
Abstract
Background
Tumor samples are unavoidably contaminated with coexisting normal cells. Here, we aimed to establish a DNA methylation marker to estimate the fraction of gastric cancer (GC) cells in any DNA sample by isolating genomic regions specifically methylated in GC cells.
Methods
Genome-wide and gene-specific methylation analyses were conducted with an Infinium HumanMethylation450 BeadChip array and by quantitative methylation-specific PCR, respectively. Purified cancer and noncancer cells were prepared by laser-capture microdissection. TP53 mutation data were obtained from our previous study using next-generation target sequencing.
Results
Genome-wide DNA methylation analysis of 12 GC cell lines, 30 GCs, six normal gastric mucosae, one sample of peripheral leukocytes, and four noncancerous gastric mucosae identified OSR2, PPFIA3, and VAV3 as barely methylated in normal cells and highly methylated in cancer cells. Quantitative methylation-specific PCR using 26 independent GCs validated that one or more of them was highly methylated in all of the GCs. Using four pairs of purified cells, we confirmed the three genes were highly methylated (85 % or more) in cancer cells and barely methylated (5 % or less) in noncancer cells. The cancer cell fraction assessed by the panel of the three genes showed good correlation with that assessed by the TP53 mutant allele frequency in 13 GCs (r = 0.77). After correction of the GC cell fraction, unsupervised clustering analysis of the genome-wide DNA methylation profiles yielded clearer clustering.
Conclusions
A DNA methylation marker—namely, the panel of the three genes—is useful to estimate the cancer cell fraction in GCs.
Similar content being viewed by others


Introduction
Extensive genomic and epigenomic analyses of a variety of human cancers, including gastric cancers (GCs), have been and are being conducted [1–4]. However, these analyses are almost always affected by contamination from coexisting normal cells in primary cancer samples. Although genomic analyses are designed to detect mutations even in a small fraction of cells, they still fail to detect gene mutations in samples with a low fraction of cancer cells [5]. Moreover, epigenomic and gene expression analyses are heavily affected by the fraction of cancer cells [6]. To overcome the contamination from normal cells, laser-capture microdissection (LCM) is conducted [7, 8]. However, LCM is labor-intensive and time-consuming, and practically impossible for diffuse-type GCs.
Without purification of cancer cells, if a fraction of cancer cells in a sample can be assessed, a sample with an extremely low fraction of cancer cells can be excluded from subsequent analyses, or the data obtained may be corrected by the fraction of cancer cells. Such assessment has been generally conducted by an expert pathologist, which is time-consuming and almost impossible for diffuse-type GCs and a large number of samples. To overcome this limitation, efforts have been made to develop molecular markers. For example, cancer-cell-specific mutations identified by a single-nucleotide polymorphism microarray and next-generation sequencing can be used to assess the fraction of cancer cells [9, 10]. However, identification of such mutations must be conducted for each sample, and there is a sizable research cost for this approach.
To overcome these issues, in our recent study, we successfully isolated CpG islands specifically methylated in esophageal squamous cell carcinoma (ESCC) cells [11]. Three genes were methylated in almost all ESCC cells, but were not methylated or were barely methylated in normal esophageal mucosae, and at least one of the three genes was methylated in virtually all of 28 ESCC cases analyzed. Therefore, a panel of the three genes was considered to be a DNA methylation marker for the fraction of cancer cells. Using the marker, we were able to correct the fraction of ESCC cells, and showed that tumor-suppressor genes were methylated in almost all cancer cells.
In this study, for GCs, we aimed to isolate a DNA methylation marker that can be used to assess the fraction of cancer cells. Different from the esophagus, isolation of such a marker is far more difficult because gastric mucosae can have very high levels of DNA methylation owing to Helicobacter pylori infection [12–15], and GC samples are contaminated with such gastric mucosae. Therefore, we paid special attention to isolation of marker genes not influenced by H. pylori infection.
