
Abstract
Background
Prediction of tissue origin of esophagogastric junction (EGJ) adenocarcinomas can be important for therapeutic decision, but no molecular marker is available. Here, we aimed to develop such a marker taking advantage of tissue-specific profiles of DNA methylation.
Methods
DNA methylation profiles of gastric adenocarcinomas (GACs) were obtained by an Infinium HumanMethylation450 BeadChip array, and those of esophageal adenocarcinoma (EACs) were obtained from the TCGA database. DNA from formalin-fixed paraffin-embedded (FFPE) samples was analyzed by bisulfite pyrosequencing.
Results
In the screening set, 51 of 145,841 CpG sites in CpG islands were methylated at significantly higher levels in 30 GACs compared to those in 30 EACs. Among them, SLC46A3 and cg09177106 were unmethylated in all the 30 EACs. Predictive powers of these two markers were successfully confirmed in an independent validation set (18 GACs and 18 EACs) (SLC46A3, sensitivity = 77.8%, specificity = 100%; cg09177106, sensitivity = 83.3%, specificity = 94.4%), and could be applied to FFPE samples (37 GACs and 18 EACs) (SLC46A3, P = 0.0001; cg09177106, P = 0.0028). On the other hand, EAC-specific markers informative in the FFPE samples could not be isolated. Using these GAC-specific markers, nine of 46 (19.6%) TCGA EGJ adenocarcinomas were predicted to be GACs.
Conclusions
Two GAC-specific markers, SLC46A3 and cg09177106, had a high specificity for identifying the tissue origin of EGJ adenocarcinoma.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Esophagogastric junction (EGJ) adenocarcinoma is a global health burden with its incidence increasing worldwide, especially in Western countries [1,2,3]. EGJ adenocarcinomas are classified into three types, Siewert types I, II, and III, based on the anatomic location of tumor epicenter [4,5,6]. Based on these subtypes, different treatment protocols are utilized. Namely, Siewert type I adenocarcinomas, mostly developed from Barrett’s esophagus, are treated as esophageal adenocarcinomas (EACs), and Siewert type III adenocarcinomas, mostly developed from proximal gastric mucosae with H. pylori-triggered inflammation, are treated as gastric adenocarcinomas (GACs) [7,8,9]. In contrast, Siewert type II adenocarcinomas are morphologically indistinguishable whether they originate from Barrett’s esophagus or the stomach, and no standard treatment strategy has been established [6, 10, 11]. Therefore, the development of a predictive marker for the tissue origin of EGJ adenocarcinomas is important, especially for Siewert type II.
To predict a tissue origin, DNA methylation profiles are known to reflect tissue types [12], and are mostly maintained during carcinogenesis [13,14,15]. For example, it is reported that tissue origin of circulating cell-free DNA can be identified using methylation profiles [16]. Also, cancer subtypes can be defined for breast cancers, colorectal cancers, and central nervous system tumors [17,18,19]. Furthermore, DNA methylation profiles can be utilized to predict tissue origins of cancers of unknown primary [14, 20]. Stratification of patients using a classifier of cancer type based on DNA methylation signatures and implementation of tumor type-specific therapy brings better survival than the use of empiric therapy [14]. This suggests that DNA methylation may also be useful for the prediction of the tissue origin of EGJ adenocarcinoma, especially Siewert type II. A decade ago, a pioneering study used 74 genes, and revealed that DNA methylation levels of GATA5 could classify EGJ adenocarcinomas into two subgroups, although distinction between GACs and EACs was unclear [21].
In this study, we aimed to develop a DNA methylation marker to predict tissue origin of EGJ adenocarcinomas analyzing DNA methylation levels of 470,870 CpG sites located on autosomes. Since our goal was to select EGJ adenocarcinomas to be treated as GACs and EGJ adenocarcinomas (e.g., Siewert type II cases) that are currently treated as EACs, we placed emphasis on reducing false-positive cases as much as possible.
Materials and methods
Tissue samples and cancer cell lines
Formalin-fixed paraffin-embedded (FFPE) tissue samples of 37 GACs in the gastric body (thus, excluding Siewert type III), and 18 EACs (including Siewert type I, or Nishi type E) were obtained at the National Cancer Center Hospital (GAC, n = 34, EAC, n = 3), the University of Tokyo Hospital (GAC, n = 3; EAC, n = 4), the Nippon Medical School Tama-Nagayama Hospital (Nippon Medical School) (EAC, n = 3), and the Cancer Institute Hospital of Japanese Foundation for Cancer Research (JFCR) (EAC, n = 8), with written informed consents [22, 23]. This study was approved by the Institutional Review Boards of the National Cancer Center (2018-024), the University of Tokyo [G3521-(21)], Nippon Medical School (A-2020-051), and the Cancer Institute Hospital of JFCR (2021-GA-1103).
