
Abstract
The simultaneous quantification of several transcripts via multiplex PCR can accelerate research in fish physiological responses to diet and enable the development of superior aquafeeds for farmed fish. We designed two multiplex PCR panels that included assays for 40 biomarker genes representing key aspects of fish physiology (growth, metabolism, oxidative stress, and inflammation) and 3 normalizer genes. We used both panels to assess the physiological effects of replacing fish meal and fish oil by terrestrial alternatives on Atlantic salmon smolts. In a 14-week trial, we tested three diets based on marine ingredients (MAR), animal by-products and vegetable oil (ABP), and plant protein and vegetable oil (VEG). Dietary treatments affected the expression of genes involved in hepatic glucose and lipid metabolism (e.g., srebp1, elovl2), cell redox status (e.g., txna, prdx1b), and inflammation (e.g., pgds, 5loxa). At the multivariate level, gene expression profiles were more divergent between fish fed the marine and terrestrial diets (MAR vs. ABP/VEG) than between the two terrestrial diets (ABP vs. VEG). Liver ARA was inversely related to glucose metabolism (gck)- and growth (igfbp-5b1, htra1b)-related biomarkers and hepatosomatic index. Liver DHA and EPA levels correlated negatively with elovl2, whereas ARA levels correlated positively with fadsd5. Lower hepatic EPA/ARA in ABP-fed fish correlated with the increased expression of biomarkers related to mitochondrial function (fabp3a), oxidative stress (txna, prdx1b), and inflammation (pgds, 5loxa). The analysis of hepatic biomarker gene expression via multiplex PCR revealed potential physiological impacts and nutrient-gene interactions in Atlantic salmon fed lower levels of marine-sourced nutrients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The future of farming of Atlantic salmon (Salmo salar) and other carnivorous fish lies in the utilization of aquafeeds with a minimum contribution from ingredients sourced in wild fish stocks. Replacement of these marine products—namely, fish meal (FM) and fish oil (FO)—by different terrestrial alternatives has been investigated, with a particular focus on their impact on fish performance and chemical composition (Oliva-Teles et al. 2015; Roques et al. 2018). Growth on experimental feeds with low marine product inclusion has been similar to that with FM/FO-based aquafeeds (Lu et al. 2015; Torrecillas et al. 2017; Beheshti Foroutani et al. 2018). However, the same studies also reported significant changes in the chemical composition of the fish, which could affect the quality of the fish as a commodity. Indeed, omics in fish nutrition research has revealed profound metabolic and physiological adjustments occurring in fish fed diets based on terrestrial products (Sitjà-Bobadilla et al. 2005; Tacchi et al. 2012; Caballero-Solares et al. 2018). Some of the affected physiological functions were related to the welfare and immune status of the fish. For example, the inclusion of plant proteins in the diet increased blood and liver redox status in gilthead sea bream (Sparus aurata; Sitjà-Bobadilla et al. 2005) and induced changes in the transcription of immune biomarker genes in the liver of Atlantic salmon (Tacchi et al. 2012; Caballero-Solares et al. 2018). Therefore, as new low-marine aquafeeds are formulated, more investigation will be required to ensure that they promote not only fish growth but also an appropriate health status.
Aquafeed producers test new formulations routinely and need cost-effective methods to evaluate their performance and the physiological effects on the fish, in addition to the measuring of phenotypic features (e.g., weight, length, hepatosomatic index, proximate body composition). High-throughput transcriptome profiling techniques such as microarray hybridizations and RNA-sequencing analyses are costly, and the analysis of resulting data requires advanced bioinformatics capacity and expertise. On the other hand, single-gene analysis via quantitative real-time reverse transcription polymerase chain reaction (singleplex qPCR) can be time-consuming depending on the number of target transcripts. Multiplexed PCR assays could become a valuable alternative to the aforementioned methodology. In brief, multiplex PCR allows the simultaneous quantitation of multiple gene transcripts, thus making gene expression analysis a much faster process compared with singleplex qPCR. This would also facilitate the addition of more biological replicates, with a relatively low expenditure of time and resources compared with high-throughput techniques.
As introduced above, many metabolic and physiological pathways impacted by dietary FM/FO replacement by terrestrial ingredients have already been identified (Sitjà-Bobadilla et al. 2005; Vilhelmsson et al. 2007; Tacchi et al. 2012; Caballero-Solares et al. 2018), although the molecular mechanisms controlling such diet effects are yet to be elucidated. Furthermore, a number of genes have been repeatedly analyzed across studies and established as informative growth-, metabolism-, stress-, and health-related biomarkers (e.g., Atlantic salmon fadsd5 and lect2 in Morais et al. (2012) and Xue et al. (2015)). Multiplex PCR panels constructed with well-characterized gene biomarkers may become a useful tool for aquafeed producers in their search for superior formulations with low content in marine ingredients. Concurrently, this analytical technique could be very useful in the fields of fish nutrition and physiology research, as reflected by the body of literature published to date (e.g., Cleveland and Weber 2011; Cleveland et al. 2012; Cleveland and Weber 2013; Kono and Korenaga 2013; Kono et al. 2013; Cleveland and Weber 2015; Manor et al. 2015).
In the present study, we developed and validated two multiplex PCR panels to evaluate the effect of terrestrial feed ingredients on the transcription of physiologically relevant biomarkers genes in the liver of Atlantic salmon.
Materials and Methods
Feeding Trial
The liver tissue samples utilized in the study were collected from Atlantic salmon smolts fed for 14 weeks with either a diet based on marine ingredients (i.e., fish meal (FM) and fish oil (FO)), referred to here as MAR; a high animal by-product/high rapeseed oil diet, referred to as ABP; and a high plant protein/high rapeseed oil diet, referred to as VEG. The formulation and nutritional composition of MAR, ABP, and VEG diets were published in previous studies (Caballero-Solares et al. 2017; Beheshti Foroutani et al. 2018; Caballero-Solares et al. 2018). These studies also described the holding and feeding conditions in detail. Briefly, PIT (passive integrated transponder)-tagged Atlantic salmon smolts (initial weight, 179 ± 29 g (mean ± standard deviation)) were allocated to 620-L tanks connected to a flow-through seawater system in the Dr. Joe Brown Aquatic Research Building (JBARB, Ocean Sciences Centre, Memorial University of Newfoundland, Canada). Each dietary treatment was randomly assigned to 4 replicate tanks, with 40 salmon in each tank. Fish were fed the experimental diets to apparent satiation twice a day for 14 weeks, after which 5 individuals/tank were euthanized via immersion in seawater containing 400 mg/L MS-222 (Syndel Laboratories, Vancouver, BC, Canada) and dissected for tissue collection.
Fish handling, euthanasia, and dissection procedures were performed following the guidelines of the Canadian Council of Animal Care (approved Memorial University Institutional Animal Care Protocol 14–71-MR).
RNA Extraction and Purification
RNA was extracted, DNaseI-treated, and purified from flash-frozen 50–100 mg liver samples following the standard procedures in the laboratory (Xu et al. 2013; Xue et al. 2015; Caballero-Solares et al. 2018). RNA integrity and purity were verified by 1% agarose gel electrophoresis and NanoDrop UV spectrophotometry (Thermo Fisher Scientific, Mississauga, ON, Canada), respectively. For all samples, no signs of RNA degradation were found (i.e., intact 28S and 18S ribosomal RNA bands), and A260/280 and A260/230 ratios were 2.1–2.3 and 1.9–2.4, respectively.
Multiplex PCR Panel Development
A list of 40 biomarker genes was selected based mainly on the results from microarray experiments previously conducted in the laboratory. Among the selected biomarkers, there were metabolism- and physiology-relevant genes that were previously microarray-identified as responsive to FO replacement by vegetable oils in the liver of Atlantic salmon (Xue et al. 2020). Other genes with similar functions were identified from a microarray study performed on the same liver RNA samples utilized here and published in Caballero-Solares et al. (2018). Singleplex qPCR data used to confirm the microarray results in Caballero-Solares et al. (2018) have been used to validate the gene expression results generated in the present study (see below). Finally, the list also included biomarker genes involved in cell redox homeostasis, detected via microarray transcriptome profiling of primary Atlantic salmon hepatocytes exposed to an exogenous oxidant (tert-Butyl hydroperoxide, tBHP; Xue et al. unpublished).