Materials and methods
GC cell lines and tissue samples
Cell lines KATOIII, MKN45, NUGC3, MKN74, and MKN7 were purchased from the Japanese Collection of Research Bioresources (Tokyo, Japan), and the AGS cell line was purchased from the American Type Culture Collection (Manassas, VA, USA). Cell lines HSC39, HSC57, 44As3, and 58As9 were gifted by K. Yanagihara from the National Cancer Center, the TMK1 cell line was gifted by W. Yasui from Hiroshima University, and the GC2 cell line was established by M. Tatematsu at Aichi Cancer Center Research Institute.
A total of 56 primary GC samples (32 intestinal type and 24 diffuse type) were collected from surgical specimens of patients who had undergone gastrectomy, and 30 of the samples were used for our previous studies [1, 16]. Genome-wide DNA methylation and TP53 mutation data of the 30 GCs were obtained from one of the studies [1]. Peripheral leukocyte samples were collected from five healthy volunteers by a centrifugation method. Gastric mucosae were collected by endoscopic biopsy from 17 healthy volunteers (11 without and six with present H. pylori infection) and from noncancerous gastric mucosae of 27 GC patients. Among the 27 noncancerous gastric mucosae, 23 (nine without and 14 with present H. pylori infection) were used for our previous study [17]. H. pylori infection status was analyzed by a serum anti-H. pylori IgG antibody test (SRL, Tokyo, Japan), rapid urease test (Otsuka, Tokushima, Japan), or culture test (Eiken, Tokyo, Japan).
All of the samples, except for those used for LCM, were stored in RNAlater (Applied Biosystems, Foster City, CA, USA), and genomic DNA was extracted by the phenol–chloroform method. LCM was performed using formalin-fixed paraffin-embedded primary GCs by a Leica LMD7000 system [7, 18]. This study was conducted with the approval of the Institutional Review Board of the National Cancer Center. Written informed consent was obtained from all individuals.
Genome-wide DNA methylation analysis
Genome-wide DNA methylation analysis was performed using an Infinium HumanMethylation450 BeadChip array (Illumina, San Diego, CA, USA), which assessed the degree of methylation of 485,512 CpG sites. The methylation level of each CpG site was obtained as a β value, which ranged from 0 (completely unmethylated) to 1 (completely methylated). We excluded 11,551 CpG sites on the sex chromosomes, and the remaining 473,961 CpG sites were used for the analysis. Genomic blocks were defined as collections of CpG sites classified by their locations against transcription start sites and CpG islands [1].
Gene-specific DNA methylation analysis
Gene-specific DNA methylation levels were analyzed by quantitative methylation-specific PCR (qMSP). For DNA from surgical specimens in RNAlater, 1 µg was digested with BamHI, treated with bisulfite, purified, and suspended in 40 µl of Tris (hydroxymethyl) aminomethane–EDTA buffer, as described in [19, 20]. For formalin-fixed paraffin-embedded samples collected by LCM, DNA extraction and bisulfite treatment was conducted with an EpiTect Plus bisulfite kit (Qiagen, Hilden, Germany). Quantitative methylation-specific PCR (qMSP) was performed by real-time PCR using primers specific to methylated or unmethylated DNA (Table S1), the bisulfite-treated DNA, and SYBR Green I (BioWhittaker Molecular Applications, Rockland, ME, USA). The number of molecules in a sample was determined by comparing its amplification with that of standard DNA samples that contained known numbers of molecules (101–106 molecules). On basis of the numbers of methylated and unmethylated molecules, a methylation level was calculated as the fraction of methylated molecules in the total number of DNA molecules (number of methylated molecules plus number of unmethylated molecules). As a fully methylated control, blood genomic DNA treated with SssI methylase (New England Biolabs, Beverly, MA, USA) was used. As a fully unmethylated control, blood genomic DNA amplified twice with Genomiphi (GE Healthcare, Piscataway, NJ, USA) was used [21].
Gene expression analysis
Complementary DNA was synthesized from 1 µg of total RNA using SuperScript III (Invitrogen, Carlsbad, CA, USA). Quantitative reverse transcription PCR was performed using SYBR Green I and an iCycler thermal cycler. The measured number of complementary DNA molecules was normalized to that of GAPDH. The primers and PCR conditions are shown in Table S1.