MKN1, SW837, DLD1, and LoVo cell lines were purchased from the Japanese Collection of Research Bioresources (Osaka, Japan). The LNCaP cell line was purchased from the American Type Culture Collection (Manassas, VA, USA). The HSC64 cell line was provided by Dr. K Yanagihara (National Cancer Center Institute Research, Tokyo, Japan). For HSC64, MKN1, SW837, DLD1, LoVo, and LNCaP cell lines, DNA was extracted and methylation profiles were analyzed by Infinium HumanMethylation450 BeadChip array (Illumina, San Diego, CA) as described previously [24, 25]. Methylation profiles of KYSE30, KYSE50, KYSE140, KYSE170, KYSE180, KYSE220, KYSE270, KYSE410, KYSE450, KYSE510, KYSE520, AGS, KATOIII, N87, HSC44, HSC59, 44As3, BT20, HCC1395, HCC1428, HCC1937, HCC1954, MCF7, and ZR-75-1 cell lines were obtained from our previous studies [24,25,26,27].
The cells were maintained in Roswell Park Memorial Institute (RPMI)-1640 medium (HSC64, MKN1, SW837, DLD1, and LNCaP) or Ham’s F12 medium (LoVo) supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin at 37 °C in a humidified atmosphere with 5% CO2.
DNA extraction
From FFPE samples, cells enriched for cancer cells were collected by macrodissection of histological sections (10 μm thick) for 37 GAC and 18 EAC adenocarcinoma FFPE samples. Dissected tissues were treated with deparaffinization solution and a proteinase K [EpiTect Plus bisulfite kit (Qiagen, Hilden, Germany)], and were purified using a Zymo-Spin I column (Zymo Research, Irvine, CA). Genomic DNA was extracted from 4 to 10 FFPE sections or 106–107 cultured cells by the phenol/chloroform method, and quantified based on the absorbance at 260 nm. For FFPE samples, 10 ng of DNA was used for the analysis of RPPH1 gene copy number by quantitative real-time PCR using SYBR Green I (BioWhittaker Molecular Applications, Rockland, MD) and primers are listed in Supplementary Table S1.
Genome-wide DNA methylation analysis
Genome-wide DNA methylation profiles using an Infinium HumanMethylation450 BeadChip array were obtained from our previous studies and public databases (Supplementary Table S2). Methylation profiles of 48 GACs were obtained from our previous studies [28, 29]. Profiles of 30 GACs, 48 EACs, and 46 EGJ adenocarcinomas were from The Cancer Genome Atlas (TCGA) database. Profiles of 16 normal gastric mucosae [8 mucosae with Helicobacter pylori (H. pylori)-infection and 8 mucosae without infection] and normal esophagus were from our previous study [30, 31]. Profiles of Barrett’s esophagus were obtained from the Gene Expression Omnibus (GEO) (accession # GSE81334). Profiles of peripheral leukocytes were from a previous study by Reinius et al. [32]. Among 485,512 probes in a BeadChip array, 470,870 CpG sites located on the autosomes were used for the analysis. β values (DNA methylation levels), ranging from 0 (complete unmethylation) to 1 (complete methylation), were obtained, and probe type difference was normalized using a web tool, MACON [33].
Bisulfite pyrosequencing
Genomic DNA (1 µg) was extracted from FFPE samples and cancer cell lines was treated with sodium bisulfite using an EZ DNA Methylation Kit (Zymo Research, Irvine, CA), and bisulfite-treated DNA was eluted in 40 μl of elution buffer. Bisulfite-treated DNA that had 50 copies of RPPH1 or more in FFPE samples (average = 137 copies; range = 50 to 628 copies) and cancer cell lines was analyzed by bisulfite pyrosequencing (PyroMark Q96 ID, Qiagen, Valencia, CA) using the listed primers (Supplementary Table S3). DNA methylation levels of target CpG sites were calculated using the PSQ Assay Design software (Qiagen). The average value of target CpG sites in a region sequenced by bisulfite pyrosequencing was obtained.
Statistical analyses
DNA methylation levels that showed the maximum Youden index (sensitivity + specificity − 1) were calculated using R version 3.6.2 with the ROCR package. The highest value was selected as a cut-off value to place emphasis on the high specificity and reduce false-positive cases as much as possible.
The correlation between bisulfite pyrosequencing and Infinium BeadChip array was analyzed by a Pearson correlation coefficient with a parametric hypothesis test. Based on the correlation analysis, a methylation level obtained by pyrosequencing was scaled to that by Infinium BeadChip array. A difference of the mean DNA methylation values was evaluated by Student’s t test. All analyses were conducted by two-sided tests and P < 0.05 was considered statistically significant.