The multiplex PCR or eXpress Profiling (XP)-PCR technique developed by Beckman Coulter is based on the use of chimeric primers to amplify DNA fragments of different sizes that are subsequently separated by capillary electrophoresis. The multiplexed analysis allows the quantification of the transcript levels of up to ~ 30 genes per panel, including biomarker and normalizer genes. The chimeric reverse and forward primers consist of a 5′-end universal sequence (5′-GTACGACTCACTATAGGGA-3′ for the reverse, 5′-AGGTGACACTATAGAATA-3′ for the forward), followed by a 3′-end gene-specific sequence. The gene-specific priming sequences were designed to amplify DNA fragments between 137 and 387 bp, with lengths differing by at least 4 bp. For the multiplex analysis, 25 ng of liver RNA template were reverse transcribed in 10 μL of reaction mixture containing 2.5 μL of kanamycin-resistance (kanr) gene RNA (internal control, pre-diluted 1:8 in 10 mM Tris-HCl, pH 8), 0.5 μL of reverse transcriptase (RT; 10 U), and 2 μL of 5X RT buffer from the GenomeLab™ GeXP Start Kit (Beckman Coulter/SCIEX), as well as 1 μL of chimeric reverse primer mix. Subsequently, 4.65 μL of cDNA were added to the PCR reaction mixture composed of 2 μL 5X PCR buffer from the GenomeLab™ GeXP Start Kit, 0.35 μL Thermo-Start Taq DNA Polymerase (Thermo Fisher Scientific), 2 μL of 25 mM MgCl2, and 1 μL of chimeric forward primer mix. RT and PCR reactions were carried out in a DNA Engine Tetrad 2 Peltier thermal cycler (Bio-Rad Laboratories), under the following programs: for the RT, 48 °C for 1 min, then 42 °C for 60 min, and finally 95 °C for 5 min; for the PCR, 1 cycle of 95 °C for 15 min, followed by 35 cycles of 94 °C for 30 s, 55 °C for 30 s, and 70 °C for 1 min. Besides the necessary substrates and salts for the reactions, the 5X RT and PCR buffers contained the reverse and forward primers (also chimeric) for kanr. Also, the 5X PCR buffer contained universal WellRED D4 dye-labeled forward and unlabeled reverse primers (homologous to the 5′-ends of the chimeric primers). After the first two PCR cycles, the amplification was taken over by the universal primers as they become predominant in the reaction mixture. Thus, the PCR yielded D4-labeled DNA fragments of different lengths, each length corresponding to a different gene. Once the PCR was concluded, the resultant solution was diluted 1:10 in 10 mM Tris-HCl, pH 8. A loading solution was thereafter prepared by mixing 1 μL of diluted PCR solution with 38.5 μL Sample Loading Solution and 0.25 μL of DNA Size Standard-400 from the GenomeLab™ GeXP Start Kit. Finally, the PCR fragments in the loading solution were separated by capillary polyacrylamide electrophoresis (Frag-3 protocol), and their fluorescence were measured using the GenomeLab™ GeXP genetic analysis system (Beckman Coulter/SCIEX).
We created 2 multiplex PCR panels that included a total of 40 genes and shared 3 normalizer genes. Primers were designed using Primer 3 v.0.4.0 software (available at (http://bioinfo.ut.ee/primer3-0.4.0/)) and quality-checked for PCR fragment size and undesired fragments. The concentration of the reverse primers was adjusted to attenuate the fluorescent signal of the highly abundant transcripts. The linear range of fluorescence detection of the GenomeLab™ GeXP genetic analysis system lies between 370 and 120,000 relative fluorescence units (rfu); however, the optimum range to measure changes in transcript abundance is 2000–50,000 rfu. The reverse primer concentration optimization, together with the pre-dilution of the resultant PCR solution, served to ensure that the amplicons’ fluorescence levels were within the optimum range and that the analysis was quantitative.
For both primer quality check and reverse primer concentration optimization, we used an RNA reference pool including an equal contribution from 12 liver samples, with 4 biological replicates (one from each quadruplicate tank) for each of the three diet groups. Table 1 shows all primers included in the 2 multiplex panels, as well as the GenBank accession numbers of the sequences used for their design, their amplicon sizes, and their functions based on gene ontology (GO) terms from Danio rerio and Homo sapiens best BLAST hits (from UniProt Knowledgebase, http://www.uniprot.org/).
Gene Expression Analysis
Individuals can exhibit different performance under the same dietary conditions, often due to variable food consumption between individuals within a tank. To minimize the impact of biological variability on our analyses, we selected samples from those fish with weight gains within one standard deviation below and above the tank mean value. The multiplex experiment included liver RNA samples of 30 individual fish (i.e., 10 MAR-, 9 ABP-, and 11 VEG-fed fish). Both multiplex panels were run on each sample as well as a no-template control in technical triplicates.
The resultant electropherograms were analyzed using the Fragment Analysis Module of the GenomeLab GeXP genetic analysis system software (Beckman Coulter/SCIEX). The obtained areas under each gene peak were thereafter normalized to kanr using the eXpress Profiler software (Beckman Coulter/SCIEX). This normalization step is intended to reduce intercapillary variation. The kanr-normalized signal levels were log2-transformed and analyzed using geNorm (qBASE plus, Biogazelle) to select the most stable normalizer genes. According to the M values calculated by geNorm, the most stable normalizer genes were eef1a1 (M = 0.213) and rpl32 (M = 0.250).
The relative quantity (RQ) of each transcript was calculated relative to a 7-point standard curve prepared with 1:2 dilutions series of a 40 ng/μL liver RNA reference pool and normalized to the geometric mean of eef1a1 and rpl32. The standard curve was also used to confirm that the amplification conditions (e.g., cDNA input, reverse primer concentration) were not limiting during the 35-cycle PCR analysis of the individual samples.
Data Transformation and Statistical Analyses
The RQ values of the different biomarker genes analyzed were log2-transformed to meet the normality assumption (checked by Kolmogorov-Smirnov test). The log2-transformed RQs (log2 RQs) were subsequently checked for outliers using Grubb’s test and analyzed through general linear models (GLM) for differences among dietary groups. Any tank-based effect was accounted for by including a random Tank factor in the model. If variances were statistically equal among dietary groups (Levene’s test for compliance with the homoscedasticity assumption), the mean values of the three dietary treatments were pairwise compared using Tukey’s post hoc test. Otherwise, if the homoscedasticity assumption was not satisfied, Games-Howell test was used for comparisons. The accepted level of significance was p < 0.05. These statistical analyses were conducted using IBM SPSS Statistics v24.0.0 (IBM Corp, Armonk, NY).
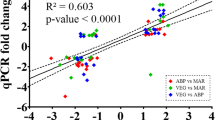
For validation purposes, we contrasted, by linear regression analysis, the present multiplex results with previously published singleplex qPCR data (Caballero-Solares et al. 2018) obtained from the same liver RNA samples. This group of singleplex qPCR-analyzed transcripts included gck, pfkfb4, adssl1a, htra1b, idi1, fabp3a, dgat2b, igmb, lect2b, eef1a1, and rpl32. For the regression analysis, fold-change values (FCs) were calculated for each dataset (i.e., singleplex and multiplex) using the formula 2A−B, where A and B are the mean log2 RQs of two dietary groups (Hori et al. 2012). For down-regulated genes, fold-change values were inverted (− 1/fold change). The significance of the regression model in predicting singleplex FCs using multiplex FCs was determined by F test. A significant correlation between both datasets was considered as proof of the validation. The linear regression analysis was performed using IBM SPSS Statistics v24.0.0.
Log2-transformed multiplex RQs were also analyzed using principal coordinates analysis (PCoA) for similarities among the different gene expression patterns observed among the salmon. We also tested for differences among gene expression patterns via permutational multivariate analysis of variance (PERMANOVA). PERMANOVA was performed with 9999 random permutations and Tank factor nested within the fixed factor Diet. The accepted level of significance was p < 0.05. Both PERMANOVA and PCoA were based on Bray-Curtis similarities of all pairwise comparisons among individuals. These analyses were performed using PRIMER 6.1.15 (Ivybridge, UK).