Genomic DNA copy number analysis
Copy number alteration (CNA) of a specific genomic region was analyzed by quantitative real-time PCR using an iCycler thermal cycler and SYBR Green I. RPPH1 was used as a control gene located on a chromosomal region with infrequent CNA [22]. The number of DNA molecules in a sample was measured for the control gene and three regions flanking the target gene (Table S1). The number of DNA molecules of the target gene was normalized to that of the control gene, and the normalized number of DNA molecules in a sample was compared with that in human leukocyte DNA to obtain the CNA. All the analysis was conducted in duplicate. A CNA (gain or loss) was defined as a twofold or greater increase or a 0.5-fold or smaller decrease.
Mutations of TP53 and mutant frequency
The TP53 mutation status and mutant frequency were obtained from our previous study [1]. Briefly, the mutation was analyzed by target sequencing using an Ion AmpliSeq cancer panel kit (Life Technologies, Carlsbad, CA, USA) and an Ion PGM next-generation sequencer.
Statistical analyses
The correlation was analyzed using Pearson’s product-moment correlation coefficients, and its P value was obtained by the parametric hypothesis test. A difference in the mean DNA methylation level was analyzed by Student’s t test. A result was considered significant when the P value was less than 0.05 by a two-sided test.
Results
Selection of regions specifically methylated in GCs by a genome-wide screening
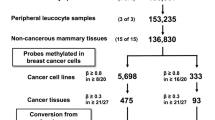
To screen specific regions not methylated in normal cells and fully methylated in GC cells using the Infinium HumanMethylation450 BeadChip array, we searched for CpG sites (1) with β ≤ 0.2 in six samples of normal gastric mucosae, one sample of peripheral leukocytes, and four samples of noncancerous mucosae, and (2) with β ≥ 0.8 in at least six of 12 GC cell lines. A total of 1,006 CpG sites were isolated from 473,961 informative CpG sites on autosomes. Then, to screen regions frequently methylated in primary GCs, CpG sites for which β ≥ 0.3 in 20 or more of 30 primary GCs [1] were searched (Fig. 1a). From the 1,006 CpG sites, 18 CpG sites derived from 16 genomic regions were isolated (Table S2). From the 16 genomic regions, PRDM16 was excluded because its gene amplification was known [23], and five other regions were also excluded because they did not have neighboring CpG islands or known genes.

Selection of specifically methylated regions by a genome-wide screening. a Specific genomic regions not methylated in normal cells and fully methylated in cancer cells were selected by a genome-wide screening using an Infinium HumanMethylation450 BeadChip array. Eighteen CpG sites derived from 16 genomic regions were isolated. b Five regions of five genes (OSR2, VAV3, PPFIA3, LTB4R2, and DIDO1) were selected because of their genomic structure and the availability of quantitative methylation-specific PCR (qMSP) primers. The genomic structure, including the location of a CpG island, transcription start site, introns, and exons, is shown at the top. The β values of the CpG sites analyzed using the bead array are shown in the middle, and the broken lines show the threshold used in the screening. A CpG map around the CpG site(s) is shown at the bottom. Vertical lines (solid or broken) show CpG sites, with broken lines showing CpG sites whose β values were measured by the bead array. Arrows show locations of primers for qMSP. M methylated, U unmethylated
For the remaining ten regions, we attempted to design primers for qMSP, and primers for both methylated and unmethylated DNA were successfully designed for five regions of five genes (OSR2, VAV3, PPFIA3, LTB4R2, and DIDO1) (Fig. 1b). To confirm the genome-wide DNA methylation data obtained by the bead array, qMSP was conducted using the 12 GC cell lines mentioned in “GC cell lines and tissue samples” and one sample of peripheral leukocytes. DIDO1 had slight methylation in the peripheral leukocytes, and was excluded from further analysis. The methylation levels of the other four genes (LTB4R2, OSR2, VAV3, and PPFIA3) obtained by qMSP were in good accordance with the bead array data (Fig. S1).