Results
Screening of GAC- and EAC-specific methylation markers
To isolate GAC-specifically methylated regions (GAC-specific markers), a genome-wide DNA methylation profile of 30 GACs and 30 EACs (the screening set) was utilized (Supplementary Table S2). 75,308 CpG sites unmethylated (average β value < 0.2) in seven types of leukocytes (Th cells, Tc cells, NK cells, B cells, monocytes, neutrophils, and eosinophils) [32], normal esophagus (n = 8), and Barrett’s esophagus (n = 8) were isolated from 145,841 CpG sites in CpG islands (Fig. 1a). A volcano plot of these 75,308 CpG sites showed that 51 CpG sites had significantly higher methylation levels in the 30 GACs than in the 30 EACs [Δβ (GAC − EAC) > 0.2, and P < 0.001] (Fig. 1b). Among them, 2 CpG sites (SLC46A3 and cg09177106) were unmethylated (β value < 0.2) in all the 30 EACs, and were considered as GAC-specific methylation markers (Supplementary Fig. S1a). Based on the maximum Youden index in the screening set, the cut-off values for SLC46A3 and cg09177106 were calculated as 0.03 and 0.061, respectively (Supplementary Fig. S1b).

Genome-wide screening of gastric adenocarcinoma (GAC)-specific and esophageal adenocarcinoma (EAC)-specific methylation markers. a Screening scheme of GAC-specific methylation markers. 75,308 CpG sites unmethylated (average β value < 0.2) in leukocytes, normal esophagus, and Barrett’s esophagus were selected from 485,512 probes. Among them, 51 CpG sites had significantly higher methylation levels in 30 GACs than in 30 EACs [Δβ (GAC − EAC) > 0.2, and P < 0.001]. Two of 51 CpG sites, SLC46A3 and cg09177106, were unmethylated in all the EACs. b Volcano plot analysis for identification of GAC-specific methylation. 51 CpG sites had significantly higher methylation levels in 30 GACs than in 30 EAC [Δβ (EAC − GAC) > 0.2, and P < 0.001]. c Screening scheme of EAC-specific methylation markers. 72,354 CpG sites unmethylated in leukocytes, gastric mucosae with H. pylori infection, and those without were selected from 485,512 probes. Among them, 51 CpG sites had significantly higher methylation levels in 30 EACs than in 30 GAC [Δβ (EAC − GAC) > 0.2, and P < 0.001]. Three of 51 CpG sites, MARCKSL1, RIC8B, and RAB11FIP3, were unmethylated in all the GACs. d Volcano plot analysis for identification of EAC-specific methylation. 51 CpG sites had significantly higher methylation levels in EACs compared to GACs
EAC-specific markers were also searched for by the same strategy. 72,354 CpG sites unmethylated (average β value < 0.2) in leukocytes, gastric mucosae without H. pylori infection (n = 8), and those with H. pylori infection (n = 8) were isolated from the 145,841 CpG sites in CpG islands (Fig. 1c). A volcano plot of these 75,354 CpG sites showed that 51 CpG sites had significantly higher methylation levels in 30 EACs than in 30 GACs [Δβ (EAC − GAC) > 0.2, and P < 0.001] (Fig. 1d). Among them, 3 CpG sites (MARCKSL1, RIC8B, and RAB11FIP3) were unmethylated in all the 30 GACs, and were considered as EAC-specific methylation markers (Supplementary Fig. S2a). Based on the maximum Youden index in the screening set, the optimal cut-off values of MARCKSL1, RIC8B, and RAB11FIP3 were calculated as 0.084, 0.143 and 0.134, respectively (Supplementary Fig. S2b).
Validation of the GAC- and EAC-specific methylation markers
To validate the predictive powers of GAC-specific markers (SLC46A3 and cg09177106) and EACs markers (MARCKSL1, RIC8B and RAB11FIP3), DNA methylation levels were analyzed by Infinium BeadChip array in an independent validation set of 18 GACs and 18 EACs. The two GAC-specific markers had significantly higher methylation levels in GACs compared to EACs (both, P < 0.001) (Fig. 2a). Using the cut-off values established in the screening set, SLC46A3 had a sensitivity of 77.8% and specificity of 100%, and cg09177106 had a sensitivity of 83.3% and specificity of 94.4% (Supplementary Table S4). Also, the three EAC-specific markers had significantly higher methylation levels in EACs compared to GACs (all three markers, P < 0.001) (Fig. 2b). Using the cut-off values established in the screening set, MARCKSL1 had a sensitivity of 100% and specificity of 88.9%, RIC8B had a sensitivity of 100% and specificity of 100%, and RAB11FIP3 had a sensitivity of 94.4% and specificity of 88.9% (Fig. 2b).