We also analyzed the relationships between liver lipid composition and the gene expression data by linear regression analysis. For the regression analysis, we only selected those ω3 and ω6 fatty acids (FAs) accounting for at least 0.5% of the total FAs in the liver. Fish weight gain and other relevant phenotypic parameters (e.g., condition factor, EPA/ARA ratio) were also included. Some of these data were previously published elsewhere (Beheshti Foroutani et al. 2018; Caballero-Solares et al. 2018). The Pearson correlation coefficients (r) resulting from the regression analyses were arranged as a correlation matrix. The different genes and phenotypic parameters were grouped and ordered by complete-linkage hierarchical clustering using r values. The correlation matrix was generated using IBM SPSS Statistics v24.0.0 and the hierarchical clustering using PRIMER 6.1.15. Liver samples for lipid composition determination were collected, processed, and analyzed as described in Hixson et al. (2017) and Beheshti Foroutani et al. (2018).
Results
Validation of Multiplex Gene Expression Data
The linear regression analysis revealed a significant correlation (F test, p < 0.0001) between multiplex and singleplex fold changes, with an r2 of 0.705 (see Supplementary Fig. S1).
Glucose Metabolism and Cell Growth-Related Genes
ABP diet down-regulated significantly the hepatic gck mRNA levels as compared with MAR and VEG diets (p < 0.05, Fig. 1a). The transcript levels of pfkfb4 followed a similar trend, but differences among diets were not statistically significant (p = 0.10, Fig. 1b). Atlantic salmon fed ABP diet also down-regulated adssl1a transcription compared with those fed MAR diet (Fig. 1c). VEG diet repressed htra1b transcription compared with MAR diet (Fig. 1f). Although not statistically significant (p = 0.06), the transcript levels of hgfa and mtor were, respectively, up- and down-regulated in the liver of salmon fed VEG diet (Fig. 1e, g). Diet did not affect igfbp-5b1 and foxa2 transcription (p > 0.05, Fig. 1d, h).

Results from the multiplex PCR analysis of transcripts related to glucose metabolism (a–b), cell growth (c–h), lipid metabolism (i–s), inflammation (t–x), and stress (y–an). Boxplots represent relative quantity (log2 RQ) medians (horizontal line), interquartile ranges (box), and minimum and maximum values (whiskers). Different letters above upper whiskers represent significant differences between diets (general linear models, Tukey’s (homogeneity of variances among groups) or Games-Howell (variances not homogenous across groups) post hoc test, p < 0.05). Transcripts previously analyzed via singleplex qPCR in Caballero-Solares et al. (2018) are indicated with an asterisk after the gene symbol. a glucokinase; b 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 4-like; c adenylosuccinate synthetase isozyme 1 C-A; d insulin-like growth factor binding protein 5b1; e hepatocyte growth factor A; f serine protease HTRA1 B; g mechanistic target of rapamycin kinase; h forkhead box protein A2; i elongation of very long chain fatty acids protein 2; j delta 5 fatty acyl desaturase; k isopentenyl-diphosphate delta-isomerase 1; l sterol regulatory element-binding protein 1; m sterol regulatory element-binding protein 2; n apolipoprotein A-I; o choline-phosphate cytidylyltransferase 1 alpha A; p liver X receptor; q peroxisome proliferator-activated receptor beta/delta A; r fatty acid-binding protein 3 A; s diacylglycerol O-acyltransferase 2 B; t immunoglobulin mu heavy chain; u lipocalin-type prostaglandin D synthase; v arachidonate 5-lipoxygenase A; w leukocyte cell-derived chemotaxin-2-like B; x glucocorticoid receptor; y thioredoxin A; z thioredoxin reductase 1 B; aa peroxiredoxin 1 B; ab catalase A; ac succinate dehydrogenase [ubiquinone] iron-sulfur subunit A; ad sestrin-1 A; ae nuclear factor (erythroid 2-related) factor 2 B; af ferritin middle subunit A; ag metallothionein B; ah cytochrome P450 3A27 A; ai heat shock protein 70 A; aj biliverdin reductase A1; ak heme oxygenase B; al cytochrome P450 1A1 A; am apoptosis regulator Bcl2-like 1; an vitellogenin
Lipid Metabolism-Related Genes
The transcript levels of elovl2, idi1, srebp1, srebp2, and apoaI were up-regulated by VEG diet compared with MAR diet (Fig. 1i, k–n). A similar trend was observed for fadsd5 mRNA levels (Fig. 1j); however, differences among diets were not statistically significant (p = 0.06). Conversely, VEG diet down-regulated dgat2b transcript levels compared with MAR diet (Fig. 1s). On the other hand, ABP diet increased pparba transcription compared with MAR diet (Fig. 1q) and that of fabp3a compared with both MAR and VEG diets (Fig. 1r). The mRNA levels of pcyt1aa and lxr were not dietarily modulated (Fig. 1o, p).
Inflammation and Oxidative Stress-Related Genes
ABP diet increased the hepatic transcript levels of igmb, pgds, txna, and prdx1b compared with MAR diet (Fig. 1t, u, y, aa) and of 5loxa compared with MAR and VEG diets (Fig. 1v). The mRNA levels of hsp70a exhibited a similar trend but were not significantly different (p = 0.09, Fig. 1ai). In contrast, nr3c1 transcript levels in fish fed VEG diet were lower than in those fed MAR diet (Fig. 1x). The other transcripts (lect2b, txnrd1b, cata, sdhba, sesn1a, nfe2l2b, ftma, mtb, cyp3a27b, blvra1, hmox1b, cyp1a1a, bcl2l1, and vtg; Fig. 1w, z, ab–ah, aj–an, respectively) were not affected by diet.
Multivariate Analyses—Similarities/Dissimilarities Among Dietary Groups
The PCoA of the resemblance among salmon based on the multiplex data explained 50.5% of the variation and separated ABP-, VEG-, and MAR-fed salmon—from the top left to the bottom right corner, respectively (Fig. 2). This segregation highly correlated (r ≥ 0.70) with fabp3a, idi1, txna, srebp1, srebp2, pgds, nr3c1, and gck. PERMANOVA found significant differences between fish fed the MAR diet and those fed the ABP or VEG diets (p = 0.004–0.008) and close to a significant difference between ABP- and VEG-fed fish (p = 0.07).

Principal coordinates analysis (PCoA) of the resemblance of salmon fed the different diets based on their gene expression patterns. Vectors indicate the association (Pearson correlation) of a given biomarker gene with the coordinates (axes x and y). Only vectors with Pearson correlation coefficients > 0.7 are shown
Multivariate Analyses—Correlation among Transcriptomic and Phenotypic Parameters
The hierarchical clustering analysis grouped the phenotypic parameters into three major clusters (Fig. 3). Weight gain (WG), 14:0, ∑SFA, and the ω3 FAs (i.e., DHA, EPA, and 22:5ω3) comprised cluster I. Condition factor (CF), hepatosomatic index (HSI), and EPA/ARA were grouped in cluster II. The other parameters, i.e., the ω6 FAs 20:3ω6 and ARA, ω6/ω3, sterol, PUFA/SFA, and DHA/EPA, comprised cluster III. Correlations among the phenotypic parameters are shown in Supplemental Fig. S2. With respect to the biomarker genes, four major clusters could be identified by hierarchical clustering (Fig. 3). The gene clusters showed no clear evidence of grouping by function (for gene-to-gene correlations information, see Supplemental Fig. S3). Instead, gene clusters showed different patterns of correlation with the phenotypic parameters. Genes in cluster I distinctively correlated negatively with ARA (significant for all), 20:3ω6 (significant for all except igfbp-5b1 and sesn1a), and ω6/ω3 (significant for htra1b, gck, and dgat2b) and showed positive correlation with WG (significant for sesn1a and dgat2b) and EPA/ARA (significant for htra1b, gck, and dgat2b). Conversely, genes in clusters II, III, and IV tended to correlate negatively with EPA/ARA and positively with 20:3ω6 and ω6/ω3. These gene-to-trait alignments were clearer for clusters III and IV, based on the higher number of significant correlations. Except for lect2b and bcl21l, all genes in cluster II showed a significant positive correlation with CF. On the other hand, cluster IV showed a characteristic pattern of negative correlation with CF (tendency, i.e., none was statistically significant), HSI, and EPA/ARA and positive correlation with ARA.