Isolation of genes not influenced by H. pylori infection
Gastric mucosae with H. pylori infection are known to have very high DNA methylation levels [12, 13]. To exclude genes influenced by H. pylori infection, the methylation levels of the four genes were analyzed in 23 gastric mucosa samples of H. pylori-positive (n = 14) and H. pylori-negative (n = 9) individuals, as well as four samples of peripheral leukocytes different from the one used for the initial screening. The LTB4R2 methylation level in the H. pylori-positive individuals was higher than that in the H. pylori-negative individuals and the four samples of peripheral leukocytes, showing that the LTB4R2 methylation level was affected by H. pylori infection. On the other hand, OSR2, VAV3, and PPFIA3 were almost unmethylated in the three groups (Fig. 2).

Isolation of genes not influenced by Helicobacter pylori infection. Methylation levels of the four genes were analyzed by quantitative methylation-specific PCR in noncancerous gastric mucosae of H. pylori-positive (n = 14) and H. pylori-negative (n = 9) individuals, as well as four samples of peripheral leukocytes. LTB4R2 was excluded because its methylation level was higher in the H. pylori-positive individuals than in the H. pylori-negative individuals
We also analyzed the expression of OSR2, VAV3, and PPFIA3 using 17 normal gastric mucosa samples of H. pylori-positive (n = 11) and H. pylori-negative (n = 6) individuals. VAV3 was highly expressed in both H. pylori-positive and H. pylori-negative gastric mucosae, whereas OSR2 and PPFIA3 were only weakly expressed (Fig. S2).
High incidence of methylation of the three genes and their specificity using LCM-purified cells
To examine the incidence of methylation of the three genes in primary GCs, we performed qMSP using 26 independent primary GCs, and observed that at least one of the three genes was methylated in all of the 26 GCs (Fig. 3a). These data showed that if these three genes were used as a panel, they would have a higher coverage (100 %) of primary GCs.

High incidence of methylation of the three genes and specificity of methylation using cells purified by laser-capture microdissection (LCM). a The incidence of hypermethylation of the three genes was analyzed in 26 independent primary gastric cancers (GCs) by quantitative methylation-specific PCR. At least one of the three genes was methylated in all of the 26 GCs. b Methylation levels of the three genes were analyzed in four primary GCs before LCM and four pairs of purified cancer and noncancer cells after LCM. At least one of the three genes was highly methylated in GC cells (more than 85 %), but all the three genes were barely methylated in noncancer cells (less than 5 %). Dotted rectangles show the panel of the three genes as a DNA methylation marker
To confirm that the three genes were highly methylated only in GC cells but not in coexisting noncancer cells, four pairs of cancer and noncancer cells were collected by LCM. We found that at least one of the three genes was highly methylated in GC cells (more than 85 %), but that all of them were barely methylated in noncancer cells (less than 5 %) (Fig. 3b). The highest methylation level of the three genes was considered to reflect the fraction of cancer cells, and we defined the panel of the three genes as a DNA methylation marker to estimate the cancer cell fraction in a GC sample.
Because DNA methylation levels of some genes can be influenced by age [24], we also analyzed the correlation between the methylation of the three genes and age. The methylation levels of the three genes were found to be independent of age (Fig. S3).
CNAs of the three genes
CNAs of a marker gene can affect the methylation level of its region in cancer samples [25]. Therefore, we analyzed CNAs of the three regions in the 20 GCs used for the bead array analysis (Fig. 4). VAV3 and PPFIA3 showed no CNAs of more than twofold or less than 0.5-fold. In contrast, OSR2 showed CNAs at low frequencies (more than twofold in one GC and less than 0.5-fold in two GCs). It was calculated that the deviation of the methylation level from the true cancer cell fraction would be 17.2 % when twofold or 0.5-fold CNA was present in cancer cells [11]. Therefore, the effect of the CNA of OSR2 was considered to be minimal in the estimation of the cancer cell fraction.

