54:739–751. https://doi.org/10.1016/0016-7037(90)90369-V
Beisner KR, Paretti NV, Tillman FD, Naftz DL, Bills DJ, Walton-Day K, Gallegos TJ (2017a) Geochemistry and hydrology of perched groundwater springs: assessing elevated uranium concentrations at Pigeon Spring relative to nearby Pigeon Mine, Arizona (USA). Hydrogeol J. https://doi.org/10.1007/s10040-016-1494-8
Beisner KR, Tillman FD, Anderson JR, Bills DJ (2017b) Geochemical characterization of groundwater discharging from springs north of the Grand Canyon, Arizona, 2009–2016. US Geol Surv Sci Inv Rep. https://doi.org/10.3133/sir20175068
Beisner KR, Solder JE, Tillman FD, Anderson JR, Antweiler RC (2020) Geochemical characterization of groundwater evolution south of Grand Canyon, Arizona (USA). Hydrogeol J. https://doi.org/10.1007/s10040-020-02192-0
Bern CR, Walton-Day K, Naftz D (2019) Improved enrichment factor calculations through principal component analysis: Examples from soils near breccia pipe uranium mines, Arizona, USA. Environ Pollut 248:90–100. https://doi.org/10.1016/j.envpol.2019.01.122
Bern CR, Campbell KM, Walton-Day K, Keith GL (2021) Data from batch experiments to test mobility of trace elements down gradient from breccia pipe uranium deposits. US Geol Surv Data Rel. https://doi.org/10.5066/P9VILVZY
Bills DJ, Flynn ME, Monroe SA (2007) Hydrogeology of the Coconino Plateau and adjacent areas, Coconino and Yavapai Counties, Arizona. US Geol Surv, Sci Inv Rep 2005–5222, p 101, https://doi.org/10.3133/sir20055222
Bills DJ, Tillman FD, Anning DW, Antweiler RC, Kraemer TF (2010) Chapter C, Historical and 2009 water chemistry of wells, perennial and intermittent streams, and springs in northern Arizona. In: Alpine AE (ed) Hydrological, geological, and biological site characterization of breccia pipe uranium deposits in Northern Arizona. U.S. Geol. Surv. Scientific Inv. Rep. 2010–5025, p 141–282, https://pubs.usgs.gov/sir/2010/5025/. Accessed 7 Dec 2020
Blowes DW, Ptacek CJ, Jambor JL, Weisener CG (2003) 9.05—The geochemistry of mine drainage. Treatise Geochm 14:204. https://doi.org/10.1016/B0-08-043751-6/09137-4
Briggs PH (2002) Chapter G. The determination of forty elements in geological and botanical samples by inductively coupled plasma-atomic emission spectrometry. In: Taggart Jr. JE (ed) Analytical methods for chemical analysis of geologic and other materials. U.S. Geol. Surv., Open-File Rep. 02–223, https://pubs.usgs.gov/of/2002/ofr-02-0223/OFR-02-0223.pdf. Accessed 11 July 2020
Briggs PH, Meier AL (2002) Chapter I. The determination of forty-two elements in geological materials by inductively coupled plasma–mass spectrometry In: Taggart Jr. JE (ed) Analytical methods for chemical analysis of geologic and other materials. U.S. Geol. Surv., Open-File Rep. 02–223, https://pubs.usgs.gov/of/2002/ofr-02-0223/OFR-02-0223.pdf. Accessed 11 July 2020
Brinton TI, Antweiler RC, Taylor HE (1995) Method for the determination of dissolved chloride, nitrate, and sulfate in natural water using ion chromatography. US Geol Surv Open-File Rep 95–426A, p 16
Brown ZA, Curry KJ (2002) Chapter R. Total carbon by combustion. In: Taggart Jr. JE (ed) Analytical methods for chemical analysis of geologic and other materials. U.S. Geol. Surv., Open-File Rep. 02–223, https://pubs.usgs.gov/of/2002/ofr-02-0223/OFR-02-0223.pdf. Accessed 11 July 2020
Brown ZA, Papp C, Brandt E, Aruscavage P (2002) Chapter S. Carbonate carbon by coulometric titration. In: Taggart Jr. JE (ed) Analytical methods for chemical analysis of geologic and other materials. U.S. Geol. Surv., Open-File Rep. 02–223, https://pubs.usgs.gov/of/2002/ofr-02-0223/OFR-02-0223.pdf. Accessed 11 July 2020