Matrix of Pearson’s correlation coefficients (r) between all the analyzed biomarker genes (rows) and a selection of phenotypic features (columns). Significant correlations are indicated by displaying the r values in the cell. The different parameters were grouped by complete-linkage hierarchical clustering (using r values) and are represented by dendrograms next to gene symbols and above phenotypic parameters. As a reference to the correlated datasets and the dietary context, the significant differences among diet groups are indicated on the right of the gene symbols for the gene expression data and below the phenotypic parameters for the diet composition (“Diet” row) and the liver/growth-related data (“Liver/Growth” row) (general linear models, Tukey’s (homogeneity of variances among groups) or Games-Howell (variances not homogenous across groups) post hoc test; p < 0.05). Significant differences are expressed as A > B, A > B,C, or A,B > C; where A, B, and C represent the parameter’s average value in the different diets or dietary groups, and the symbol > means “greater than”. WG, weight gain; SFA, saturated fatty acids; DHA, docosahexaenoic acid (22:6ω3); EPA, eicosapentaenoic acid (20:5ω3); CF, condition factor (body mass/length3); HSI, hepatosomatic index [100 × (liver mass/body mass)]; ARA, arachidonic acid (20:4ω6); PUFA, polyunsaturated fatty acids; N/A, not applicable; NS, non-significant differences
Regardless of the hierarchical clustering analysis, some genes and phenotypic parameters showed correlation patterns that are worth highlighting (Fig. 3). First, several genes in clusters II, III, and IV showed negative correlations with DHA, EPA, and 22:5ω3. Curiously, hmox1b in cluster IV deviated from the common pattern and was found to be correlated positively with DHA, EPA, and 22:5ω3. Transcripts related to fatty acid (i.e., elovl2, fabp3a, and pparba) and cholesterol (i.e., idi1, srebp1, and srebp2) metabolism tended to show negative correlations with SFA and ω3 FAs and positive correlations with ω6 FAs and ω6/ω3. Biomarker genes involved in inflammation (i.e., pgds) and redox homeostasis (i.e., prdx1b and txna) showed the same trend.
Discussion
Multiplex PCR platforms have demonstrated their potential for research in fish physiology and immunology (Cleveland and Weber 2011; Cleveland et al. 2012; Cleveland and Weber 2013; Kono and Korenaga 2013; Kono et al. 2013; Cleveland and Weber 2015; Manor et al. 2015). In the present study, the two multiplex panels based on previous microarray analyses were capable of detecting changes in multiple pathways and processes that are key for Atlantic salmon growth and welfare. Indeed, the multivariate analyses effectively segregated the different dietary groups, both visually (PCoA) and statistically (PERMANOVA). Moreover, the gene expression results revealed that differences were more pronounced between the marine (MAR) and the terrestrial (ABP and VEG) diets than between the two terrestrial diets, thus indicating that the dietary lipid source dominated over the protein source in the transcriptomic changes observed. Relevant performance/phenotypic differences were found among dietary groups—including ABP vs. VEG—that associated with certain gene expression profiles. These relations and their physiological implications are discussed below.
Glucose Metabolism and Cell Growth and Proliferation
The liver of Atlantic salmon fed ABP diet showed repressed transcription of genes involved in the glycolytic pathway (gck and pfkfb4). Our previous microarray and singleplex qPCR results indicated likewise (Caballero-Solares et al. 2018), but here we confirmed the inverse relation between gck and ω6 FAs in the liver of salmon. High dietary ω6/ω3 ratios have been associated with insulin resistance in mammals (Storlien et al. 1997; Poudyal et al. 2011). Conversely, ω3 FAs improve glucose utilization and reduce insulin resistance in rats (Poudyal et al. 2011). As reviewed by Poudyal et al. (2011) and Scorletti and Byrne (2013), glycolytic and lipogenic enzymes in mammals are transcriptionally co-regulated by PPARs and SREBP1. Research in fish nutrition revealed that SREBP1a activates the transcription of gck and pfkfb1 in gilthead seabream (Metón et al. 2006; Egea et al. 2008). In contrast to SREBP1, SREBP2 predominantly regulates genes involved in cholesterol homeostasis (Shimomura et al. 1997; Horton et al. 2002; Horton et al. 2003). Here, srebp1 and srebp2 transcription was up-regulated by VEG diet, which correlated positively with the higher liver ω6/ω3 ratios and pfkfb4 (not gck) mRNA levels found in salmon fed this diet. However, neither gck nor pfkfb4 showed a positive association with any of the hepatic ω6 FA parameters analyzed. This is largely due to MAR-fed salmon as their livers showed distinctive high gck and pfkfb4 transcript levels and low ARA, 20:3ω6, and ω6/ω3 values. As discussed in Caballero-Solares et al. (2018), MAR diet had a higher carbohydrate content than ABP and VEG, which could have contributed to gck and pfkfb4 up-regulation. Contrary to previous studies reporting that the activation of mammalian PPARB improves glucose utilization through gck up-regulation (Liu et al. 2010), our study found gck and pparba mRNA levels to be negatively correlated. The higher pparba mRNA levels in ABP-fed fish were the predominant driver of this relationship. Lower feed intake had previously been argued to be responsible for gck and pfkfb4 down-regulation in the fish fed ABP diet (Caballero-Solares et al. 2018). However, in view of pparba positive correlation with liver ω6 FA levels (i.e., 20:3ω6 and ω6/ω3), and likewise for srebp1 and srebp2, the influence of diet-related changes on liver lipid metabolism should also be considered. In summary, the expression patterns observed here for gck, pfkfb4, srebp1, srebp2, and pparba might reflect the myriad of specific and shared mechanisms ruling fish glucose and lipid metabolism upon different dietary conditions (i.e., diet FA profile, diet carbohydrate content, and feed intake).
The transcription of gck correlated positively with that of the cell growth and proliferation-related genes igfbp-5b1 and htra1b. IGFBP5 regulates IGF-I signaling in mammals and fish (Duan and Xu 2005; Macqueen et al. 2013; Cleveland and Weber 2015), and igfbp-5b1 down-regulation has been associated with a camelina-induced reduction in Atlantic salmon growth (Xue et al. 2015). In the present study, igfbp-5b1 mRNA levels did not show significant differences among diets nor correlate with WG. Nevertheless, igfbp-5b1 did correlate positively with htra1b, which encodes a protease known to cleave IGFBP5 in mammals (Clausen et al. 2011). Given that htra1b transcription was down-regulated in the VEG-fed fish compared with those fed the MAR diet (both in the present study and in Caballero-Solares et al. (2018)), a dietary impact on free IGF-I concentrations (and activity, as discussed in Cleveland and Weber (2015)) can be suspected.
Despite not correlating significantly with WG, gck, igfbp-5b1, and hgfa correlated positively with the increased HSI in salmon fed MAR diet compared with those fed VEG diet. Given WG similarities between MAR and VEG-fed fish, higher HSI in MAR-fed fish could be due to increased deposition of hepatic glycogen (since glucose phosphorylation by GCK initiates the glycogenesis) and/or promotion of cell proliferation. On the other hand, gck, igfbp-5b1, and htra1b mRNA levels were inversely correlated with the hepatic ARA levels, which, in turn, were negatively correlated with HSI. Previous studies found increasing dietary ARA levels to reduce HSI in fish (Xu et al. 2010; Shahkar et al. 2016; Torrecillas et al. 2018). In the present study, although MAR diet had higher ARA content than ABP diet, ABP-fed fish presented higher ARA concentration in the liver. Interestingly, liver ARA levels correlated negatively with dgat2b and hence with the synthesis of triacylglycerol. Suppressing dgat2b expression reduced hepatic steatosis in rats (Choi et al. 2007), while Torrecillas et al. (2018) linked lower HSI in European sea bass with an ARA-induced reduction of hepatocyte lipid vacuolization. Taken together, our results point to MAR diet promoting liver tissue development and accumulation of energy storage products (i.e., glycogen and lipids).
Lipid Metabolism
The fatty acid profile of VEG diet seemed to stimulate fatty acid elongation and desaturation, as suggested by elovl2 and fadsd5 results (significant up-regulation in elovl2; close to significant (p = 0.06) in fadsd5). Desaturation and elongation of PUFAs by FADSD5 (or FADS2D5; Betancor et al. (2014)) and ELOVL2 have been described in Atlantic salmon (Hastings et al. 2004; Morais et al. 2009), thus demonstrating the enzymatic capacity to synthesize EPA, DHA, and ARA. At the gene expression level, low dietary levels of EPA and DHA induce elovl2 and fadsd5 transcription in the liver of Atlantic salmon (Betancor et al. 2014; Xue et al. 2015, 2020). Here, a slight, 1.3 fold induction of elovl2 (statistically significant) and fadsd5 (p = 0.06) was observed when comparing VEG-fed and MAR-fed salmon, which is close to that found by Hixson et al. (2017) between the fish fed control fish oil (FO)-based and the high-vegetable oil (VO) diets. Also, the differences in EPA and DHA levels between control FO-based and high-VO diets were comparable between the present study and Hixson et al. (2017). Yet changes in hepatic elovl2 and fadsd5 mRNA levels were not significant in Hixson et al. (2017), which is likely due to differences in the design of the experiment and the statistical analysis: the present compares 3 diets with contrasted formulations, while Hixson et al. (2017) tested 10 diets with more gradual variation in their FA composition.