Copy number alterations (CNAs) of the three genes. CNA of the three genes was analyzed by real-time PCR of the 20 gastric cancers (GCs) used for the bead array analysis. Significant CNA (gain or loss) was defined as a twofold or greater increase or a 0.5-fold or smaller decrease, respectively. Only OSR2 showed CNAs at low frequencies (twofold or greater in one GC; 0.50-fold or smaller in two GCs)
Correlation between the cancer cell fraction estimated by DNA methylation and that estimated by a genetic alteration
To evaluate the accuracy of the DNA methylation marker, 13 GCs with TP53 mutation were identified among the 30 GCs used for the bead array analysis, and the cancer cell fraction estimated by the marker was compared with the TP53 mutant frequency. A high correlation between the two methods was observed (r = 0.77, P < 0.001; Fig. 5). This result showed that the cancer cell fraction estimated by the DNA methylation marker accurately reflected the true fraction of cancer cells in a tumor sample.

Correlation between the cancer cell fraction estimated by DNA methylation and that estimated by a genetic alteration. The cancer cell fraction estimated by the DNA methylation marker was compared with the TP53 mutant frequency. A high correlation between the two methods was observed (r = 0.77, P < 0.001)
Application of the DNA methylation marker to correction of the bead array data
We applied the DNA methylation marker to correct the influence of contamination by normal cells in the data from the epigenomic analysis. For the 30 primary GCs used for the bead array analysis, we measured the fraction of cancer cells using the marker, and corrected the bead array data by division with the evaluated fraction. Unsupervised hierarchical clustering analysis was conducted using 263 genomic blocks selected because their downstream genes were silenced by aberrant methylation [1] (Fig. 6b). Compared with the heatmap before the correction (Fig. 6a), two samples, S20T and S22T, moved from the CpG island methylator phenotype (CIMP)-negative group to the CIMP-high group. The cancer cell fraction in these two samples was less than 20 % (Fig. 3a). After exclusion of these two samples and correction of the methylation levels, the clustering of the CIMP-high, CIMP-moderate, CIMP-low, and CIMP-negative GCs became much clearer (Fig. 6c). From these data, we concluded that the DNA methylation marker could be used to identify and exclude samples with an extremely low fraction of cancer cells, and to correct the molecular data.

Application of the DNA methylation marker to the correction of the bead array data. a Unsupervised hierarchical clustering analysis of the 30 primary gastric cancers using DNA methylation profiles of 263 genomic blocks. b Two samples surrounded by a red square (S20T and S22T) moved from the CpG island methylator phenotype (CIMP)-negative group to the CIMP-high group after the Infinium HumanMethylation450 BeadChip array data had been corrected by the DNA methylation marker. c After exclusion of two samples with a low fraction of cancer cells, a heatmap using the corrected bead array data showed a much clearer clustering of CIMP-high, CIMP-moderate, CIMP-low, and CIMP-negative gastric cancers
Discussion
We successfully established a panel of three genes (OSR2, VAV3, and PPFIA3) as a marker to estimate the fraction of cancer cells in primary GCs. Using the DNA methylation marker, we were also able to identify and exclude samples with a low fraction of cancer cells, and to correct the methylation levels by the fraction of cancer cells. After this, the genome-wide DNA methylation profiles yielded clearer clustering of CIMP by unsupervised hierarchical clustering analysis. This is the first molecular marker for the cancer cell fraction in GC.
The DNA methylation marker has the advantages of simplicity without the need for experienced pathologists or paired normal samples, compared with microscopic examination and genomic alterations. Also, the DNA methylation marker is likely to have a broad coverage in primary GCs because the DNA methylation marker was methylated in 100 % of the 26 primary GCs used for validation. Further, we were easily able to use the DNA methylation marker to assess the cancer cell fraction, even in diffuse-type GCs, for which even an expert pathologist has difficulty in estimating the cancer cell fraction. Finally, since the methylation levels of the three genes were independent of age, this marker was regarded to be useful to estimate the cancer cell fraction irrespective of age.