Validation of GAC-specific and EAC-specific methylation markers. a Methylation levels of GAC-specific markers, SLC46A3 and cg09177106, in the validation set. SLC46A3 and cg09177106 had significantly higher methylation levels in 18 GACs than in 18 EACs. b Methylation levels of EAC-specific markers, MARCKSL1, RIC8B, and RAB11FIP3, in the validation set. MARCKSL1, RIC8B, and RAB11FIP3, had significantly higher methylation levels in 18 EACs than in 18 GACs. Box plots represent mean methylation levels. Error bars represent SD. Two-sided Student’s t test was conducted
Application to FFPE samples
Implementation of a cost-effective method, for example pyrosequencing, is essential for clinical application. In addition, since most clinical samples are FFPE samples, we decided to establish a pyrosequencing system for the GAC- and EAC-specific markers for FFPE samples. We attempted to design pyrosequencing primers for the five potential markers, and those for the two GAC-specific markers (SLC46A3 and cg09177106) and one (MARCKSL1) of the three EAC-specific markers were successfully developed (Supplementary Table S3). Consistency between the methylation levels obtained by the pyrosequencing and those by the Infinium BeadChip array was analyzed using DNA from 30 cell lines. Using 17–18 cell lines with known methylation levels by Infinium BeadChip array in our previous studies [24,25,26,27] and obtained in this study, methylation levels of the two GAC-specific markers and one EAC-specific marker were measured by pyrosequencing. Correlation coefficients between the values obtained by the two methods were 0.947 for SLC46A3 (P < 0.001), 0.935 for cg09177106 (P < 0.001) (Supplementary Fig. S3a), and 0.934 for MARCKSL1 (P < 0.001) (Supplementary Fig. S3b).
DNA methylation levels of the GAC- and EAC-specific markers were measured in 37 GAC and 18 EAC FFPE samples by pyrosequencing. Two GAC-specific markers had higher methylation levels in GACs compared to EACs (SLC46A3, P = 0.0001; cg09177106, P = 0.0028) (Fig. 3a). Analysis of the Cancer Cell Line Encyclopedia database (www.broadinstitute.org/ccle) showed that SLC46A3 methylation and mRNA expression are strongly negatively correlated in 831 cancer cell lines (R = − 0.592, P = 9.8 × 10–80) (Supplementary Fig. S4) [34]. In contrast, the EAC-specific marker, MARCKSL1, did not show any difference between the GAC and EAC FFPE samples (Fig. 3b).

Application of GAC- specific and EAC-specific methylation markers to FFPE samples. a Methylation levels of GAC-specific markers, SLC46A3 and cg09177106, in the FFPE samples. SLC46A3 and cg09177106 had significantly higher methylation levels in 20 GACs than in 7 EACs. b Methylation levels of one EAC-specific marker, MARCKSL1, in the FFPE samples. Methylation levels of MARCKSL1 were not different between FFPE GAC and EAC samples. Box plots represent mean methylation levels. Error bars represent SD. Two-sided Student’s t test was conducted
Prediction of tissue origin of EGJ adenocarcinoma by the GAC-specific markers
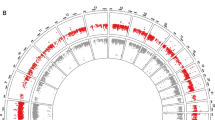
First, DNA methylation levels of the GAC-specific markers, SLC46A3 and cg09177106, were analyzed in GACs in Western countries. Both markers also had high methylation levels in GACs in Western countries similar to cases of Japanese GACs (Supplementary Figure S5). Then, to further evaluate the clinical utility, a TCGA cohort (n = 46) of EGJ adenocarcinomas in Western countries was assessed for the fraction of GACs by the GAC-specific markers. SLC46A3 and cg09177106 had high methylation levels in six and four EGJ adenocarcinomas, respectively (one EGJ adenocarcinoma was overlapped). Finally, 9 of 46 EGJ adenocarcinomas (19.6%) were predicted to be GACs (Fig. 4a, b). This result indicated that most EGJ adenocarcinomas in Western countries are from the esophagus.

Prediction of tissue origin of EGJ adenocarcinomas by two GAC-specific markers, SLC46A3 and cg09177106. a DNA methylation levels of SLC46A3 in 46 TCGA EGJ adenocarcinomas. Six of 46 (13.0%) TCGA EGJ adenocarcinomas were predicted to be GACs (fresh-frozen samples, cut-off value = 0.03). b DNA methylation levels of cg09177106 in 46 TCGA EGJ adenocarcinomas. Four of 46 (8.7%) TCGA EGJ adenocarcinomas were predicted to be GACs (fresh-frozen samples, cut-off value = 0.061)
Discussion
The tissue origin of EGJ adenocarcinomas was predicted by utilizing DNA methylation profiles. Although the determination of the treatment protocols for Siewert type II adenocarcinomas is still controversial between Eastern and Western countries [35, 36], the predictive marker developed here might be useful to determine the treatment strategy. Since the two GAC-specific markers, SLC46A3 and cg09177106, have a very high specificity for detection of GACs in EGJ adenocarcinomas, once identified as GACs, patients would have benefitted on the treatment strategy of gastric adenocarcinomas. To clinically utilize these markers, we expect that future studies will clarify the sensitivity and specificity of the markers in FFPE samples.