Cleveland CC, Hinck JE, Lankton JS (2019) Assessment of chronic low-dose elemental and radiological exposures of biota at the Kanab North uranium mine site in the Grand Canyon watershed. Integr Environ Assess Manag 15:112–125. https://doi.org/10.1002/ieam.4095
Cleveland D, Hinck JE, Lankton JS (2021) Elemental and radionuclide exposures and uptakes by small rodents, invertebrates, and vegetation at active and post-production uranium mines in the Grand Canyon watershed. Chemosphere. https://doi.org/10.1016/j.chemosphere.2020.127908
Cowie R, Williams MW, Wireman M, Runkel RL (2014) Use of natural and applied tracers to guide targeted remediation efforts in an acid mine drainage system, Colorado Rockies, USA. Water 6:745–777. https://doi.org/10.3390/w6040745
Dixit S, Hering JG (2003) Comparion of Arsenic(V) and Arsenic(III) sorption onto iron oxide minerals: implications for arsenic mobility. Environ Sci Technol 37:4182–4189. https://doi.org/10.1021/es030309t
Dong W, Brooks SC (2006) Determination of the formation constants of ternary complexes of uranyl and carbonate with alkaline earth metals (Mg2+, Ca2+, Sr2+, and Ba2+) using anion exchange method. Environ Sci Technol 40(15):4689–4695. https://doi.org/10.1021/es0606327
Dzombak DA, Morel FMM (1990) Surface complexation modeling: hydrous ferric oxide. Wiley, New York
Eberl D (2003) A user's guide to ROCKJOCK - A program for determining quantitative mineralogy from powder x-ray diffraction data. US Geol Surv Open-File Rep 03–78:41 p., https://pubs.usgs.gov/of/2003/of03-078/. Accessed 11 July 2020
España JS, Pamo EL, Santofimia E, Aduvire O, Reyes J, Barettino D (2005) Acid mine drainage in the Iberian Pyrite Belt (Odiel river watershed, Huelva, SW Spain): Geochemistry, mineralogy and environmental implications. Appl Geochemistry 20:1320–1356. https://doi.org/10.1016/j.apgeochem.2005.01.011
Fernándes-Martínez A, Charlet L (2009) Selenium environmental cycling and bioavailability: a structural chemist point of view. Rev Environ Sci Biotechnol 8:81–110. https://doi.org/10.1007/s11157-009-9145-3
Filzmoser P, Hron K, Reimann C (2009) Principal component analysis for compositional data with outliers. Environmetrics 20:621–632. https://doi.org/10.1002/env.966
Goldberg S, Su C, Forster HS (1998) Sorption of molybdenum on oxides, clay minerals and soils. In: Jenne EA (ed) Adsorption of metals by geomedia. Academic Press, San Diego, pp 401–426
Grand Canyon Wildlands Council (2004) Biological inventory and assessment of ten South Rim springs in Grand Canyon National Park—Revised final report, Report №. Grand Canyon Wildlands Council Inc., National Park Service Contract WPF-230, Flagstaff, Arizona, p 62
Guo X, Wu Z, He M, Meng X, Jin X, Qiu N, Zhang J (2014) Adsorption of antimony onto iron oxyhydroxides: adsorption behavior and surface structure. J Hazard Mater 276:339–345. https://doi.org/10.1016/j.jhazmat.2014.05.025
Hinck JE, Linder G, Otton JK, Finger SE, Little E, Tillit DE (2013) Derivation of soil-screening thresholds to protect the chisel-toothed kangaroo rat from uranium mine waste in northern Arizona. Arch Environ Contam Toxicol 65:332–344. https://doi.org/10.1007/s00244-013-9893-5
Hinck JE, Linder G, Darrah AJ, Drost CA, Duniway MC, Johnson MJ, Méndez-Harclerode FM, Nowak EM, Valdez EW, van Riper IIIC, Wolff S (2014) Exposure pathways and biological receptors: Baseline data for the Canyon Uranium Mine, Coconino County, Arizona. J Fish Wildl Manag 5(2):422–440. https://doi.org/10.3996/052014-JFWM-039
Hinck JE, Cleveland D, Brumbaugh WG, Linder G, Lankton J (2017) Pre-mining trace element and radiation exposure to biota from a breccia pipe uranium mine in the Grand Canyon (Arizona, USA) watershed. Environ Monit Assess 189:56. https://doi.org/10.1007/s10661-017-5765-1