As expected, based on previous studies (Turchini et al. 2009; Xue et al. 2020), hepatic EPA and DHA levels reflected those of the diets, which translated into a negative correlation with elovl2. In contrast, liver ARA and precursor 20:3ω6 levels correlated positively with fadsd5. Increased liver ARA levels in fish fed the VEG, and ABP diets, despite consuming diets with less ARA, are in agreement with previous studies in Atlantic salmon (Tocher et al. 1997; Jordal et al. 2007). Similar to those studies, the high-VO diets in our study were more abundant in precursor linoleic acid (LNA, 18:2ω6) than the FO-based diet. As illustrated in Katan et al. (2019), dietary LNA is largely converted into ARA in the liver of Atlantic salmon. Considering the above, it appears that the higher LNA content of ABP and VEG diets enhanced the synthesis of ARA in the liver of salmon, regardless of dietary ARA concentration. However, the interaction of dietary ARA and ω6/ω3 modulatory effects on Atlantic salmon liver FA metabolism remains unclear.
Liver DHA and EPA showed association with almost half of the analyzed biomarker genes, thus suggesting that most biological processes studied here were affected by these nutrients. Continuing with those involved in lipid metabolism, both DHA and EPA were negatively correlated with the transcription factors srebp1 and srebp2. DHA and EPA were previously reported to down-regulate srebp1 in Atlantic salmon in vitro (SHK-1 cell line; Minghetti et al. (2011)) and in vivo (liver; Hixson et al. (2017)). In mammals, SREBP1 up-regulates the transcription of genes involved in FA desaturation and elongation (Horton et al. 2003). However, as previously stated, SREBP2 predominantly regulates genes involved in cholesterol biosynthesis (Shimomura et al. 1997; Horton et al. 2002, 2003). To explain the negative correlation with liver EPA and DHA levels shared by srebp1 and srebp2, it should be noted that marine ingredient replacement by plant products jointly affects dietary levels of DHA, EPA, and cholesterol. In fact, the present and previous studies suggest that Atlantic salmon SREBP1 and SREBP2 play similar roles to those of mammals. First, our correlation analyses revealed fadsd5 and elovl2 to be more strongly related to srebp1 than to srebp2 (r, 0.57–0.67 (srebp1) vs 0.41–0.42 (srebp2)). Second, previous studies showed Atlantic salmon srebp2 to be induced by low cholesterol/high phytosterol levels in plant-based feeds (Leaver et al. 2008; Kortner et al. 2012; Liland et al. 2013; Kortner et al. 2014). In the present study, srebp2 mRNA levels were strongly correlated with idi1 (r = 0.81), a transcript encoding an enzyme involved in cholesterol biosynthesis. The correlation between srebp1 and idi1 was not as strong (r = 0.69). We found no correlation of srebp1, srebp2, and idi1 with liver sterol levels. However, lipid class separation by thin-layer chromatography does not discriminate between cholesterol and phytosterols. Therefore, the sterol concentration data presented here may not reflect diet-related changes in cholesterol biosynthesis rates in salmon livers. Finally, VEG diet up-regulated apoaI transcription (as compared with MAR diet), which could be interpreted as a promotion of lipoprotein assembly (mainly high-density lipoproteins (HDLs)) and lipid transport. Similar results and explanation were reported by Gu et al. (2013) in their study on the effects of plant meal-based diets on the liver of Atlantic salmon. In support of this hypothesis, apoaI mRNA levels correlated positively with liver sterol levels. Since hepatic cholesterol synthesis was also enhanced by VEG, as suggested by idi1 transcript levels, it would appear that the liver metabolic machinery adapted to lower dietary cholesterol intake to maintain cholesterol levels.
Liver DHA and EPA levels correlated negatively with pparba mRNA levels, which were up-regulated by ABP diet compared with MAR diet. In turn, pparba up-regulation correlated negatively with WG, as well as with other phenotypic parameters such as 14:0, 22:5ω3, and EPA/ARA. With respect to 14:0, the activation of mammalian PPARB was found to reduce hepatic SFA levels while increasing those of monounsaturated FAs (Liu et al. 2010). As in mammals, Atlantic salmon PPARB has been suggested to up-regulate genes involved in beta-oxidation (Torstensen et al. 2009). Despite the above, and that FA membrane-mitochondria transport seemed to be up-regulated in ABP-fed fish (i.e., higher fabp3a transcription compared with MAR), our previous study found no dietary-induced changes in the expression of beta-oxidation-related transcripts (acyl-coenzyme A oxidase 1 and carnitine palmitoyltransferase 1; Caballero-Solares et al. (2018)). Nevertheless, changes in hepatic beta-oxidation activity may not agree with that observed at the transcriptomic level. As inferred from Kjær et al. (2008), damaged mitochondrial function due to oxidative stress is another factor to consider when discussing beta-oxidation in the liver of Atlantic salmon. Indeed, FABP3 overexpression has been associated with mitochondrial dysfunction in murine cells (Song et al. 2012). In this regard, it should be noted that pparba and fabp3a mRNA levels correlated positively with those of txna and prdx1b. TXN and PRDX proteins are well known for remediating oxidative stress in mammals (Nordberg and Arnér 2001) and fish (Pacitti et al. 2014). These gene expression patterns suggest interactions between oxidative stress-induced mitochondrial injury and cell apoptosis (i.e., mitochondrial permeability transition) and inflammation (Kowaltowski et al. 2001; Tschopp 2011).
Inflammation and Stress
We previously presented evidence that ABP diet promoted the transcription of the pro-apoptosis biomarker growth arrest and DNA damage-inducible protein GADD45 beta (gadd45b; Caballero-Solares et al. 2018). In the present study, the mRNA levels of the anti-apoptosis biomarker bcl2l1 (Eimon and Ashkenazi 2010) were not significantly affected by diet but correlated significantly and positively with those of txna and prdx1b, which were significantly up-regulated by ABP diet. Similar expression patterns of gadd45b and bcl2l1 may suggest a tight balance between pro- and anti-apoptosis regulators. With respect to the connection between cellular oxidative stress (i.e., txna, prdx1b) and apoptosis (i.e., bcl2l1), the literature on Atlantic salmon nutrigenomics indicates that specific diet formulations can increase cell oxidative stress and apoptosis. In Kjær et al. (2008), excessive dietary DHA and EPA levels caused mitochondrial membrane disruption, oxidative stress, and increased apoptosis in the liver of Atlantic salmon. Excessive dietary DHA and EPA levels would not have caused the aforementioned gene expression changes, as hepatic DHA and EPA levels were lower in the ABP-fed fish compared with the MAR-fed fish, clearly reflecting diet composition. Consequently, liver DHA levels were inversely proportional to the transcript levels of most oxidative stress (i.e., prdx1b, cata, blvra1, sdhba, txnrd1b, txna) and apoptosis biomarkers (bcl2l1). Moreover, DHA and EPA levels in the present experimental diets were 17–55 times lower than in those tested in Kjær et al. (2008). On the other hand, despite its similarity with ABP diet in dietary and hepatic DHA and EPA levels, VEG diet did not elicit such transcriptomic changes, thus discarding FM replacement by plant alternatives as the cause, contrary to previous findings in Atlantic salmon (Tacchi et al. 2012). To our knowledge, the effects of animal by-products in aquafeeds on the hepatic fish transcriptome had not been investigated prior to Caballero-Solares et al. (2018). Given that ABP appeared to reduce feed intake, which is most likely what lowered weight gain (Beheshti Foroutani et al. 2018; Caballero-Solares et al. 2018), this question would be best addressed as in Skugor et al. (2011). Skugor et al. (2011) found that both dietary soybean meal inclusion and feed restriction similarly induced the transcription of pro-apoptotic genes and repressed that of relevant oxidative stress genes in the liver of Atlantic salmon. In mice, caloric restriction is known to increase hepatic apoptosis but—as opposed to our results—decrease oxidative stress (Gredilla and Barja 2005; Spindler and Dhahbi 2007). To this latter contradiction must be added the fact that, while anti-inflammatory properties were attributed to caloric restriction in mice (Cao et al. 2001), the ABP-fed fish in the present study showed increased mRNA levels of various pro-inflammatory biomarker genes (i.e., 5loxa, pgds, and igmb). Therefore, there is no substantial evidence for dietary DHA and EPA levels, or feed intake, to have caused the apparent increase in oxidative stress, apoptosis, and inflammation in the liver of the ABP-fed fish. Instead, our correlation data lead our attention to the ω6 FAs.