The marker CpG site of SLC46A3 was located in its TSS200 region within a CpG island, and thus, its methylation was expected to be associated with its expression. The negative correlation between its methylation and expression in the Cancer Cell Line Encyclopedia database supported the expectation. In general, promoter CpG islands of genes with low expression are more likely to be methylated than those with high expression [30]. SLC46A3 methylation in GACs, but not in EACs, may have been caused by its low expression in the stomach and high expression in Barrett’s esophagus, which is considered as an origin of EACs [37]. In contrast, cg09177106 was located in a CpG island out of a genic region. Therefore, its methylation cannot be connected to expression of a specific gene, and the mechanism of how it is specifically methylated in GAC remains unknown.
Based on the markers isolated, the TCGA EGJ adenocarcinomas were actually mixtures of GACs and EACs, supporting a previous finding that Siewert type II EGJ adenocarcinomas are possibly a mixture of types I and III adenocarcinomas [38]. However, the fraction of GACs in TCGA EGJ adenocarcinomas was only 19.6%, indicating that most EGJ adenocarcinomas in Western countries were derived from the esophagus. Considering the sensitivity of the two markers (SLC46A3, 77.8%; cg09177106, 83.3%), the actual GAC fraction can be estimated to be around 25% in the TCGA cohort. It has been reported that Siewert type II adenocarcinoma patients in South Korea have a similar prognosis with type III, most of which are GACs [39]. The high incidence of GAC among the EGJ adenocarcinomas in Eastern countries was considered to be due to the high incidence of H. pylori infection [40, 41], and the high incidence of EAC in Western countries was considered to be due to a high incidence of gastroesophageal reflux disease and Barrett’s esophagus [42, 43].
Although the correlation of methylation levels between pyrosequencing and Infinium BeadChip array was high for one EAC-specific markers, MARCKSL1 (CHR1, 32802033), there was no difference in the methylation levels between EACs and GACs in the FFPE samples. This suggested that the marker CpG site is susceptible to DNA degradation in the FFPE samples. To address this issue, we used our previous study on the analysis of fresh-frozen and FFPE samples of the same specimen, and MARCKSL1 turned out to show a significant difference between the two types of samples. In contrast, the other two GAC-specific markers, SLC46A3 and cg09177106, presented consistent values (Supplementary Table. S5). Therefore, the probe of MARCKSL1 (CHR1, 32802033) was considered to be susceptible to DNA degradation during formalin fixation.
As alternative EAC-specific markers, screening of candidate markers using CpG sites located outside of CpG islands was also conducted. One CpG site (DAXX, cg03369465) was identified as a candidate EAC-specific marker (screening set, P = 7.2 × 10–6; validation set, P = 7.1 × 10–6) (Supplementary Fig. S6). However, DNA methylation levels of its adjacent CpG sites were discordant with that of cg03369465. Such discordant methylation levels in closely neighboring probes are often caused by unstable DNA methylation values of an unreliable probe, and we therefore decided to discard this probe.
Although both GAC-specific markers, SLC46A3 and cg09177106, had high specificity in both screening and validation sets, the sensitivity is still insufficient to be applied as a routine standard to identify the tissue origin of EGJ adenocarcinomas. Therefore, further screening of additional markers, which can be combined with markers identified in this study, will be necessary. To conduct additional screening, an increase in sample number, especially for EACs, is critical. Once a sufficient number of EAC samples are collected in future studies, efforts to identify additional combination markers should be made.
In conclusion, two GAC-specific DNA methylation markers, SLC46A3 and cg09177106, had high specificity for predicting the tissue origin of EGJ adenocarcinomas. This is further expected to be important for determining the therapeutic strategy for EGJ adenocarcinoma patients, especially for Siewert type II adenocarcinomas.
Availability of data and materials
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Botterweck AA, Schouten LJ, Volovics A, Dorant E, van Den Brandt PA. Trends in incidence of adenocarcinoma of the oesophagus and gastric cardia in ten European countries. Int J Epidemiol. 2000;29:645–54.
Yamashita H, Seto Y, Sano T, Makuuchi H, Ando N, Sasako M. Results of a nation-wide retrospective study of lymphadenectomy for esophagogastric junction carcinoma. Gastric Cancer. 2017;20:69–83.
Mariette C, Piessen G, Briez N, Gronnier C, Triboulet JP. Oesophagogastric junction adenocarcinoma: which therapeutic approach? Lancet Oncol. 2011;12:296–305.
Rüdiger Siewert J, Feith M, Werner M, Stein HJ. Adenocarcinoma of the esophagogastric junction: results of surgical therapy based on anatomical/topographic classification in 1,002 consecutive patients. Ann Surg. 2000;232:353–61.