Hron K, Templ M, Filzmoser P (2010) Imputation of missing values for compositional data using classical and robust methods. Comput Stat Data Anal 54:3095–3107. https://doi.org/10.1016/j.csda.2009.11.023
Klymus KE, Richter CA, Thompson N, Hinck JE (2017) Metabarcoding of environmental DNA samples to explore the use of uranium mine containment ponds as a water source for wildlife. Diversity 9:54. https://doi.org/10.3390/d9040054
Lamothe PJ, Meier AL, Wilson SA (2002) Chapter H. The determination of forty four elements in aqueous samples by inductively coupled plasma – mass spectrometry. In: Taggart Jr. JE (ed) Analytical methods for chemical analysis of geologic and other materials. U.S. Geol. Surv, Open-File Rep. 02–223, https://pubs.usgs.gov/of/2002/ofr-02-0223/OFR-02-0223.pdf. Accessed 11 July 2020
Mathisen MB, Wilson V, Woods JL (2017) Technical report on the Canyon Mine, Coconino County, Arizona, U.S.A. NI 43–101 Report:https://www.energyfuels.com/pinyon-plain-mine. Accessed 3 May 2020
Meier AL, Slowik T (2002) Chapter K. Rare earth elements by inductively coupled plasma-mass spectrometry In: Taggart Jr. JE (ed) Analytical methods for chemical analysis of geologic and other materials. U.S. Geol. Surv., Open-File Rep. 02–223, https://pubs.usgs.gov/of/2002/ofr-02-0223/OFR-02-0223.pdf. Accessed 11 July 2020
Minter KM, Jannik GT, Hinck JE, Cleveland CC, Kubilius WP, Kuhne WW (2019) Biota dose assessment of small rodents sampled near breccia pipe uranium mines in the Grand Canyon Watershed. Health Phys 117:20–27. https://doi.org/10.1097/HP.0000000000001041
Naftz D, Walton-Day K (2016) Establishing a pre-mining geochemical baseline at a uranium mine near Grand Canyon National Park, USA. Geoderma Reg 7:76–92. https://doi.org/10.1016/j.geodrs.2016.01.004
Nordstrom DK (2011) Hydrogeochemical processes governing the origin, transport and fate of major and trace elements from mine wastes and mineralized rock to surface waters. Appl Geochem 26:1777–1791. https://doi.org/10.1016/j.apgeochem.2011.06.002
Nordstrom DK, Alpers CN (1997) Geochemistry of acid mine waters. In: Plumlee GS, Logsdon MJ, Filipek LF (eds), The environmental geochemistry of mineral deposits: part a: processes, techniques, and health issues Society of Economic Geologists, https://doi.org/10.5382/Rev.06.06
Otton JK, Van Gosen BS (2010) Uranium resource availability in breccia pipes in northern Arizona. In: Alpine AE (ed), Hydrological, Geological, and Biological Site Characterization of Breccia Pipe Uranium Deposits in Northern Arizona. U.S. Geol. Surv. Sci. Inv. Rep. 2010–5025, p 23–39
Parkhurst D, Appelo C (1999) User's guide to PHREEQC (version 2)—A computer program for speciation, batch-reaction, one-dimensional transport, and inverse geochemical calculations. US Geol Survey Water-Resour Inv Rep, 99–4259
R Core Team (2021) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/. Accessed 4 May 2020
Runkel RL, Kimball BA, McKnight DM, Bencala KE (1999) Reactive solute transport in streams: A surface complexation approach for trace metal sorption. Water Resour Res 35:3829–3840. https://doi.org/10.1029/1999WR900259
Singer PC, Stumm W (1970) Acidic mine drainage: the rate-determining step. Science 167:1121–1123. https://doi.org/10.1126/science.167.3921.1121
Smith KS (1999) Metal soprtion on mineral surfaces: an overview with examples relating to mineral deposits. In: Plumlee GS, Logsdon MJ (eds) The environmental geochemistry of mineral deposits, part a: processes, techniques, and health issues. Society of Economic Geologists Inc., pp 162–182
Solder JE, Beisner KR, Anderson J, Bills DJ (2020) Rethinking groundwater flow on the South Rim of the Grand Canyon, USA: characterizing recharge sources and flow paths with environmental tracers. Hydrogeol J 28:1593–1613. https://doi.org/10.1007/s10040-020-02193-z