The pro-inflammatory properties of ABP diet were previously reported (Caballero-Solares et al. 2017, 2018). The present multiplex PCR data served to confirm these previous findings since ABP diet up-regulated the transcription of genes involved in the synthesis of eicosanoids (5loxa, pgds) and igmb (IgM+ B cells were found to drive peritoneal inflammatory response in rainbow trout (Castro et al. 2017; Caballero-Solares et al. 2018)). Furthermore, the up-regulation of all three genes correlated significantly with pro-inflammatory hepatic FA profiles, such as low EPA/ARA and high ω6/ω3 and 20:3ω6 levels. Low hepatic EPA/ARA levels would promote the synthesis of pro-inflammatory eicosanoids (i.e., ARA-derived eicosanoids) by 5LOX and PGDS (Wall et al. 2010). Interestingly, the oxidative stress-related genes prdx1b and txna showed a similar correlation to these FA parameters. Also, the inflammation-related genes 5loxa, pgds, and igmb correlated positively with those representing oxidative stress (i.e., prdx1b and txna), apoptosis (i.e., bcl2l1), and mitochondrial function (i.e., fabp3a). A growing body of evidence links ARA to cellular oxidative stress and apoptosis in mammals (Wolf and Laster 1999; Brash 2001; Chen and Chang 2009; Pompeia et al. 2012; Shen et al. 2018). According to the mammalian literature, ARA-induced cell death would result from the depolarization of mitochondria by activation of mitogen-activated protein kinases (p38α MAPK and JNK) and the promotion of reactive oxygen species (ROS) production. In contrast to mammals, to the best of our knowledge, the role of ARA in the regulation of cellular redox homeostasis and cell death has yet to be investigated in fish.
Conclusions
In the present study, we analyzed the effects of terrestrial feed ingredients on the transcript levels of 40 physiologically relevant biomarker genes in the liver of Atlantic salmon using multiplex PCR assays. The endpoint multiplex PCR assays were optimized to ensure the quantitative accuracy of the analysis, which was validated by correlation analysis with singleplex qPCR data (r2 = 0.705; p < 0.0001). Our multiplex PCR data showed ingredients of terrestrial origin modulated metabolism, growth-related mechanisms, inflammation, and oxidative stress in the liver of Atlantic salmon. Additionally, correlations between transcript abundance and response parameters such as HSI and liver ARA, DHA, and EPA levels indicated physiological impacts of nutrient-gene interactions induced by the terrestrial feed ingredients. For example, the association of DHA, EPA, and ARA with gene biomarkers of oxidative stress and apoptosis suggested terrestrial ingredients may have significant effects on health status. Investigating numerous gene biomarkers using multiplex PCR provided the capacity to quickly and cost-effectively determine how multiple physiological processes respond to dietary modulation. Utilizing these assays in future research will allow us to identify gaps of knowledge in fish physiology (e.g., effects of animal by-products on liver function) and draw hypotheses for future studies (e.g., ARA induction of cell death via oxidative stress).
Data Availability
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Change history
06 August 2021
A Correction to this paper has been published: https://doi.org/10.1007/s10126-021-10051-6
References
Beheshti Foroutani M, Parrish CC, Wells J, Taylor RG, Rise ML, Shahidi F (2018) Minimizing marine ingredients in diets of farmed Atlantic salmon (Salmo salar): effects on growth performance and muscle lipid and fatty acid composition. PLoS One 13:e0198538
Betancor MB, Howarth FJE, Glencross BD, Tocher DR (2014) Influence of dietary docosahexaenoic acid in combination with other long-chain polyunsaturated fatty acids on expression of biosynthesis genes and phospholipid fatty acid compositions in tissues of post-smolt Atlantic salmon (Salmo salar). Comp Biochem Physiol B Biochem Mol Biol 172-173:74–89
Brash AR (2001) Arachidonic acid as a bioactive molecule. J Clin Invest 107:1339–1345
Caballero-Solares A, Hall JR, Xue X, Eslamloo K, Taylor RG, Parrish CC, Rise ML (2017) The dietary replacement of marine ingredients by terrestrial animal and plant alternatives modulates the antiviral immune response of Atlantic salmon (Salmo salar). Fish Shellfish Immunol 64:24–38
Caballero-Solares A, Xue X, Parrish CC, Foroutani MB, Taylor RG, Rise ML (2018) Changes in the liver transcriptome of farmed Atlantic salmon (Salmo salar) fed experimental diets based on terrestrial alternatives to fish meal and fish oil. BMC Genomics 18:796
Cao SX, Dhahbi JM, Mote PL, Spindler SR (2001) Genomic profiling of short- and long-term caloric restriction effects in the liver of aging mice. Proc Natl Acad Sci U S A 98:10630–10635
Castro R, Abós B, González L, Granja AG, Tafalla C (2017) Expansion and differentiation of IgM+ B cells in the rainbow trout peritoneal cavity in response to different antigens. Dev Comp Immunol 70:119–127
Chen KC, Chang LS (2009) Arachidonic acid-induced apoptosis of human neuroblastoma SK-N-SH cells is mediated through mitochondrial alteration elicited by ROS and Ca2+−evoked activation of p38α MAPK and JNK1. Toxicology 262:199–206
Choi CS, Savage DB, Kulkarni A, Yu XX, Liu ZX, Morino K, Kim S, Distefano A, Samuel VT, Neschen S, Zhang D, Wang A, Zhang XM, Kahn M, Cline GW, Pandey SK, Geisler JG, Bhanot S, Monia BP, Shulman GI (2007) Suppression of diacylglycerol acyltransferase-2 (DGAT2), but not DGAT1, with antisense oligonucleotides reverses diet-induced hepatic steatosis and insulin resistance. J Biol Chem 282:22678–22688
Clausen T, Kaiser M, Huber R, Ehrmann M (2011) HTRA proteases: regulated proteolysis in protein quality control. Nat Rev Mol Cell Biol 12:152–162
Cleveland BM, Weber GM (2011) Effects of sex steroids on indices of protein turnover in rainbow trout (Oncorhynchus mykiss) white muscle. Gen Comp Endocrinol 174:132–142
Cleveland BM, Weber GM (2013) Effects of triploidy on growth and protein degradation in skeletal muscle during recovery from feed deprivation in juvenile rainbow trout (Oncorhynchus mykiss). Comp Biochem Physiol A Mol Integr Physiol 166:128–137
Cleveland BM, Weber GM (2015) Effects of sex steroids on expression of genes regulating growth-related mechanisms in rainbow trout (Oncorhynchus mykiss). Gen Comp Endocrinol 216:103–115
Cleveland BM, Kenney PB, Manor ML, Weber GM (2012) Effects of feeding level and sexual maturation on carcass and fillet characteristics and indices of protein degradation in rainbow trout (Oncorhynchus mykiss). Aquaculture 338–341:228–236
Duan C, Xu Q (2005) Roles of insulin-like growth factor (IGF) binding proteins in regulating IGF actions. Gen Comp Endocrinol 142:44–52
Egea M, Metón I, Córdoba M, Fernández F, Baanante IV (2008) Role of Sp1 and SREBP-1a in the insulin-mediated regulation of glucokinase transcription in the liver of gilthead sea bream (Sparus aurata). Gen Comp Endocrinol 155:359–367
Eimon PM, Ashkenazi A (2010) The zebrafish as a model organism for the study of apoptosis. Apoptosis 15:331–349
Gredilla R, Barja G (2005) Minireview: the role of oxidative stress in relation to caloric restriction and longevity. Endocrinology 146:3713–3717
Gu M, Kortner TM, Penn M, Hansen AK, Krogdahl Å (2013) Effects of dietary plant meal and soya-saponin supplementation on intestinal and hepatic lipid droplet accumulation and lipoprotein and sterol metabolism in Atlantic salmon (Salmo salar L.). Br J Nutr 111:432–444