Siewert JR, Stein HJ. Carcinoma of the gastroesophageal junction—classification, pathology and extent of resection. Dis Esophagus. 1996;9:173–82.
Kurokawa Y, Takeuchi H, Doki Y, Mine S, Terashima M, Yasuda T, et al. Mapping of lymph node metastasis from esophagogastric junction tumors: a prospective nationwide multicenter study. Ann Surg. 2021;274:120–7.
Ajani JA, D’Amico TA, Bentrem DJ, Chao J, Corvera C, Das P, et al. Esophageal and esophagogastric junction cancers, version 2.2019, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2019;17:855–83.
McColl KE, Going JJ. Aetiology and classification of adenocarcinoma of the gastro-oesophageal junction/cardia. Gut. 2010;59:282–4.
Lutz MP, Zalcberg JR, Ducreux M, Adenis A, Allum W, Aust D, et al. The 4th St. Gallen EORTC Gastrointestinal Cancer Conference: controversial issues in the multimodal primary treatment of gastric, junctional and oesophageal adenocarcinoma. Eur J Cancer. 2019;112:1–8.
Giacopuzzi S, Bencivenga M, Weindelmayer J, Verlato G, de Manzoni G. Western strategy for EGJ carcinoma. Gastric Cancer. 2017;20:60–8.
Hölscher AH, Law S. Esophagogastric junction adenocarcinomas: individualization of resection with special considerations for Siewert type II, and Nishi types EG, E=G and GE cancers. Gastric Cancer. 2020;23:3–9.
Schultz MD, He Y, Whitaker JW, Hariharan M, Mukamel EA, Leung D, et al. Human body epigenome maps reveal noncanonical DNA methylation variation. Nature. 2015;523:212–6.
Hovestadt V, Jones DT, Picelli S, Wang W, Kool M, Northcott PA, et al. Decoding the regulatory landscape of medulloblastoma using DNA methylation sequencing. Nature. 2014;510:537–41.
Moran S, Martínez-Cardús A, Sayols S, Musulén E, Balañá C, Estival-Gonzalez A, et al. Epigenetic profiling to classify cancer of unknown primary: a multicentre, retrospective analysis. Lancet Oncol. 2016;17:1386–95.
Suzuki H, Maruyama R, Yamamoto E, Kai M. DNA methylation and microRNA dysregulation in cancer. Mol Oncol. 2012;6:567–78.
Moss J, Magenheim J, Neiman D, Zemmour H, Loyfer N, Korach A, et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat Commun. 2018;9:5068.
Holm K, Hegardt C, Staaf J, Vallon-Christersson J, Jönsson G, Olsson H, et al. Molecular subtypes of breast cancer are associated with characteristic DNA methylation patterns. Breast Cancer Res. 2010;12:R36.
Bormann F, Rodríguez-Paredes M, Lasitschka F, Edelmann D, Musch T, Benner A, et al. Cell-of-origin DNA methylation signatures are maintained during colorectal carcinogenesis. Cell Rep. 2018;23:3407–18.
Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, et al. DNA methylation-based classification of central nervous system tumours. Nature. 2018;555:469–74.
Varadhachary GR, Raber MN. Cancer of unknown primary site. N Engl J Med. 2014;371:757–65.
Wang X, Kang GH, Campan M, Weisenberger DJ, Long TI, Cozen W, et al. Epigenetic subgroups of esophageal and gastric adenocarcinoma with differential GATA5 DNA methylation associated with clinical and lifestyle factors. PLoS ONE. 2011;6:e25985.
Kitagawa Y, Uno T, Oyama T, Kato K, Kato H, Kawakubo H, et al. Esophageal cancer practice guidelines 2017 edited by the Japan Esophageal Society: part 1. Esophagus. 2019;16:1–24.
Japanese Classification of Esophageal Cancer, 11th Edition: part II and III. Esophagus 2017;14:37–65.
Iwabu J, Yamashita S, Takeshima H, Kishino T, Takahashi T, Oda I, et al. FGF5 methylation is a sensitivity marker of esophageal squamous cell carcinoma to definitive chemoradiotherapy. Sci Rep. 2019;9:13347.
Zong L, Hattori N, Yoda Y, Yamashita S, Takeshima H, Takahashi T, et al. Establishment of a DNA methylation marker to evaluate cancer cell fraction in gastric cancer. Gastric Cancer. 2016;19:361–9.
Yamashita S, Hattori N, Fujii S, Yamaguchi T, Takahashi M, Hozumi Y, et al. Multi-omics analyses identify HSD17B4 methylation-silencing as a predictive and response marker of HER2-positive breast cancer to HER2-directed therapy. Sci Rep. 2020;10:15530.
Zong L, Hattori N, Yasukawa Y, Kimura K, Mori A, Seto Y, et al. LINC00162 confers sensitivity to 5-Aza-2’-deoxycytidine via modulation of an RNA splicing protein, HNRNPH1. Oncogene. 2019;38:5281–93.