The correlation of the cancer cell fraction estimated by the DNA methylation marker with TP53 mutant frequency was high (r = 0.77, P < 0.001). However, in two samples, the cancer cell fraction estimated by the marker was twice as large as that estimated by the TP53 mutant frequency. Since loss of heterozygosity can coexist with a mutation of TP53 in GCs, we speculated that the discrepancy between the two methods in the two GC samples might have been caused by the loss of heterozygosity of TP53.
Gastric mucosae, especially when infected with H. pylori, can have very high levels of DNA methylation, so we paid special attention to isolation of marker genes in this study. The panel of the three genes was not affected by H. pylori infection because the genes were barely methylated in H. pylori-positive mucosae. Only two samples in H. pylori-negative individuals had a high methylation of VAV3 or PPFIA3, respectively. One possible reason for detection of such high methylation levels in H. pylori-negative samples is that these two samples were contaminated with cancer cells because they were resected from samples from GC patients. Another possible reason is that they were methylated in noncancer cells during past H. pylori infection.
A CNA can affect the methylation level of a marker gene. Therefore, we analyzed the CNAs of the three genes in 20 primary GCs used for the bead array analysis, and found CNAs of the three genes had little influence on the estimation of the cancer cell fraction. Regarding the expression of the three marker genes, only VAV3 was highly expressed in normal gastric mucosae. The region of VAV3, for which DNA methylation was analyzed, was outside the nucleosome-free region, suggesting that its transcription is not necessarily suppressed by the methylation.
In summary, a DNA methylation marker—namely, the panel of the three genes—was isolated, and was shown to be qualified to estimate the cancer cell fraction in GCs. Application of the marker to correction of the bead array data showed promising results for improving the accuracy of molecular analysis. The DNA methylation marker is expected to be useful in many aspects of GC research.
References
Kim JG, Takeshima H, Niwa T, Rehnberg E, Shigematsu Y, Yoda Y, et al. Comprehensive DNA methylation and extensive mutation analyses reveal an association between the CpG island methylator phenotype and oncogenic mutations in gastric cancers. Cancer Lett. 2013;330:33–40.
Letouze E, Martinelli C, Loriot C, Burnichon N, Abermil N, Ottolenghi C, et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell. 2013;23:739–52.
Mudbhary R, Hoshida Y, Chernyavskaya Y, Jacob V, Villanueva A, Fiel MI, et al. UHRF1 overexpression drives DNA hypomethylation and hepatocellular carcinoma. Cancer Cell. 2014;25:196–209.
Oue N, Mitani Y, Motoshita J, Matsumura S, Yoshida K, Kuniyasu H, et al. Accumulation of DNA methylation is associated with tumor stage in gastric cancer. Cancer. 2006;106:1250–9.
Roma C, Esposito C, Rachiglio AM, Pasquale R, Iannaccone A, Chicchinelli N, et al. Detection of EGFR mutations by TaqMan mutation detection assays powered by competitive allele-specific TaqMan PCR technology. Biomed Res Int. 2013;2013:385087.
Meyerson M, Gabriel S, Getz G. Advances in understanding cancer genomes through second-generation sequencing. Nat Rev Genet. 2010;11:685–96.
Joseph A, Gnanapragasam VJ. Laser-capture microdissection and transcriptional profiling in archival FFPE tissue in prostate cancer. Methods Mol Biol. 2011;755:291–300.
Lin J, Marquardt G, Mullapudi N, Wang T, Han W, Shi M, et al. Lung cancer transcriptomes refined with laser capture microdissection. Am J Pathol. 2014;184(11):2868–84.
Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–21.
McFadden DG, Papagiannakopoulos T, Taylor-Weiner A, Stewart C, Carter SL, Cibulskis K, et al. Genetic and clonal dissection of murine small cell lung carcinoma progression by genome sequencing. Cell. 2014;156:1298–311.