Hastings N, Agaba MK, Tocher DR, Zheng X, Dickson CA, Dick JR, Teale AJ (2004) Molecular cloning and functional characterization of fatty acyl desaturase and elongase cDNAs involved in the production of eicosapentaenoic and docosahexaenoic acids from α-linolenic acid in Atlantic salmon (Salmo salar). Mar Biotechnol 6:463–474
Hixson SM, Parrish CC, Xue X, Wells JS, Collins SA, Anderson DM, Rise ML (2017) Growth performance, tissue composition, and gene expression responses in Atlantic salmon (Salmo salar) fed varying levels of different lipid sources. Aquaculture 467:76–88
Hori TS, Gamperl A, Booman M, Nash GW, Rise ML (2012) A moderate increase in ambient temperature modulates the Atlantic cod (Gadus morhua) spleen transcriptome response to intraperitoneal viral mimic injection. BMC Genomics 13:431
Horton JD, Goldstein JL, Brown MS (2002) SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 109:1125–1131
Horton JD, Shah NA, Warrington JA, Anderson NN, Park SW, Brown MS, Goldstein JL (2003) Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci U S A 100:12027–12032
Jordal AEO, Lie Ø, Torstensen BE (2007) Complete replacement of dietary fish oil with a vegetable oil blend affect liver lipid and plasma lipoprotein levels in Atlantic salmon (Salmo salar L.). Aquac Nutr 13:114–130
Katan T, Caballero-Solares A, Taylor RG, Rise ML, Parrish CC (2019) Effect of plant-based diets with varying ratios of ω6 to ω3 fatty acids on growth performance, tissue composition, fatty acid biosynthesis and lipid-related gene expression in Atlantic salmon (Salmo salar). Comp Biochem Physiol Part D Genomics Proteomics 30:290–304
Kjær MA, Todorčević M, Torstensen BE, Vegusdal A, Ruyter B (2008) Dietary n-3 HUFA affects mitochondrial fatty acid β-oxidation capacity and susceptibility to oxidative stress in Atlantic salmon. Lipids 43:813–827
Kono T, Korenaga H (2013) Cytokine gene expression in CD4 positive cells of the Japanese pufferfish, Takifugu rubripes. PLoS One 8:e66364
Kono T, Takayama H, Nagamine R, Korenaga H, Sakai M (2013) Establishment of a multiplex RT-PCR assay for the rapid detection of fish cytokines. Vet Immunol Immunopathol 151:90–101
Kortner TM, Gu J, Krogdahl Å, Bakke AM (2012) Transcriptional regulation of cholesterol and bile acid metabolism after dietary soyabean meal treatment in Atlantic salmon (Salmo salar L.). Br J Nutr 109:593–604
Kortner TM, Björkhem I, Krasnov A, Timmerhaus G, Krogdahl Å (2014) Dietary cholesterol supplementation to a plant-based diet suppresses the complete pathway of cholesterol synthesis and induces bile acid production in Atlantic salmon (Salmo salar L.). Br J Nutr 111:2089–2103
Kowaltowski AJ, Castilho RF, Vercesi AE (2001) Mitochondrial permeability transition and oxidative stress. FEBS Lett 495:12–15
Leaver MJ, Villeneuve LA, Obach A, Jensen L, Bron JE, Tocher DR, Taggart JB (2008) Functional genomics reveals increases in cholesterol biosynthetic genes and highly unsaturated fatty acid biosynthesis after dietary substitution of fish oil with vegetable oils in Atlantic salmon (Salmo salar). BMC Genomics 9:299
Liland NS, Espe M, Rosenlund G, Waagbø R, Hjelle JI, Lie Ø, Fontanillas R, Torstensen BE (2013) High levels of dietary phytosterols affect lipid metabolism and increase liver and plasma TAG in Atlantic salmon (Salmo salar L.). Br J Nutr 110:1958–1967
Liu S, Hatano B, Zhao M, Yen CC, Kang K, Reilly SM, Gangl M, Gorgun C, Balschi JA, Ntambi JM, Lee CH (2010) Role of peroxisome proliferator-activated receptor delta/beta in hepatic metabolic regulation. J Biol Chem 286:1237–1247
Lu F, Haga Y, Satoh S (2015) Effects of replacing fish meal with rendered animal protein and plant protein sources on growth response, biological indices, and amino acid availability for rainbow trout Oncorhynchus mykiss. Fish Sci 81:95–105
Macqueen DJ, Garcia De La Serrana D, Johnston IA (2013) Evolution of ancient functions in the vertebrate insulin-like growth factor system uncovered by study of duplicated salmonid fish genomes. Mol Biol Evol 30:1060–1076
Manor ML, Cleveland BM, Brett Kenney P, Yao J, Leeds T (2015) Differences in growth, fillet quality, and fatty acid metabolism-related gene expression between juvenile male and female rainbow trout. Fish Physiol Biochem 41:533–547
Metón I, Egea M, Anemaet IG, Fernández F, Baanante IV (2006) Sterol regulatory element binding protein-1a transactivates 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase gene promoter. Endocrinology 147:3446–3456
Minghetti M, Leaver MJ, Tocher DR (2011) Transcriptional control mechanisms of genes of lipid and fatty acid metabolism in the Atlantic salmon (Salmo salar L.) established cell line, SHK-1. BBA-Mol Cell Biol L 1811:194–202
Morais S, Monroig O, Zheng X, Leaver MJ, Tocher DR (2009) Highly unsaturated fatty acid synthesis in Atlantic salmon: characterization of ELOVL5- and ELOVL2-like elongases. Mar Biotechnol 11:627–639
Morais S, Taggart J, Guy D, Bell J, Tocher DR (2012) Hepatic transcriptome analysis of inter-family variability in flesh n-3 long-chain polyunsaturated fatty acid content in Atlantic salmon. BMC Genomics 13:410
Nordberg J, Arnér ESJ (2001) Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic Biol Med 31:1287–1312
Oliva-Teles A, Enes P, Peres H (2015) Replacing fishmeal and fish oil in industrial aquafeeds for carnivorous fish. In: Davis DA (ed) Feed and feeding practices in aquaculture. Elsevier, Boston
Pacitti D, Wang T, Martin SAM, Sweetman J, Secombes CJ (2014) Insights into the fish thioredoxin system: expression profile of thioredoxin and thioredoxin reductase in rainbow trout (Oncorhynchus mykiss) during infection and in vitro stimulation. Dev Comp Immunol 42:261–277
Pompeia C, Freitas JJS, Kim JS, Zyngier SB, Curi R (2012) Arachidonic acid cytotoxicity in leukocytes: implications of oxidative stress and eicosanoid synthesis. Biol Cell 94:251–265
Poudyal H, Panchal SK, Diwan V, Brown L (2011) Omega-3 fatty acids and metabolic syndrome: effects and emerging mechanisms of action. Prog Lipid Res 50:372–387
Roques S, Deborde C, Richard N, Skiba-Cassy S, Moing A, Fauconneau B (2018) Metabolomics and fish nutrition: a review in the context of sustainable feed development. Rev Aquac 12:261–282
Scorletti E, Byrne CD (2013) Omega-3 fatty acids, hepatic lipid metabolism, and nonalcoholic fatty liver disease. Annu Rev Nutr 33:231–248
Shahkar E, Yun H, Lee S, Kim DJ, Kim SK, Lee BI, Bai SC (2016) Evaluation of the optimum dietary arachidonic acid level and its essentiality based on growth and non-specific immune responses in Japanese eel, Anguilla japonica. Aquaculture 452:209–216
Shen Z, Ma Y, Ji Z, Hao Y, Yan X, Zhong Y, Tang X, Ren W (2018) Arachidonic acid induces macrophage cell cycle arrest through the JNK signaling pathway. Lipids Health Dis 17:26
Shimomura I, Shimano H, Horton JD, Goldstein JL, Brown MS (1997) Differential expression of exons 1a and 1c in mRNAs for sterol regulatory element binding protein-1 in human and mouse organs and cultured cells. J Clin Invest 99:838–845
Sitjà-Bobadilla A, Peña-Llopis S, Gómez-Requeni P, Médale F, Kaushik S, Pérez-Sánchez J (2005) Effect of fish meal replacement by plant protein sources on non-specific defence mechanisms and oxidative stress in gilthead sea bream (Sparus aurata). Aquaculture 249:387–400
Skugor S, Grisdale-Helland B, Refstie S, Afanasyev S, Vielma J, Krasnov A (2011) Gene expression responses to restricted feeding and extracted soybean meal in Atlantic salmon (Salmo salar L.). Aquac Nutr 17:505–517
Song GX, Shen YH, Liu YQ, Sun W, Miao LP, Zhou LJ, Liu HL, Yang R, Kong XQ, Cao KJ, Qian LM, Sheng YH (2012) Overexpression of FABP3 promotes apoptosis through inducing mitochondrial impairment in embryonic cancer cells. J Cell Biochem 113:3701–3708