Kim JG, Takeshima H, Niwa T, Rehnberg E, Shigematsu Y, Yoda Y, et al. Comprehensive DNA methylation and extensive mutation analyses reveal an association between the CpG island methylator phenotype and oncogenic mutations in gastric cancers. Cancer Lett. 2013;330:33–40.
Yoda Y, Takeshima H, Niwa T, Kim JG, Ando T, Kushima R, et al. Integrated analysis of cancer-related pathways affected by genetic and epigenetic alterations in gastric cancer. Gastric Cancer. 2015;18:65–76.
Yamashita S, Nanjo S, Rehnberg E, Iida N, Takeshima H, Ando T, et al. Distinct DNA methylation targets by aging and chronic inflammation: a pilot study using gastric mucosa infected with Helicobacter pylori. Clin Epigenetics. 2019;11:191.
Yamashita S, Kishino T, Takahashi T, Shimazu T, Charvat H, Kakugawa Y, et al. Genetic and epigenetic alterations in normal tissues have differential impacts on cancer risk among tissues. Proc Natl Acad Sci U S A. 2018;115:1328–33.
Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlén SE, Greco D, et al. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS ONE. 2012;7:e41361.
Iida N, Okuda Y, Ogasawara O, Yamashita S, Takeshima H, Ushijima T. MACON: a web tool for computing DNA methylation data obtained by the illumina infinium human DNA methylation BeadArray. Epigenomics. 2018;10:249–58.
Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–7.
Parry K, Ruurda JP, van der Sluis PC, van Hillegersberg R. Current status of laparoscopic transhiatal esophagectomy for esophageal cancer patients: a systematic review of the literature. Dis Esophagus. 2017;30:1–7.
Kurokawa Y, Sasako M, Sano T, Yoshikawa T, Iwasaki Y, Nashimoto A, et al. Ten-year follow-up results of a randomized clinical trial comparing left thoracoabdominal and abdominal transhiatal approaches to total gastrectomy for adenocarcinoma of the oesophagogastric junction or gastric cardia. Br J Surg. 2015;102:341–8.
Que J, Garman KS, Souza RF, Spechler SJ. Pathogenesis and cells of origin of Barrett’s esophagus. Gastroenterology. 2019;157:349-364.e1.
Reynolds JV, Ravi N, Muldoon C, Larkin JO, Rowley S, O’Byrne K, et al. Differential pathologic variables and outcomes across the spectrum of adenocarcinoma of the esophagogastric junction. World J Surg. 2010;34:2821–9.
Suh YS, Han DS, Kong SH, Lee HJ, Kim YT, Kim WH, et al. Should adenocarcinoma of the esophagogastric junction be classified as esophageal cancer? A comparative analysis according to the seventh AJCC TNM classification. Ann Surg. 2012;255:908–15.
Erőss B, Farkas N, Vincze Á, Tinusz B, Szapáry L, Garami A, et al. Helicobacter pylori infection reduces the risk of Barrett’s esophagus: a meta-analysis and systematic review. Helicobacter. 2018;23:e12504.
Ford AC, Yuan Y, Forman D, Hunt R, Moayyedi P. Helicobacter pylori eradication for the prevention of gastric neoplasia. Cochrane Database Syst Rev. 2020;7:Cd005583.
Rustgi AK, El-Serag HB. Esophageal carcinoma. N Engl J Med. 2014;371:2499–509.
Dong J, Buas MF, Gharahkhani P, Kendall BJ, Onstad L, Zhao S, et al. Determining risk of Barrett’s esophagus and esophageal adenocarcinoma based on epidemiologic factors and genetic variants. Gastroenterology. 2018;154:1273-1281.e3.
Acknowledgements
Sample collection was supported by the National Cancer Center Biobank, Tokyo, Japan. The results here are partly based on data generated by the TCGA Research Network (https://www.cancer.gov/tcga) and the Cancer Cell Line Encyclopedia database (www.broadinstitute.org/ccle). Chun-Dong Zhang (CZ) was partly supported by the China Scholarship Council (Number 201908050148) and the Japan China Sasakawa Medical Fellowship (Number 2017816).
Funding
This research was partly supported by AMED under Grant Number JP20cm0106451 to TU and JP20ck0106552 to TU.
Author information
Authors and Affiliations
Contributions
Concept and design: CZ, HT, and TU; collections of samples: CZ, HT, SS, SY, HA, HY, TU, HM, HK, YI, and MW; molecular analysis: CZ, HT, SY, YL, NH, and MF; writing manuscript: CZ, HT, and TU; all authors read, revised, and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interests
The authors state no competing interests regarding this work.