Stumm W, Morgan JJ (1995) Aquatic chemistry: chemical equilibria and rates in natural waters. Wiley-Interscience, New York
Tobin BW, Springer AE, Kreamer DK, Schenk E (2018) Review: the distribution, flow, and quality of Grand Canyon Springs, Arizona (USA). Hydrogeol J 26:721–732. https://doi.org/10.1007/s10040-017-1688-8
U.S. Department of the Interior (2012) Record of decision: northern Arizona withdrawal; Mohave and Coconino counties, Arizona. https://eplanning.blm.gov/public_projects/nepa/103221/149482/183513/N._AZ_Record_of_Decision.pdf. Accessed 19 Mar 2020
U.S. Geological Survey (variously dated) National field manual for the collection of water-quality data: U.S. Geological Survey Techniques of Water-Resources Investigations. Book 9, A1-A10 http://pubs.water.usgs.gov/twri9A. Accessed 24 Aug 2016
Van Gosen BS, Johnson MR, Goldman MA (2016) Three GIS datasets defining areas permissive for the occurrence of uranium-bearing, solution-collapse breccia pipes in northern Arizona and southeast Utah. US Geol Surv Data Rel. https://doi.org/10.5066/F76D5R3Z
Van Gosen BS, Benzel WM, Campbell KM (2020a) Geochemical and x-ray diffraction analyses of drill core samples from the Canyon uranium-copper deposit, a solution-collapse breccia pipe, Grand Canyon area, Coconino County, Arizona. US Geol Surv Data Rel: https://doi.org/10.5066/P9UUILQI
Van Gosen BS, Benzel WM, Kane TJ, Lowers HA (2020b) Geochemical and mineralogical analyses of uranium ores from the Hack II and Pigeon deposits, solution-collapse breccia pipes, Grand Canyon region, Mohave and Coconino Counties, Arizona, USA. US Geol Surv Data Rel. https://doi.org/10.5066/P9VM6GKF
Walton-Day K, Mills TJ (2015) Hydrogeochemical effects of a bulkhead in the Dinero mine tunnel, Sugar Loaf mining district, near Leadville, Colorado. Appl Geochem 62:61–74. https://doi.org/10.1016/j.apgeochem.2015.03.002
Wazne M, Korfiatis GP, Meng X (2003) Carbonate effects on hexavalent uranium adsorption by iron oxyhydroxide. Environ Sci Technol 37:3619–3624. https://doi.org/10.1021/es034166m
Wenrich KJ (1985) Mineralization of breccia pipes in northern Arizona. Econ Geol 80:1722–1735
Wenrich KJ, Van Gosen BS, Finch WI (1996) Chapter 31: Solution-collapse breccia pipe U deposits In: du Bray EA (ed), Preliminary compilation of descriptive geoenvironmental mineral deposit models. U.S. Geol. Surv., Open-File Rep. 95–0831, p 244–251, https://pubs.usgs.gov/of/1995/ofr-95-0831/CHAP31.pdf. Accessed 17 June 2016
Acknowledgements
This work was supported by the U.S. Geological Survey, Environmental Health Program in the Ecosystems Mission Area as part of the Grand Canyon Uranium Studies. Robert Runkel and Rebecca Lambert provided helpful comments on earlier versions of the paper. We thank Kim Beisner, Bill Benzel, Dale Brokaw, Tyler Kane, Abby Keith, and David Naftz for assistance and discussions that improved this work. Joe Whelan archived the ore samples used in the study. Blaine McCleskey conducted ion chromatograph analyses. We thank the Grand Canyon National Park for permission to collect samples in the Park and Ben Tobin for logistical support. Any use of trade, firm, or product names is for descriptive purposes only and does not imply endorsement by the U.S. Government.
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bern, C.R., Campbell, K.M., Walton-Day, K. et al. Laboratory Simulation of Groundwater Along Uranium-Mining-Affected Flow Paths Near the Grand Canyon, Arizona, USA. Mine Water Environ 41, 370–386 (2022). https://doi.org/10.1007/s10230-022-00872-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10230-022-00872-9