Spindler SR, Dhahbi JM (2007) Conserved and tissue-specific genic and physiologic responses to caloric restriction and altered IGFI signaling in mitotic and postmitotic tissues. Annu Rev Nutr 27:193–217
Storlien LH, Kriketos AD, Calvert GD, Baur LA, Jenkins AB (1997) Fatty acids, triglycerides and syndromes of insulin resistance. Prostaglandins Leukot Essent Fat Acids 57:379–385
Tacchi L, Secombes C, Bickerdike R, Adler M, Venegas C, Takle H, Martin S (2012) Transcriptomic and physiological responses to fishmeal substitution with plant proteins in formulated feed in farmed Atlantic salmon (Salmo salar). BMC Genomics 13:363
Tocher DR, Bell JG, Dick JR, Sargent JR (1997) Fatty acyl desaturation in isolated hepatocytes from Atlantic salmon (Salmo salar): stimulation by dietary borage oil containing γ-linolenic acid. Lipids 32:1237–1247
Torrecillas S, Robaina L, Caballero MJ, Montero D, Calandra G, Mompel D, Karalazos V, Kaushik S, Izquierdo MS (2017) Combined replacement of fishmeal and fish oil in European sea bass (Dicentrarchus labrax): production performance, tissue composition and liver morphology. Aquaculture 474:101–112
Torrecillas S, Betancor MB, Caballero MJ, Rivero F, Robaina L, Izquierdo M, Montero D (2018) Supplementation of arachidonic acid rich oil in European sea bass juveniles (Dicentrarchus labrax) diets: effects on growth performance, tissue fatty acid profile and lipid metabolism. Fish Physiol Biochem 44:283–300
Torstensen BE, Nanton DA, Olsvik PA, Sundvold H, Stubhaug I (2009) Gene expression of fatty acid-binding proteins, fatty acid transport proteins (cd36 and FATP) and β-oxidation-related genes in Atlantic salmon (Salmo salar L.) fed fish oil or vegetable oil. Aquac Nutr 15:440–451
Tschopp J (2011) Mitochondria: sovereign of inflammation? Eur J Immunol 41:1196–1202
Turchini GM, Torstensen BE, Ng WK (2009) Fish oil replacement in finfish nutrition. Rev Aquac 1:10–57
Vilhelmsson OT, Martin SAM, Médale F, Kaushik SJ, Houlihan DF (2007) Dietary plant-protein substitution affects hepatic metabolism in rainbow trout (Oncorhynchus mykiss). Br J Nutr 92:71–80
Wall R, Ross RP, Fitzgerald GF, Stanton C (2010) Fatty acids from fish: the anti-inflammatory potential of long-chain omega-3 fatty acids. Nutr Rev 68:280–289
Wolf LA, Laster SM (1999) Characterization of arachidonic acid-induced apoptosis. Cell Biochem Biophys 30:353–368
Xu H, Ai Q, Mai K, Xu W, Wang J, Ma H, Zhang W, Wang X, Liufu Z (2010) Effects of dietary arachidonic acid on growth performance, survival, immune response and tissue fatty acid composition of juvenile Japanese seabass, Lateolabrax japonicus. Aquaculture 307:75–82
Xu Q, Feng CY, Hori TS, Plouffe DA, Buchanan JT, Rise ML (2013) Family-specific differences in growth rate and hepatic gene expression in juvenile triploid growth hormone (GH) transgenic Atlantic salmon (Salmo salar). Comp Biochem Physiol Part D Genomics Proteomics 8:317–333
Xue X, Hixson SM, Hori TS, Booman M, Parrish CC, Anderson DM, Rise ML (2015) Atlantic salmon (Salmo salar) liver transcriptome response to diets containing Camelina sativa products. Comp Biochem Physiol Part D Genomics Proteomics 14:1–15
Xue X, Hall JR, Caballero-Solares A, Eslamloo K, Taylor RG, Parrish CC, Rise ML (2020) Liver transcriptome profiling reveals that dietary DHA and EPA levels influence suites of genes involved in metabolism, redox homeostasis, and immune function in Atlantic salmon (Salmo salar). Mar Biotechnol 22:263–284
Funding
This study was conducted within the Biomarker Platform for Commercial Aquaculture Feed Development project, a Genomic Applications Partnership Program (GAPP no. 6604) project, funded by the Government of Canada through Genome Canada and Genome Atlantic. Cargill Innovation (formerly EWOS Innovation) provided support to the study in the form of salary for RGT and the production of the experimental feeds. Innovate NL (no. 5404-1019-107) and Atlantic Canada Opportunities Agency (ACOA no. 206200) funded the purchase of the instrument used for the multiplex PCR analyses (GenomeLab GeXP genetic analysis system (Beckman Coulter/SCIEX)). Research programs at MLR’s laboratory are also supported by a Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grant (341304-2012). The authors would like to thank the Dr. Joe Brown Aquatic Research Building (JBARB) staff for their assistance with fish husbandry and sampling. We are also grateful to Jeanette Wells for providing the liver lipid composition and to Cara Kirkpatrick (Genome Atlantic, Halifax, Canada) for helping as the Program Manager of this project.
Author information
Authors and Affiliations
Contributions
ACS had the lead role in the design and execution of the gene expression experiment, as well as the analysis of the results and the writing of the manuscript. MLR, CCP, and RGT designed the experimental diets. RGT led the team at Cargill Innovation (formerly EWOS Innovation) that elaborated the diets. ACS, XX, and MBF took part in the collection of the tissue samples for gene expression and lipid composition analyses. ACS, BMC, XX, MLR, and RGT collaborated in the design of the two multiplex PCR panels. BMC trained ACS in the multiplex PCR technique. MBF conducted the liver lipid composition analyses under CCP guidance. MLR and CCP helped with the data analysis and interpretation. MLR supervised the experimentation, result analyses, and the preparation of this manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
RGT was employed by Cargill Innovation at the time this research was conducted. RGT, in the representation of Cargill Innovation, participated in the formulation of the experimental diets and the design of the trial. RGT also collaborated in the design of the multiplex panels and reviewed the manuscript, but had no role in the design of the gene expression experiment, the data collection and analysis, preparation of the manuscript, and the decision to publish. The corresponding author has full access to all the data in the study and takes complete responsibility for the integrity of the data and the accuracy of its analysis. All authors declare that they have no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised due to a retrospective Open Access order.
Electronic supplementary material
ESM 1
Scatter plot of fold-changes between diets calculated using log2-transformed multiplex (x axis) and singleplex (y axis) relative quantity (log2 RQ) gene expression values. Each dot represents either an ABP vs MAR, VEG vs MAR, or VEG vs ABP comparison for a given gene. (PNG 124 kb)
ESM 2
Matrix of Pearson’s correlation coefficients (r) among the selected phenotypic features. Significant correlations are indicted by displaying the r values in the cell. Despite being redundant, half of each correlation matrix was not omitted to facilitate its interpretation. (PNG 380 kb)
ESM 3
Matrix of Pearson’s correlation coefficients (r) among the analyzed biomarker genes. Significant correlations are indicted by displaying the r values in the cell. Despite being redundant, half of each correlation matrix was not omitted to facilitate its interpretation. (PNG 1447 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Caballero-Solares, A., Xue, X., Cleveland, B.M. et al. Diet-Induced Physiological Responses in the Liver of Atlantic Salmon (Salmo salar) Inferred Using Multiplex PCR Platforms. Mar Biotechnol 22, 511–525 (2020). https://doi.org/10.1007/s10126-020-09972-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10126-020-09972-5