Ethics approval and consent to participate
The study was performed according to ethics approval and consent. The study was approved by the Institutional Review Boards of the National Cancer Center (2018-024), The University of Tokyo [G3521-(21)], Nippon Medical School (A-2020-051), and the Cancer Institute Hospital of Japanese Foundation for Cancer Research (2021-GA-1103). All procedures in this study were conducted in accordance with the Declaration of Helsinki.
Consent for publication
Informed consent for publication was obtained from all participants.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
10120_2021_1252_MOESM1_ESM.pptx
Supplementary file1 Supplementary Figure S1 Methylation status of gastric adenocarcinoma (GAC)-specific methylation markers in the screening set. (a) Methylation levels of GAC-specific methylation markers, SLC46A3 and cg09177106, in GACs, EACs, leukocytes (LK), normal esophagus mucosae (EM), and Barrett's esophagus (BE). GAC-specific markers had higher methylation levels in GACs, but not in others. (b) ROC analysis of the GAC-specific markers. SLC46A3 had a sensitivity of 56.7% and a specificity of 100% (cut-off value = 0.1), and cg09177106 had a sensitivity of 43.3% and a specificity of 93.3% (cut-off value = 0.1) using cut-off values determined in the screening set. Box plots represent mean methylation levels. Error bars represent SD. Two-sided Student's t test (*P < 0.001) was conducted. Supplementary Figure S2 Methylation status of esophageal adenocarcinoma (EAC)-specific methylation markers in the screening set. (a) Methylation levels of EAC-specific methylation markers, MARCKSL1, RIC8B and RAB11FIP3, in GACs, EACs, leukocytes (LK), gastric mucosae without H. pylori infection (Hp-), and those with (Hp+). EAC-specific markers had higher methylation levels in EACs, but not in others. (b) ROC analysis of the EAC-specific markers. MARCKSL1 had a sensitivity of 80.0% and a specificity of 96.7% (cut-off value = 0.1), RIC8B had a sensitivity of 100% and a specificity of 100% (cut-off value = 0.106), and RAB11FIP3 had a sensitivity of 96.7% and a specificity of 100% (cut-off value = 0.116) using cut-off values determined in the screening set. Box plots represent mean methylation levels. Error bars represent SD. Two-sided Student's t test (*P < 0.001) was conducted. Supplementary Figure S3 The correlations between the methylation levels obtained by the Infinium BeadChip assay and bisulfite pyrosequencing in cancer cell lines. (a) Correlation was analyzed in 17 cell lines, and the correlation coefficients between the two methods were 0.947 for SLC46A3 (P < 0.001), and 0.935 for cg09177106 (P < 0.001). (b) The correlation was analyzed in 18 cell lines, and the correlation coefficient between the two methods was 0.934 for MARCKSL1 (P < 0.001). Supplementary Figure S4 The correlations between SLC46A3 DNA methylation (RRBS) and its mRNA expression (RNA-seq) in 831 cancer cell lines from the Cancer Cell Line Encyclopedia database [correlation coefficient (R) = −0.592; P = 0.0001]. Supplementary Figure S5 Comparison of Japanese GACs, western GACs and EACs by two GAC-specific methylation markers, SLC46A3 and cg09177106. SLC46A3 and cg09177106 had significantly higher methylation levels in Japanese GACs and western GACs (white people, n = 30) than in EACs. Two-sided student’s t test was conducted. Supplementary Figure S6 Genome-wide screening of gastric adenocarcinoma (GAC)-specific and esophageal adenocarcinoma (EAC)-specific methylation markers genomic regions other than CpG islands. (a) Screening scheme of GAC-specific methylation markers. 32,055 CpG sites located in non-CpG islands unmethylated (average β value < 0.2) in leukocytes, normal esophagus, and Barrett’s esophagus were selected from 485,512 probes. Among them, 10 CpG sites had significantly higher methylation levels in 30 GACs than in 30 EACs [Δβ (GAC−EAC) > 0.2, and P < 0.001]. No CpG site was unmethylated in any of the EACs. (b) Screening scheme of EAC-specific methylation markers. 33,639 CpG sites located in non-CpG islands unmethylated in leukocytes, gastric mucosae with H. pylori infection, and those without were selected from 485,512 probes. Among them, 16 CpG sites had significantly higher methylation levels in 30 EACs than in 30 GACs [Δβ (EAC−GAC) > 0.2, and P < 0.001]. One of 16 CpG sites, DAXX, were unmethylated in all the GACs. (c) DNA methylation levels of two adjacent CpG sites and the target probe, DAXX, in the screening set. DNA methylation levels of adjacent CpG sites were similarly low in both EACs and GACs. (PPTX 240 KB)
Rights and permissions
About this article

Cite this article
Zhang, CD., Takeshima, H., Sekine, S. et al. Prediction of tissue origin of adenocarcinomas in the esophagogastric junction by DNA methylation. Gastric Cancer 25, 336–345 (2022). https://doi.org/10.1007/s10120-021-01252-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10120-021-01252-y