Takahashi T, Matsuda Y, Yamashita S, Hattori N, Kushima R, Lee YC, et al. Estimation of the fraction of cancer cells in a tumor DNA sample using DNA methylation. PLoS One. 2013;8:e82302.
Maekita T, Nakazawa K, Mihara M, Nakajima T, Yanaoka K, Iguchi M, et al. High levels of aberrant DNA methylation in Helicobacter pylori-infected gastric mucosae and its possible association with gastric cancer risk. Clin Cancer Res. 2006;12:989–95.
Ushijima T, Hattori N. Molecular pathways: involvement of Helicobacter pylori-triggered inflammation in the formation of an epigenetic field defect, and its usefulness as cancer risk and exposure markers. Clin Cancer Res. 2012;18:923–9.
Kang GH, Lee S, Cho NY, Gandamihardja T, Long TI, Weisenberger DJ, et al. DNA methylation profiles of gastric carcinoma characterized by quantitative DNA methylation analysis. Lab Invest. 2008;88:161–70.
Lu ZM, Zhou J, Wang X, Guan Z, Bai H, Liu ZJ, et al. Nucleosomes correlate with in vivo progression pattern of de novo methylation of p16 CpG islands in human gastric carcinogenesis. PLoS One. 2012;7:e35928.
Yoda Y, Takeshima H, Niwa T, Kim JG, Ando T, Kushima R, et al. Integrated analysis of cancer-related pathways affected by genetic and epigenetic alterations in gastric cancer. Gastric Cancer 2015;18:65–76.
Okochi-Takada E, Hattori N, Tsukamoto T, Miyamoto K, Ando T, Ito S, et al. ANGPTL4 is a secreted tumor suppressor that inhibits angiogenesis. Oncogene. 2014;33:2273–8.
Tretiakova M, Hart J. Laser microdissection for gene expression study of hepatocellular carcinomas arising in cirrhotic and non-cirrhotic livers. Methods Mol Biol. 2011;755:233–44.
Yamashita S, Takahashi S, McDonell N, Watanabe N, Niwa T, Hosoya K, et al. Methylation silencing of transforming growth factor-β receptor type II in rat prostate cancers. Cancer Res. 2008;68:2112–21.
Wanajo A, Sasaki A, Nagasaki H, Shimada S, Otsubo T, Owaki S, et al. Methylation of the calcium channel-related gene, CACNA2D3, is frequent and a poor prognostic factor in gastric cancer. Gastroenterology. 2008;135:580–90.
Asada K, Ando T, Niwa T, Nanjo S, Watanabe N, Okochi-Takada E, et al. FHL1 on chromosome X is a single-hit gastrointestinal tumor-suppressor gene and contributes to the formation of an epigenetic field defect. Oncogene. 2013;32:2140–9.
Kolacsek O, Krizsik V, Schamberger A, Erdei Z, Apati A, Varady G, et al. Reliable transgene-independent method for determining Sleeping Beauty transposon copy numbers. Mob DNA. 2011;2:5.
Man TK, Lu XY, Jaeweon K, Perlaky L, Harris CP, Shah S, et al. Genome-wide array comparative genomic hybridization analysis reveals distinct amplifications in osteosarcoma. BMC Cancer. 2004;4:45.
Maegawa S, Hinkal G, Kim HS, Shen L, Zhang L, Zhang J, et al. Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res. 2010;20:332–40.
Robinson MD, Stirzaker C, Statham AL, Coolen MW, Song JZ, Nair SS, et al. Evaluation of affinity-based genome-wide DNA methylation data: effects of CpG density, amplification bias, and copy number variation. Genome Res. 2010;20:1719–29.
Acknowledgment
This work was supported by the Applied Research for Innovative Treatment of Cancer (H26-019) from the Ministry of Health, Labour and Welfare.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zong, L., Hattori, N., Yoda, Y. et al. Establishment of a DNA methylation marker to evaluate cancer cell fraction in gastric cancer. Gastric Cancer 19, 361–369 (2016). https://doi.org/10.1007/s10120-015-0475-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10120-015-0475-2